Introduction
Chronic myeloid leukemia (CML) is a clonal disease
of pluripotent hematopoietic stem cells that is characterized by
the Philadelphia (Ph) chromosome with the reciprocal translocation
t(9;22)(q34;q11). At diagnosis, the majority of cases (85%) exhibit
the typical Ph chromosome as the only cytogenetic finding. A few
cases either harbor variant translocations [t(v;22)] or cryptic
rearrangement (1).
However, additional chromosome abnormalities (ACAs)
are a recurring event associated with clonal evolution. An extra
Ph, +8, i(17q) and +19 have been described as the most common
secondary changes (known as the major route), whereas others
infrequent changes have been labeled as the minor route. The major
route of ACAs is usually detected during disease progression into
the accelerated phase or blast crisis (2). At diagnosis, this has been documented
with low frequency (<5% of cases) (3). These aberrations have been reported as
an independent prognostic factor with a negative impact on the
cytogenetic and molecular response (4,5). The
t(1;11)(q21;q23) translocation has been described at diagnosis in
certain cases of pediatric acute myeloid leukemia or myelomonocytic
leukemia, with a poor prognosis, but the subgroup with this
rearrangement suggests an uncertain impact in the outcome of acute
leukemia (6).
The present study reports a rare case of CML with a
cryptic Ph chromosome and a t(1;11)(q21;q23) translocation at
diagnosis, with unfavourable responses, even though the patient
received appropriate treatments. Furthermore, a T315I mutation was
later detected in the patient.
Case report
In June 2000, a 47-year-old female with a CML
diagnosis was referred to the Hematology Research Institute
‘Mariano R. Castex’, National Academy of Medicine (Buenos Aires,
Argentina) for further studies and treatment. The clinical records
of the patient indicated the onset of the disease in 1998, when the
patient was 45 years old, with a cytogenetic study showing the
translocation t(1;11)(q21;q23). The patient was treated with
hydroxyurea (1 g/day).
In the first consultation at the current institute,
the patient presented with fatigue, weakness, fever and
splenomegaly (4 cm below the costal margin). Laboratory results
were as follows: Hematocrit, 36% (normal range, 36–46%);
haemoglobin, 11 g/dl (normal range, 11.5–15.5 g/dl); white blood
cells (WBC), 11.2×109/l (normal range,
4.5–10.5×109/l); platelets, 5.90×1011/l
(normal range, 1.5–4.0×1011/l); reticulocytes, 1.2%
(normal range, 0.9–5.5%); LDH, 205 U/l (normal range, 100–200 U/l).
Bone marrow biopsy revealed hyper-cellularity with granulocytic
hyperplasia, which was consistent with a diagnosis of chronic phase
CML.
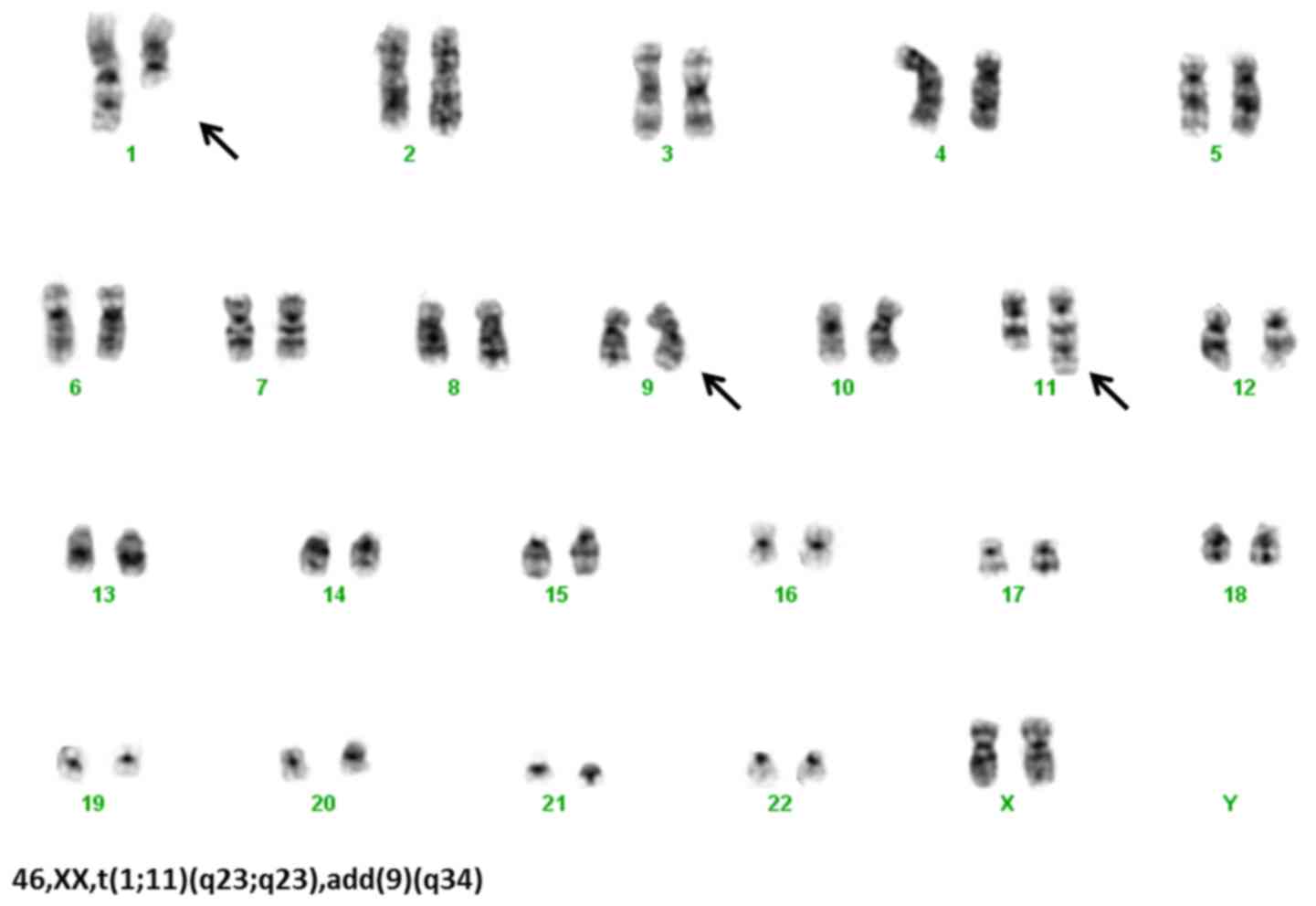
The cytogenetic analysis by G banding (7) confirmed t(1;11)(q21;q23), add(9)(q34) and a cryptic Ph chromosome (Fig. 1) [detected by fluorescence in
situ hybridization (FISH) simple fusion using DAKO DNA Probes;
Dako, Glostrup, Denmark]. The molecular analysis by reverse
transcription-polymerase chain reaction (RT-PCR) (8) showed the chimeric breakpoint cluster
region-Abelson murine leukemia viral oncogene homolog 1
(BCR-ABL1) mRNA transcripts. To determine whether the
chromosome marker t(1;11) was of constitutional origin, a
phytohemaglutinin-stimulated peripheral blood culture was
performed. A normal karyotype was detected, which excluded the
constitutional origin and indicated clonality.
The patient started treatment with hydroxyurea (1
and 2 g on alternate days) and interferon α (4.5 MU/day) until 2002
when interferon therapy was discontinued due to intolerance. With
these treatments, the patient only achieved a complete
haematological response. On September 2005, the patient started
treatment with imatinib (400 mg/day) and mild adverse events were
observed, including facial and lower limb edema, and a minor
cutaneous rash.
The patient achieved a hematological response
without a cytogenetic and molecular response after 8 months of
therapy. In October 2006, a laboratory analysis showed the
following: Hematocrit, 41.2%; hemoglobin, 12.5%; WBC,
9.3×109/l; and platelets, 9.50×1011/l. The
dose of imatinib was increased to 600 mg/day and then to 800 mg/day
in 2007 for better control of the thrombocytosis and to enhance the
responses.
In May 2009, due to increased platelets counts of
1.39×1012/l and the absence of a major cytogenetic
response, the treatment was switched to 800 mg/day nilotinib. After
15 months of treatment, the patient achieved a complete
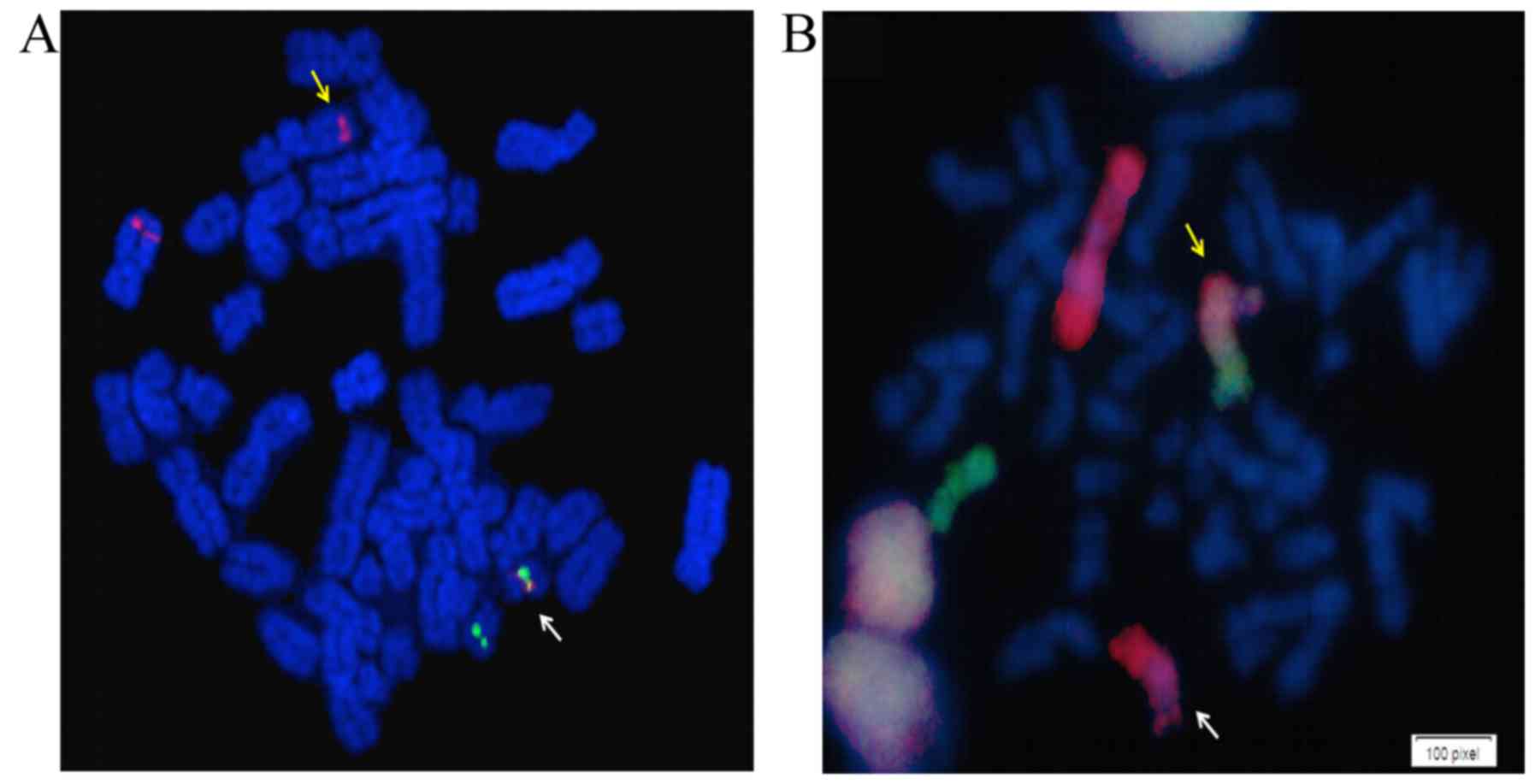
hematological response, and FISH studies showed 69% of cells
positive for BCR-ABL1 (BCR-ABL1 dual fusion-dual
color, LIVe Probes; Lexel, Buenos Aires, Argentina) (Fig. 2A). Quantification by RT-quantitative
(q)PCR (MolecularMD® One-Step qRT-PCR BCR-ABL kit),
according to the International Scale, gave a result of 8.5%,
representing a minimal molecular response.
In October 2010, following adequate treatment
adherence, the patient maintained the complete hematological
response and exhibited reduced levels of BCR-ABL1
transcripts at 0.84% (minor molecular response). Treatment was
sustained between 2011 and 2013 with good tolerance, but without
significant changes in cytogenetic and molecular responses (values
range from 20.0–24.5% of Ph-positive cells by FISH analysis and
3.23–5.00% of BCR-ABL1 transcripts by RT-qPCR). At the end
of 2013, pegylated interferon (80 µcg/15 days) was added to
treatment with nilotinib in order to improve responses, but not
better outcome was observed.
In 2014, the patient's condition worsened, with
levels of BCR-ABL1 transcript reaching of 12.3% and with 71%
Ph-positive cells, as determined by FISH. The treatment was
switched to dasatinib (50 mg/day). In June 2015, cytogenetic
control follow-up showed the karyotype 46,XX,t(1;11)(q21;q23),
add(9)(q34) in all metaphases and
RT-qPCR detected 100% of BCR-ABL1 transcripts.
An obstructed carotid artery was diagnosed during
the control follow-up. After 2 months, the screening for mutations
in the ABL1 gene by direct sequencing (9) detected the T315I mutation. Treatment
with tyrosine kinase inhibitor (TKI) was suspended. A carotid
artery angioplasty was performed and omacetaxine was prescribed at
an induction dose (1.25 mg/m2) by subcutaneous injection
twice daily for 14 consecutive days. Grade 3 anemia and grade 2
thrombocytopenia were observed, and the patient received blood red
cells transfusions. With a delay of 6 days, a second cycle was
administered at the same dosage but for 9 days only. To date, the
patient has progressed to blast crisis and is receiving
transfusions and supportive care for febrile neutropenia, and
chemotherapy is under evaluation. Bone marrow transplantation was
not an option, as the only sibling available as a donor for stem
cell transplant was unfit due to histocompatibility and limiting
comorbidities.
In order to assess if MLL [also known as
lysine methyltransferase 2A (KMT2A)] was affected by the
t(1;11)(q21;q23) translocation, FISH was performed using a dual
color-split signal probes (Cytocell Aquarius probes; Cytocell,
Cambridge, UK). The pattern of signal observed was not consistent
with alterations in this gene (data not shown), indicating that the
MLL gene was not split by the t(1;11) translocation.
Finally, to determine if the cryptic Ph chromosome
was the result of a variant translocation, involving chromosomes 1,
9, 11 and 22, a whole chromosome painting (WCP) of chromosomes 1
and 11 was performed (WCP LIVe Probes; Lexel), as previously
described (10). The results
indicated that the t(1;11)(q21;q23) translocation and cryptic Ph
chromosome had an independent origin (Fig. 2B). The FISH analysis could not
determine the nature of the extra material in add(9q).
The experiments performed in this study were
approved by the Ethics Committee of the Institutes of the National
Academy of Medicine (Buenos Aires, Argentina) and the patient
provided written informed consent for the publication of the
study.
Discussion
In CML patients, ACAs in Ph-positive cells affect
the progression and response to treatment according to the
chromosome aberration and time of appearance. The European
LeukemiaNet guidelines suggest that the presence of ACAs at
diagnosis may represent a ‘warning’ feature, requiring careful
monitoring (11). ACAs at diagnosis
have been observed in ~5% of cases and are associated with clonal
evolution, the mechanism of resistance and an adverse prognosis
under treatment with imatinib (3). A
previous study showed that the presence of ACAs in the early
chronic phase was one of the independent adverse predictors for
progression within a year (12).
Fabarius et al reported that only the major route ACAs [+8,
+Ph and i(17q)] at diagnosis are associated with a negative impact
on progression-free and overall survival times (13). However, the impact of ACAs at
diagnosis and their prognostic significance are issues that remain
under discussion.
The present study reports the case of a CML patient
who presented with a reciprocal translocation between chromosomes 1
and 11 [t(1;11)(q21;q23)], add(9q34) and a cryptic Ph chromosome at
diagnosis. Genetic alterations in 11q23 are frequent in
hematological malignancies, particularly in acute leukemia
(6). These alterations affect the
MLL gene in ~85% of cases and are associated with an adverse
prognosis (14). In CML, alterations
involving 11q23 and affecting MLL are rare, and have been
reported only sporadically (15–19).
However, this translocation could affect other genes such as CBL,
generating a new chimeric gene that could deregulate the cell cycle
and affect the treatment response (20,21).
Through use of BCR-ABL1 dual fusion-dual
color FISH probes (LIVe Probes; Lexel), as previously described
(22), the present study found the
absence of a second fusion signal on der (9), keeping only the residual signal of the
ABL1 gene, which is indicative of the deletion of the
BCR sequence on der (9)
(Fig. 2A). This deletion was
confirmed by molecular analysis of the reciprocal transcript
ABL1-BCR (data not shown). Deletions on der (9) have been reported in several studies in
association with a bad response prior to the TKI era (6,11,23).
One of the most important mechanisms of resistance
is via acquired mutations in the tyrosine kinase domain of
ABL1. The T315I mutation was detected in the present patient
during the last year of follow-up. This mutation is associated with
the resistance to the majority of TKIs (24,25).
Although a long overall survival time (>15 years)
was experienced in the present study, the CML patient has since
progressed to a blast crisis. At present, the patient is receiving
transfusions and supportive care for severe febrile neutropenia.
Throughout the course of the disease, only a hematological response
was achieved, with an occasional partial cytogenetic response and a
lack of a molecular response, despite an increased dose of imatinib
(from 400 to 600 to 800 mg/day) and a switch to second-generation
TKIs (nilotinib and dasatinib). In the early stages, the
t(1;11)(q21;q23) may have played a negative role in the outcome.
Whether this atypical translocation may trigger the emergence of
the T315I mutation CML is a matter for further investigation.
Acknowledgements
This study was supported by the National Council of
Scientific and Technical Research (CONICET), the National Agency
for Scientific and Technological Promotion (ANPCyT) and Roemmers
Foundation.
References
|
1
|
Kurzrock R, Kantarjian HM, Druker BJ and
Talpaz M: Philadelphia chromosome-positive leukemias: From basic
mechanisms to molecular therapeutics. Ann Intern Med. 138:819–830.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heim S and Mitelman F: Chronic Myeloid
LeukemiaCancer cytogenetics. 3rd. Wiley Blackwell, John Wiley &
Sons, Inc.; Hoboken, NJ: pp. 179–208. 2009
|
|
3
|
Luatti S, Castagnetti F, Marzocchi G,
Baldazzi C, Gugliotta G, Iacobucci I, Specchia G, Zanatta F,
Rege-Cambrin G, Mancini M, et al: Additional chromosomal
abnormalities in Philadelphia-positive clone: Adverse prognostic
influence on frontline imatinib therapy: A GIMEMA. Working Party on
CML analysis. Blood. 120:761–767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sokal JE, Gomez GA, Baccarani M, Tura S,
Clarkson BD, Cervantes F, Rozman C, Carbonell F, Anger B and
Heimpel H: Prognostic significance of additional cytogenetic
abnormalities at diagnosis of Philadelphia chromosome-positive
chronic granulocytic leukemia. Blood. 72:294–298. 1998.
|
|
5
|
Baccarani M, Cortes J, Pane F,
Niederwieser D, Saglio G, Apperley J, Cervantes F, Deininger M,
Gratwohl A, Guilhot F, et al: Chronic myeloid leukemia: An update
of concepts and management recommendations of European LeukemiaNet.
J Clin Oncol. 27:6041–6051. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
De Braekeleer E, Meyer C, Douet-Guilbert
N, Morel F, Le Bris MJ, Berthou C, Arnaud B, Marschalek R, Férec C
and De Braekeleer M: Complex and cryptic chromosomal rearrangements
involving the MLL gene in acute leukemia: A study of 7 patients and
review of the literature. Blood Cells Mol Dis. 44:268–274. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Czepulkowski B, Bhatt B and Rooney D:
Analysis of Chromosomes from Bone marrow and Leukaemic Blood. In:
Human cytogenetics. A practical approachMalignancy and acquired
abnormalities. 2. 2nd. Oxford University Press; New York, NY: pp.
1–25. 1992
|
|
8
|
van Dongen JJ, Macintyre EA, Gabert JA,
Delabess E, Rossi V, Saglio G, Gottardi E, Rambaldi A, Dotti G,
Griesinger F, et al: Standardized RT-PCR analysis of fusion gene
transcripts from chromosome aberrations in acute leukemia for
detection of minimal residual disease. Report of the BIOMED-1
Concerted Action: Investigation of minimal residual disease in
acute leukemia. Leukemia. 13:1901–1928. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance STI-571
Cancer Therapy caused by BCR ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stedum SV and King W: Basic FISH
Techniques and Troubleshooting. In: Molecular cytogenetics:
Protocols and applicationsMethods in Molecular Biology. Fan YS:
204. Humana Press; Totowa, NJ: pp. 51–67. 2002, PubMed/NCBI
|
|
11
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marin D, Milojkovic D, Olavarria E,
Khorashad JS, de Lavallade H, Reid AG, Foroni L, Rezvani K, Bua M,
Dazzi F, et al: European LeukemiaNet criteria for failure or
suboptimal response reliably identify patients with CML in early
chronic phase treated with imatinib whose eventual outcome is poor.
Blood. 112:4437–4444. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fabarius A, Kalmanti L, Dietz CT, Lauseker
M, Rinaldetti S, Haferlach C, Göhring G, Schlegelberger B,
Jotterand M, Hanfstein B, et al: Impact of unbalanced minor route
versus major route karyotypes at diagnosis on prognosis of CML. Ann
Hematol. 94:2015–2024. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rowley JD: The Critical role of chromosome
translocations in human leukemias. Annual Review of Genetics.
32:495–519. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Collongen Rame MA: The t(1;11)(q21;q23).
Atlas Genet Cytogenet Oncol Haematol. 10:125–126. 2006.
|
|
16
|
Sessarego M, Frassoni F, Defferrari R,
Bacigalupo A, Fugazza G, Mareni C, Bruzzone R, Dejana A and Ajmar
F: Karyotype evolution of Ph positive chronic myelogenous leukemia
patients relapse in advanced phases of the disease after allogeneic
bone marrow transplantation. Cancer Genet Cytogenetic. 57:69–78.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dastugue N, Duchayne E, Huguet F, Demur C,
Plaisancie H, Calvas P, Bourrouillou G, Pris J and Colombies P:
t(9;11)(p22;q23) translocation in blastic phase of chronic myeloid
leukemia. Cancer Genet Cytogenet. 63:37–42. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Ritterbach J, Harbott J, Schroyens
W, Lohmeyer J, Pralle H and Lampert F: Blastic phase chronic
myeloid leukemia with a four-break rearrangement:
t(11;9)(9;22)(q23;p22;q34;q11). Cancer Genet Cytogenet. 68:131–134.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Otero L, Moellmann AC, Pombo-de-Oliveira
MS, Ornellas MH, Pires V, Bouzas LF and Tde Fernandez S: Additional
t(1;11)(p21;q23) with mixed lineage leukemia rearrangement in
T-blastic crisis of a Ph-positive chronic myeloid leukemia. Eur J
Haematol. 79:179–181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mikhail FM, Sinha KK, Saunthararajah Y and
Nucifora G: Normal and transforming functions of RUNX1: A
perspective. J Cell Physiol. 207:582–593. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Klampfl T, Milosevic JD, Puda A,
Schönegger A, Bagienski K, Berg T, Harutyunyan AS, Gisslinger B,
Rumi E, Malcovati L, et al: Complex patterns of chromosome 11
aberrations in myeloid malignancies Target CBL, MLL, DDB1 and LMO2.
PLoS One. 8:e778192013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dewald G: Interphase fish studies of
chronic myeloid leukemia. In: Molecular cytogenetics: Protocols and
applicationsMethods in molecular biology. Fan YS: 204. Humana
Press; Totowa, NJ: pp. 311–342. 2002, PubMed/NCBI
|
|
23
|
Huntly BJ, Bench A and Green AR: Double
Jeopardy from a single translocation: Deletions of the derivative
chromosome 9 in chronic myeloid leukemia. Blood. 102:1160–1168.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Soverini S, Colarossi S, Gnani A, Rosti G,
Castagnetti F, Poerio A, Iacobucci I, Amabile M, Abruzzese E,
Orlandi E, et al: Contribution of ABL kinase domain mutations to
imatinib resistance in different subsets of Philadelphia-positive
patients: By the GIMEMA 213 Working Party on Chronic Myeloid
Leukemia. Clin Cancer Res. 12:7374–7379. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Redaelli S, Piazza R, Rostagno R,
Magistroni V, Perini P, Marega M, Gambacorti-Passerini C and
Boschelli F: Activity of bosutinib, dasatinib, and nilotinib
against 18 imatinib-resistant bcr/abl mutants. J Clin Oncol.
27:469–471. 2009. View Article : Google Scholar : PubMed/NCBI
|