Introduction
Tongue squamous cell carcinoma is one of the most
common cancers in the neck and head (1), which often results in short survival and
poor prognosis, even after surgery and chemotherapy. Previous
studies have revealed that TSCC is associated with a high incidence
of tobacco abuse. However, the mechanism for how tobacco promotes
the progression of TSCC is still unclear.
Nicotine, the addictive component in cigarettes, has
been shown to promote tumor cell proliferation and metastasis by
promoting cell-cycle progression, epithelial-to-mesenchymal
transition (EMT), migration, invasion, angiogenesis, and
anti-apoptosis through several signaling pathways (2). Nicotine is thought to promote tumor
progression by binding to nicotinic acetylcholine receptors and
stimulate multiple cancer-promoting signaling cascades, such as
VEGFR, JAK/STAT, PI3K/Akt, and Ras/Raf/MEK/ERK signaling pathways
(3–5).
Specifically, it is been shown that nicotine stimulation can induce
proliferation, angiogenesis and EMT in non-small cell lung cancer
cells and esophageal squamous cell cancer (6–8). Nicotine
can even promote EMT via Wnt/β-catenin signaling in normal human
airway epithelial cells and human alveolar interstitial fibroblasts
(9,10). In these recent studies, nicotine has
been found to contribute to the progression and metastasis of tumor
cells or normal cells by activation of several signaling
pathways.
So far several studies have investigated the
physiologic nicotine concentration of tobacco consumers and find
that nicotine is present in the plasma and saliva at levels ranging
from 0.03 to 0.5 µM (11), and 0.6 µM
to 10 mM (12), respectively.
Epithelial cells act as a mechanical barrier to reduce a portion of
the nicotine and make it impossible to reach 10 mM in the tongue
tissue. In many experiments about nicotine stimulation to lung
tissue, various concentrations of nicotine have been used (6,7,9,10,13,14). They
chose 1, 5, 0.08 or 6 µM to conduct subsequent functional
experiments. Combining the physiological plasma and saliva nicotine
concentration and above previous studies about the effects of
nicotine to lung tissue, we carefully selected to use nicotine from
0.1 to 10 µM in our study.
The effects of nicotine stimulation on TSCC are
unknown, and the underlying mechanism is not clear. According to
previous studies, the activation of canonical Wnt/β-catenin
signaling promotes invasion, proliferation and anti-apoptosis in
TSCC cells (15–17). Thus we have attempted to explore the
possible connection between nicotine stimulation and activation of
the Wnt/β-catenin pathway in TSCC. In addition, the PI3 K/AKT
pathway can be activated after nicotine stimulation, and the
Wnt/PCP pathway can crosstalk with the PI3k/AKT pathway and
Wnt/β-catenin pathway (18).
Therefore, we aimed to determine whether Wnt/PCP pathway is also
affected by nicotine stimulation in TSCC.
Materials and methods
Antibodies and reagents
Rabbit monoclonal β-catenin, Ror2 (tyrosine-protein
kinase transmembrane receptor Ror2) and c-Myc antibodies were
purchased from Abcam Inc. (Cambridge, MA, USA; cat. nos. ab32572,
ab92379 and ab32072). Rabbit monoclonal phospho-Akt (Ser473) and
phospho-GSK-3β antibodies were from Cell Signaling Technologies
(Beverly, MA, USA; cat. nos. 4060 and 5558). Rabbit polyclonal
E-cadherin, Wnt5a and c-jun antibodies were obtained from Wanlei
Bio (Shenyang, China; cat. nos. wl01482, wl0198 and wl0219a). The
α7 nAChR antibody was from Santa Cruz Biotechnology, Inc. (Dallas,
Texas, USA; cat. no. sc-5544). Goat monoclonal HRP-conjugated
antibody against GAPDH and goat anti-rabbit HRP-conjugated
secondary antibodies were obtained from Dingguo Biological
Technology (Beijing, China; cat. no. SH-0031). Nicotine was
purchased from Sigma-Aldrich (St. Louis, MO, USA). α-Bungarotoxin
(α-BTX), a specific antagonist of α7 nAChR, was purchased from
Tocris (Bristol, UK).
Tissue specimens
Tongue squamous cell carcinoma tissue specimens were
collected at Stomatology Hospital of Shandong University (Jinan,
China). Upon recruitment, informed consent was obtained from each
subject. This study was approved by the Institutional Research
Ethics Committee of School of Stomatology, Shandong University.
Cell culture
The tongue cancer cell line Cal27 was obtained from
American Type Culture Collection (ATCC; Manassas, VA, USA). Cal27
has been identified by the measurement of short tandem repeats. The
cells were passaged approximately 6 times in our laboratory during
a period of fewer than 6 months after reidentification. Cal27 was
cultured in Dulbecco's modified Eagle's medium (DMEM) (Hyclone,
Logan, UT, USA) containing 10% (v/v) fetal bovine serum (FBS) and
1% penicillin-streptomycin. Cal27 cells were cultured at 37°C in a
humidified atmosphere of 95% air and 5% CO2.
Cell proliferation assay
Cells were cultured in 96-well tissue culture plates
(3×103 cells/well) with 10% FBS for 24 h. Then, the cells were
exposed to 0.1 µM nicotine for 24, 48, 72, 96 and 120 h. Cell
proliferation was measured by a CCK-8 assay (19). Briefly, 10 µl CCK-8 solution was added
to each well, and the plates were incubated for additional 2 h. The
absorbance was measured using a spectrometer at a wave length of
450 nm. Cell proliferation was also assessed using the clonogenic
formation assay (20). Briefly, 1,000
Cal27 cells were seeded per well of a 6-well plate. Cells were
treated with the indicated concentration of nicotine with or
without α-BTX for one week to determine the impact of nicotine on
proliferation. After one week, colonies were
paraformaldehyde-fixed, and the number of colonies was determined
after crystal violet staining as previously described (20).
Wound healing assay
Cells (5×105 cells/well) were seeded in 6-well
plates and allowed to attach to 80% confluence. Cell monolayers
were wounded by scratching with 200-µl pipette tips before being
washed twice with phosphate-bufferred saline (PBS) to remove
floating cells. Cells in the each well were subsequently exposed to
serum-free DMEM with or without nicotine for up to 24 h. Cells were
photographed at ×100 magnification under a phase-contrast
microscope at each time-point. The healing area at different times
was measured using Image-Pro Plus 6.0 software.
Transwell invasion assay
Cal27 (2×104 cells/0.4 ml) cells were seeded in the
upper chamber of the Transwell inserts (8-µm pore size) pre-coated
with Matrigel (both from Corning, Corning, NY, USA) and exposed to
FBS-free medium with or without 10 µM nicotine. Medium containing
10% FBS was placed in the lower chamber, and cells for each
treatment were incubated for 24 h at 37°C in a humidified
atmosphere with 95% air and 5% CO2. Then, the
non-invasive cells in the upper chamber were removed with a cotton
swab, and the invaded cells were fixed with 4% formaldehyde for 15
min and then stained with 0.1% crystal violet in 0.01 M PBS for 15
min after being washed with PBS. The number of cells that
penetrated the membrane was counted, and images were captured under
a light microscope at a magnification of ×200, as previously
described (21).
TOP/FOP flash assay
TOP/FOP flash assay is a luciferase reporter assay
which is used to assess the activity of β-catenin and T-cell factor
(TCF) signaling. The cells (3×104 cells/well in 24-well plates)
were transfected with 0.1 g of TOPflash or FOPflash (Upstate
Biotechnology, Lake Placid, NY, USA) and 5 ng of pRL-SV40 (Promega,
Madison, WI, USA) using Lipofectamine 2000 reagent (Invitrogen).
After 24 h, the cells were switched to normal complete medium with
or without 10 µm nicotine for another 24 h. Then, the cells were
lysed, and the luciferase activity was determined according to the
manufacturer's recommendations. The luciferase activity of each
sample was normalized with its respective Renilla luciferase
activity.
Western blot
Cells were harvested and lysed, and protein
concentration was determined by the BCA assay. Equal quantities of
protein were separated on 10% SDS-polyacrylamide gels and
transferred onto polyvinylidene difluoride (PVDF) membranes
(Millipore, Bedford, MA, USA). After blocking in 5% fat-free dry
milk in Tween-20 Tris-buffered saline (TBST) for one hour, the
membranes were probed with antibody against β-catenin (1:5,000),
Ror2 (1:2,000), c-Myc (1:1,000), E-cadherin (1:3,000), Wnt5a
(1:1,000), c-jun (1:500), p-Akt (1:1,000), p-GSK3β (1:1,000), GAPDH
(1:20,000) and α7 nAChR (1:1,000), washed in TBST and then
incubated with HRP-conjugated secondary antibody (1:20,000).
Finally, protein bands were detected using the Immobilon western
chemiluminescent HRP substrate kit.
Immunofluorescence and
Immunohistochemical staining
Immunohistochemistry (IHC) analysis was performed to
investigate the expression of β-catenin and Ror2 in different
samples of human tongue cancer. Briefly, the sections were
deparaffinized in xylene, hydrated through a graded alcohol series,
and washed with PBS. Antigen retrieval was performed by treatment
with 10 mM sodium citrate buffer (Zhongshan, Beijing, China) in a
pressure cooker for 5 min. The activity of endogenous tissue
peroxidase was blocked with 3% H2O2
(Zhongshan) for 30 min. After pretreatment with normal goat serum
(Zhongshan) for 30 min to block nonspecific binding, the sections
were incubated with β-catenin (1:500) and Ror2 (1:250) at 4°C
overnight. Sections treated with PBS instead of the primary
antibody were used as negative controls. The sections were
incubated with HRP-conjugated goat-anti-rabbit secondary antibody
for 30 min, followed by reaction with diaminobenzidine, and
counterstaining with Mayer hematoxylin. For evaluation of Ror2
(cytoplasmic and membranous staining), c-jun and Wnt5a (cytoplasmic
and nuclear staining), we used a semiquantitative approach based on
staining intensity (SI) and percentage of positive cells (PP), to
create the immunoractive score (IRS) as follows: IRS=SIxPP, for
each sample, as previously described (22). Intensity was scored as follows: 0, no
staining; 1, weakly positive; 2, moderately positive; and 3,
strongly positive. The scoring of the staining pattern was based on
the percentage of positive tumor cells: 0, 0–5%; 1, 6–25%; 2,
26–50%; and 3, 51–100%). The IRS score ranged from 0 to 9. To
evaluation of β-catenin expression, we used a method described by
Maruyama et al (23). Normal
expression was defined as positive membrane staining seen in
>70% cells, otherwise, it was deemed as a deletion of membrane
expression. Positive cytoplasmic and nuclear expression was defined
when staining was observed in >10% cells. Deletion of membrane
expression and positive cytoplasmic and nuclear expression were
proposed as defined abnormal expression.
For immunofluorescence assays, cells were seeded on
glass coverslips in a 6-well plate for 24–48 h with or without
nicotine stimulation. Then, cells were fixed with paraformaldehyde
for 15 min at room temperature and washed with PBS. After blocking
with normal goat serum, cells were incubated with associated
antibody (β-catenin, 1:200) overnight at 4°C. Then, slides were
washed and incubated with FITC-conjugated goat anti-rabbit IgG for
1 h at room temperature. Slides were washed with PBS again before
being stained with DAPI and examined with a fluorescence
microscope.
Statistical analysis
All statistical analyses were performed using SPSS
version 19.0 (SPSS, Chicago, IL, USA). Student's t-test or analysis
of variance was used to compare group distributions. All results
were expressed as mean ± standard deviation (SD). A value of
P<0.05 was considered statistically significant.
Results
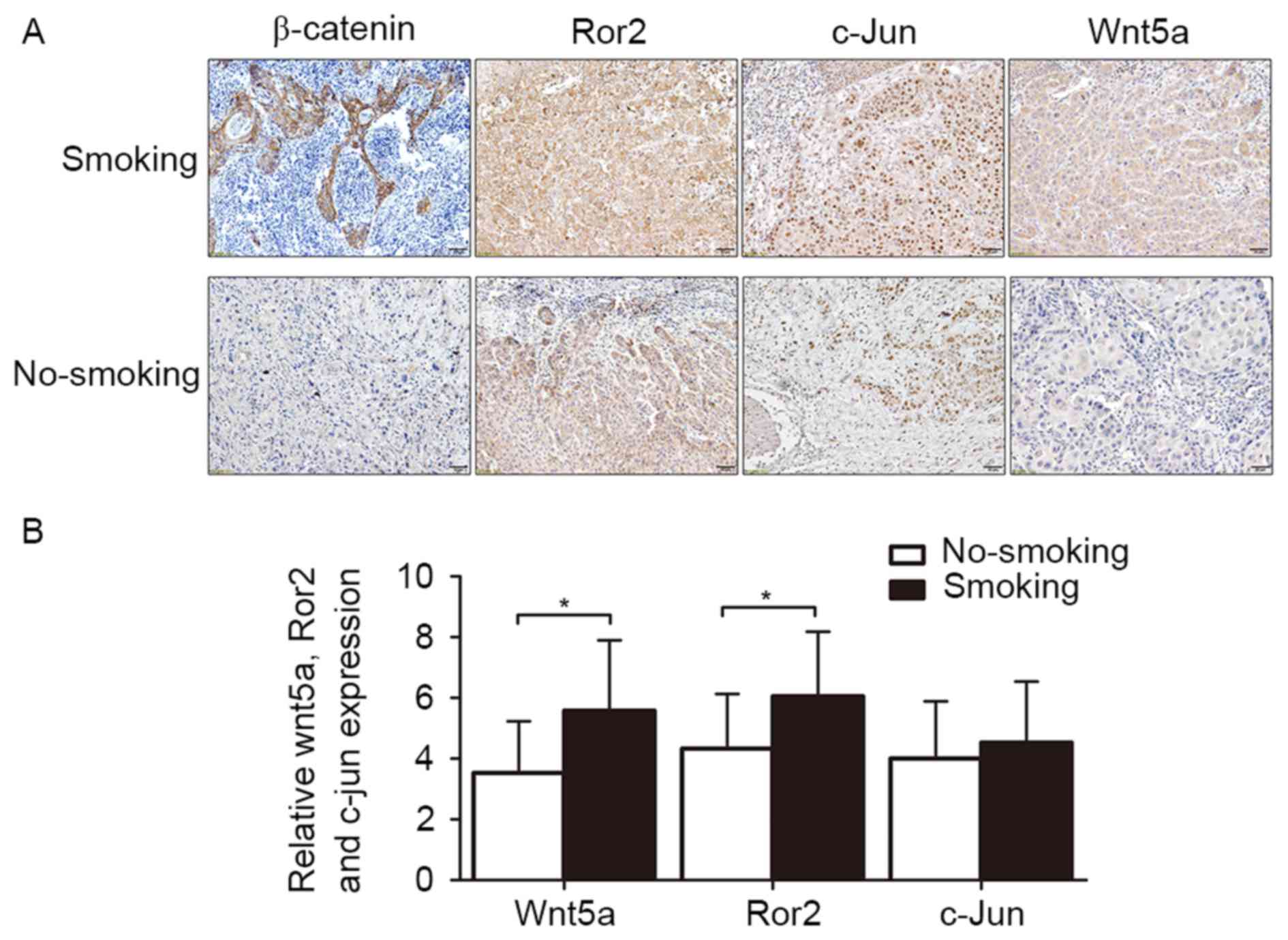
The expression levels of β-catenin,
Wnt5a, and Ror2 are associated with smoking history in TSCC
patients
A previous study has reported that cigarette smoke
extract can activate Wnt/β-catenin pathway in vitro
(21) and promote the expression of
Wnt5a in vivo (24). To
investigate relationship between smoking history and the expression
levels of β-catenin, Wnt5a, c-jun and Ror2 in human TSCC, IHC was
performed to determine expression levels of β-catenin, Wnt5a, c-jun
and Ror2 in paraffin-embedded TSCC tissues (smoking patients, n=18;
non-smoking patients, n=15). IHC staining revealed that Wnt5a
(IRS=5.58±2.32) and Ror2 (IRS=6.05±2.13) were significantly higher
in tumor tissues of smoking patients than those (IRS=3.53±2.17) and
(IRS=4.33±2.02) of non-smoking patients (P=0.013 and P=0.019)
(Fig. 1). In smoking group, 89.47%
(16/18) of patients had abnormal expression of β-catenin but only
53.33% (8/15) in non-smoking group (P=0.018). In addition, the
expression of c-jun were not significantly associated with a
smoking history (IRS=4.53±2.01 vs. IRS=4.00±1.89, P=0.442). This
result suggests the elevated expression of β-catenin, Wnt5 and Ror2
are potentially connected to smoking history in TSCC patients.
Based on the IHC results, we presume that Wnt/β-catenin pathway and
Wnt/PCP pathway in TSCC cells may be overactivated by nicotine
stimulation as well in vitro.
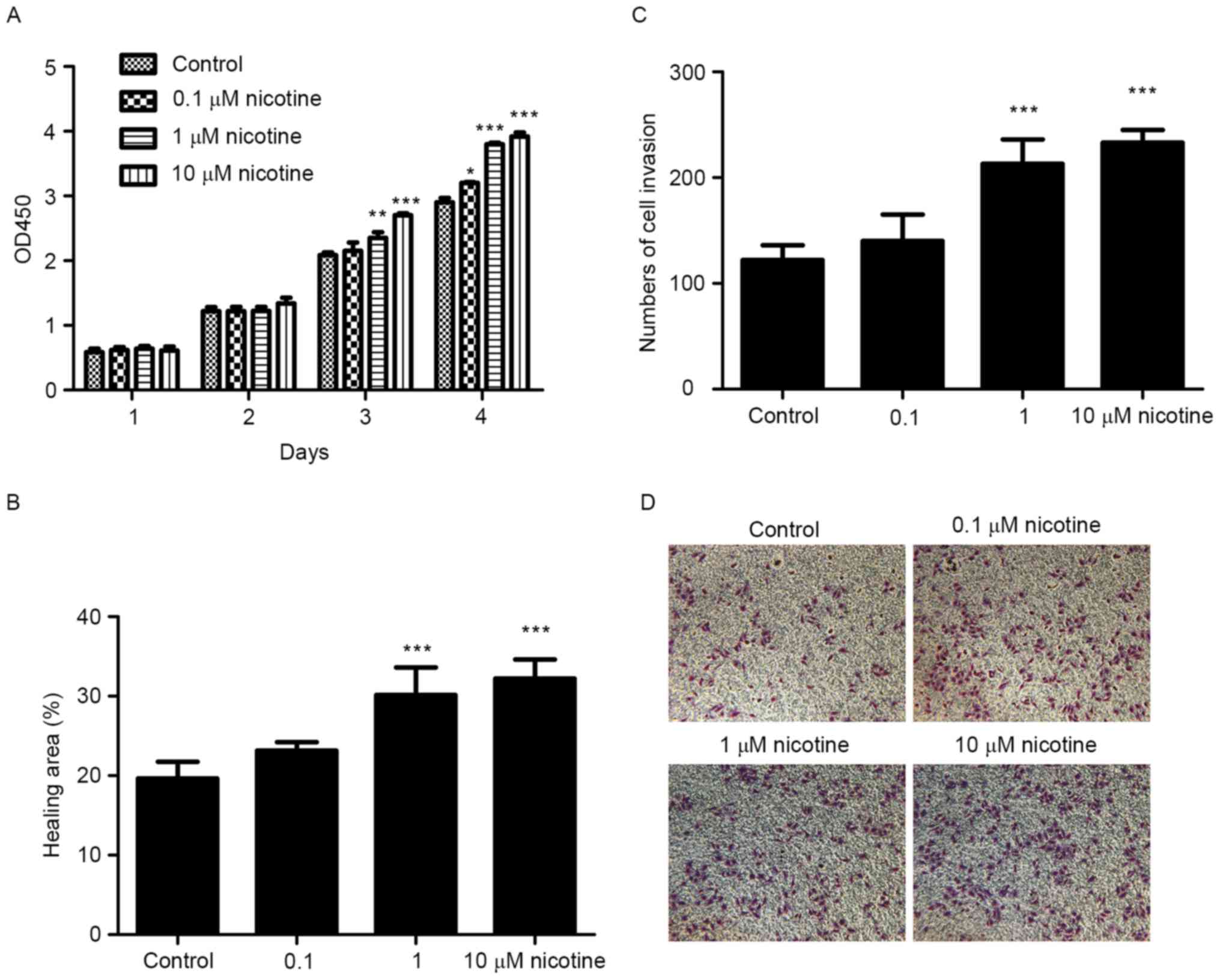
Nicotine stimulation promotes
proliferation, migration and invasion of Cal27 cells
To investigate the effects of nicotine on
proliferation, we treated cells with different concentrations of
nicotine and measured the change of cell proliferation by CCK-8
assay. As expected, the proliferation of Cal27 cells was promoted
by nicotine in a time and dose dependent manner (Fig. 2A). When incubated for 72 h, the number
of cells in the 10 µM nicotine group was significantly greater than
in that in the control group (2.71±0.03 vs. 2.08±0.09, P<0.001).
In the wound healing assay, nicotine stimulation reduced the time
of scratch healing (Fig. 2B). For
example, at 12 h 32.2±2.43% of the wound was healed in the 10 µM
nicotine group, but only 19.67±2.08% was healed in the control
group in Cal27 cells (P<0.001). Similarly, cells' invasion
ability after nicotine stimulation was improved as determined by
Transwell invasion assays (Fig. 2C).
These results suggest that nicotine can promote proliferation,
migration and invasion of Cal27 cells in vitro.
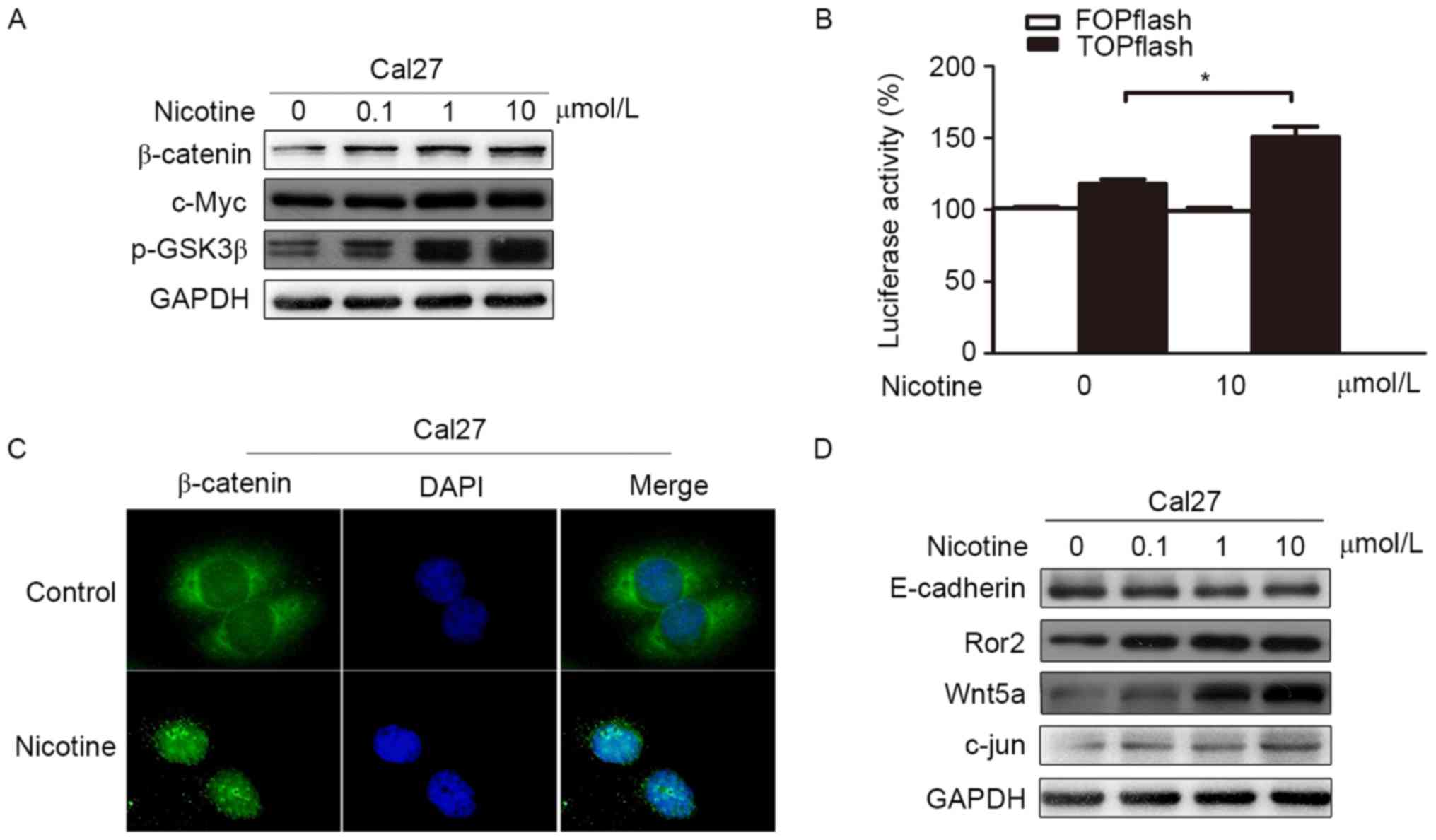
Nicotine can activate Wnt/β-catenin
and the Wnt/PCP pathways in Cal27
To test our hypothesis, cells were stimulated by
nicotine in vitro, and expression levels of related proteins
were determined by western blot analysis and immunofluorescence. We
found that the expression levels of total β-catenin and nuclear
β-catenin were increased in Cal27 after nicotine simulation
(Fig. 3A and C), and western blot
analysis detected that p-GSK3β was increased as well (Fig. 3A). The expression level of c-Myc, a
downstream target gene of Wnt/β-catenin pathway, was increased in a
dose-dependent manner upon nicotine stimulation (Fig. 3A). Additionally, we found that
Wnt/β-catenin signaling activity was substantially higher in Cal27
cells treated with nicotine compared to the control group, as
determined using the TCF-dependent TOPflash reporter (Fig. 3B). Taken together, these results
suggest that the Wnt/β-catenin pathway is activated by nicotine
stimulation.
Western blot analysis also detected that levels of
Ror2, Wnt5a and c-jun, a downstream effector of the Wnt/PCP
pathway, were significantly increased in the nicotine stimulation
condition in a dose-dependent manner (Fig. 3D). Therefore, in the presence of
nicotine, the Wnt/PCP pathway is also activated in Cal27 cells
in vitro.
Nicotine may promote progression of
Cal27 cells by activating the Wnt/β-catenin and Wnt/PCP pathways
through α7 nAChR
Our previous research indicated that Wnt/PCP pathway
can facilitate migration and invasion of Cal27 cells (25). Previous studies showed that nicotine
can promote proliferation and invasion in several cancers via the
Wnt/β-catenin pathway (9,13). In this study, we examined if the
activation of Wnt/β-catenin and Wnt/PCP pathways by nicotine
involved α7 nAChR in Cal27 cells. We found that the upregulation of
β-catenin, p-GSK-3β, Wnt5a, Ror2 and c-jun was reversed when
co-treated with the α7 nAChR inhibitor α-BTX (Fig. 4A) in Cal27 cells. In addition, the
results of colony formation, Transwell and migration assays
revealed that the stimulating effects of nicotine on proliferation,
invasion, and migration of Cal27 cells could be antagonized if
co-treated with α-BTX. The number of colonies formed in the
nicotine group was 318.66±29.67, while only 212.54±24.03 colonies
formed in the control group (P=0.009) and the nicotine-induced
proliferation was reversed when co-treated by α-BTX (224.32±28.15,
P=0.016) (Fig. 4B). In the wound
healing assay, at 12 h, 37.2±2.21% of the wound was healed in the
nicotine group, while only 29.21±1.18% was healed in the α-BTX
co-treated group in Cal27 cells (P=0.005) (Fig. 4C). Based on the results above, the
change of biological characteristics of Cal27 cells by nicotine
stimulation may be explained by the activation of the Wnt/β-catenin
and Wnt/PCP pathways, a process that requires α7 nAChR.
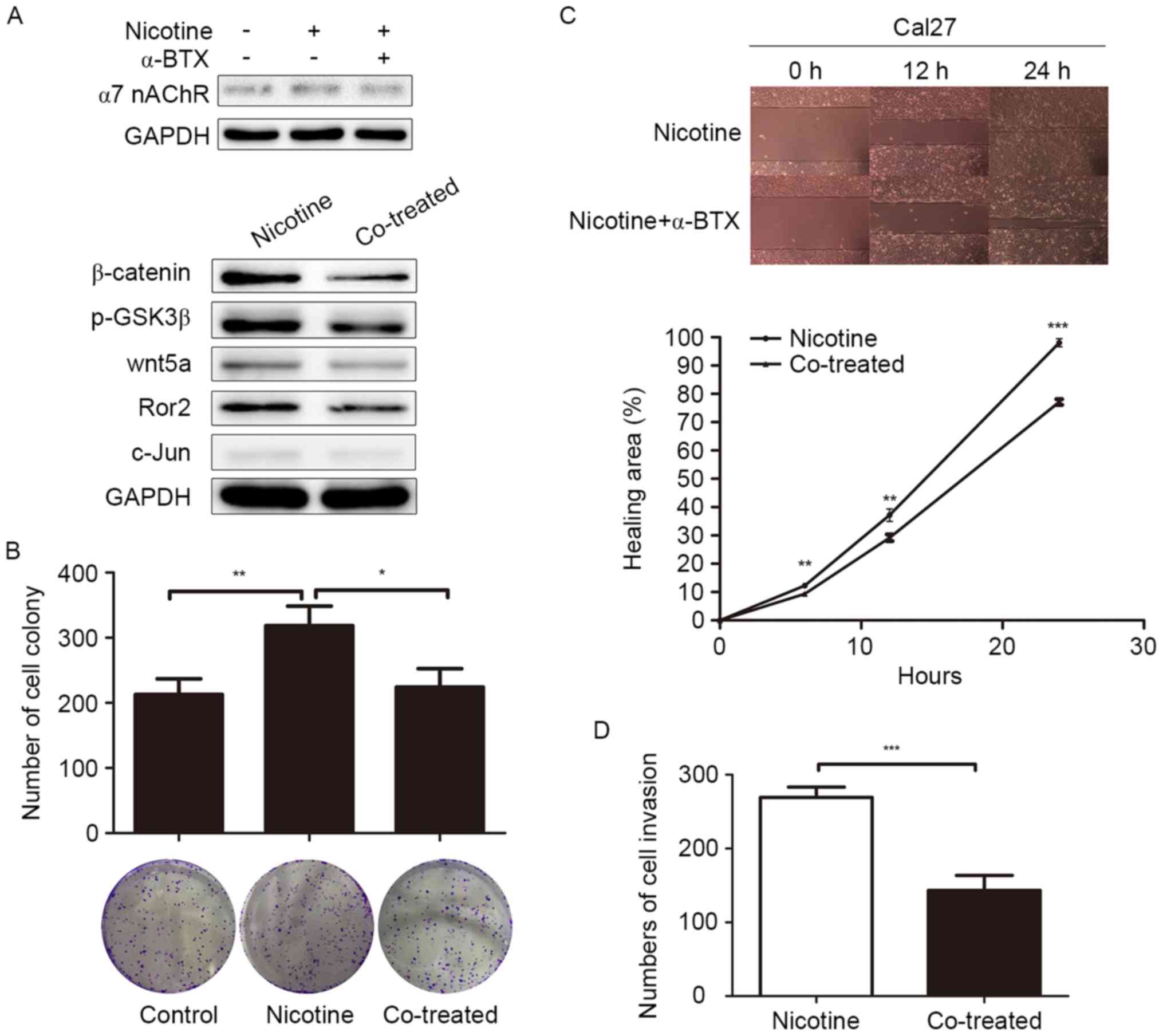 | Figure 4.Effect of α-BTX on the
nicotine-induced proliferation, migration and invasion of Cal27
cells. (A) α-BTX (0.1 µM) reversed the nicotine-induced
upregulation of β-catenin, p-GSK3β, Wnt5a, Ror2 and c-jun in
western blot assays. (B) Representative images of a colony-forming
assay of Cal27 cells, which were cultured in the presence of
nicotine at the indicated concentrations with or without α-BTX (0.1
µM) or in DMSO as a control. (C) Representative images of wound
healing of Cal27 in 10 µM nicotine with or without α-BTX (0.1 µM)
at indicated time-point. Student's t-test was used to
compare the speed of wound healing between the nicotine stimulation
group and the control group (original magnification, ×100). (D)
Images of the Transwell invasion of Cal27 after different
treatments were analyzed using Image-Pro Plus 6.0 software. The
cells were allowed to invade through a layer of Matrigel for 24 h.
The number of cells invaded was counted and expressed as the mean ±
SD (nicotine group vs. co-treated group: 269.33±14.05 vs.
143.36±20.21, P=0.001) (original magnification, ×100) (*P<0.05,
**P<0.01, ***P<0.001). |
Additionally, we have found that E-cadherin
expression was decreased (Fig. 2D),
which could explain the aggressive migration of Cal27 in the
presence of nicotine.
Discussion
Tongue squamous cell carcinoma is one of the most
common and malignant cancers affecting the oral cavity. After lymph
node or distant metastasis, TSCC patients will have a poor
prognosis even after systemic therapy (26). Therefore, it is critical to explore
and understand the mechanism of TSCC development and metastasis,
which may help us to interrupt tongue cancer progression.
In our study, we determined that nicotine
stimulation could promote the proliferative and invasive abilities
of TSCC cells in accordance with a previous study (27). These findings suggest that TSCC
patients with a history of smoking should receive specific care and
should be encouraged to quit smoking.
It is well known that metastasis of TSCC is closely
related to EMT (28). EMT facilitates
tumor cell migration, invasion and metastasis, and may also be a
major mechanism of tumor progression (29). Previous findings have demonstrated
that a decrease in E-cadherin expression is one of the hallmarks of
EMT, which is closely associated with recurrence and survival in
TSCC patients (30,31). In addition, previous studies have
proved that Wnt/β-catenin pathway is directly connected to EMT in
TSCC cells (32,33). In our study, we have found that
nicotine can reduce the expression level of E-cadherin and activate
Wnt/β-catenin pathway in vitro. Combination treatment with
nicotine and α-BTX abrogated the increased effects of
nicotine-induced migration and invasion in Cal27. Thus EMT
involving activation of Wnt/β-catenin pathway may be the molecular
mechanisms of nicotine-induced metastasis in Cal27 cells and may be
ubiquitous in smoking-related cancers.
The Wnt/β-catenin pathway has been also found to be
connected to promote proliferation in non-small cell lung cancer
after nicotine stimulation (6,9,10,13,34,35).
We first found that nicotine can also activate the Wnt/β-catenin
pathway in the TSCC cell line Cal27 in vitro. It suggests
that nicotine-induce activation of Wnt/β-catenin pathway may be a
general molecular signaling effect in different cancer cells. We
investigated expression levels of β-catenin in samples of TSCC
patients with or without a smoking history. We found that aberrant
expression of β-catenin is more common in patients with a smoking
history. Based on these findings, nicotine-induce aberrant
activation of β-catenin may play a significant role in
smoking-related TSCC progression.
Overexpression of Wnt5a has recently been linked to
melanoma, osteosarcoma, renal cell carcinoma, prostate cancer,
breast cancer and TSCC. Wnt5a is reported to promote invasion of
certain types of cancer cells, including HeLa cervical cancer
cells, A549 lung cancer cells, and KKLS gastric cancer cells
(36). In addition, Ror2 is closely
associated with the degree of malignancy in oral epithelial tissue
(37). In our study, we found that
the expression levels of Wnt5a and Ror2 were increased by nicotine
stimulation in Cal27 cells in vitro, as well as cell
migration and invasion. In addition our results indicate that
aberrant expressions of Ror2 and Wnt5a are more common in patients
with a smoking history, which are consistent with previous reports
(24). According to the results
above, we presume that the increased migration and invasion of
Cal27 cells after nicotine stimulation may be relevant for the
upregulation of Wnt5a and Ror2 which needs further research.
In conclusion, we have demonstrated the effects of
nicotine stimulation on Cal27 cells, promoting their proliferation
and invasion in vitro. Therefore, nicotine may be a possible
substance in cigarette smoke that aggressively promotes TSCC and
leads to a poor prognosis. We also observed activation of
Wnt/β-catenin and upregulation of Ror2 and Wnt5a. These molecular
changes may be a common mechanism that induces poor prognosis in
other smoking-related cancers. It may be a potential molecular
target to slow the development of some smoking-related cancers and
improve their prognosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (no. 81402298). The study was
also supported by the Young Scholars Program of Shandong
University.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schaal C and Chellappan SP:
Nicotine-mediated cell proliferation and tumor progression in
smoking-related cancers. Mol Cancer Res. 12:14–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh S, Pillai S and Chellappan S:
Nicotinic acetylcholine receptor signaling in tumor growth and
metastasis. J Oncol. 2011:4567432011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schuller HM: Is cancer triggered by
altered signalling of nicotinic acetylcholine receptors? Nat Rev
Cancer. 9:195–205. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carlisle DL, Liu X, Hopkins TM, Swick MC,
Dhir R and Siegfried JM: Nicotine activates cell-signaling pathways
through muscle-type and neuronal nicotinic acetylcholine receptors
in non-small cell lung cancer cells. Pulm Pharmacol Ther.
20:629–641. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi J, Liu F, Zhang W, Liu X, Lin B and
Tang X: Epigallocatechin-3-gallate inhibits nicotine-induced
migration and invasion by the suppression of angiogenesis and
epithelial-mesenchymal transition in non-small cell lung cancer
cells. Oncol Rep. 33:2972–2980. 2015.PubMed/NCBI
|
|
7
|
Zhao Y, Zhou W, Xue L, Zhang W and Zhan Q:
Nicotine activates YAP1 through nAChRs mediated signaling in
esophageal squamous cell cancer (ESCC). PLoS One. 9:e908362014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoneyama R, Aoshiba K, Furukawa K, Saito
M, Kataba H, Nakamura H and Ikeda N: Nicotine enhances hepatocyte
growth factor-mediated lung cancer cell migration by activating the
α7 nicotine acetylcholine receptor and phosphoinositide
kinase-3-dependent pathway. Oncol Lett. 11:673–677. 2016.PubMed/NCBI
|
|
9
|
Zou W, Zou Y, Zhao Z, Li B and Ran P:
Nicotine-induced epithelial-mesenchymal transition via
Wnt/β-catenin signaling in human airway epithelial cells. Am J
Physiol Lung Cell Mol Physiol. 304:L199–L209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakurai R, Cerny LM, Torday JS and Rehan
VK: Mechanism for nicotine-induced up-regulation of Wnt signaling
in human alveolar interstitial fibroblasts. Exp Lung Res.
37:144–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Russell MA, Jarvis M, Iyer R and
Feyerabend C: Relation of nicotine yield of cigarettes to blood
nicotine concentrations in smokers. Br Med J. 280:972–976. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Benowitz NL: Cigarette smoking and
nicotine addiction. Med Clin North Am. 76:415–437. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu W, Yi D, Guo J, Xiang Z, Deng L and He
L: Nuciferine, extracted from Nelumbo nucifera Gaertn,
inhibits tumor-promoting effect of nicotine involving Wnt/β-catenin
signaling in non-small cell lung cancer. J Ethnopharmacol.
165:83–93. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiang T, Fei R, Wang Z, Shen Z, Qian J and
Chen W: Nicotine enhances invasion and metastasis of human
colorectal cancer cells through the nicotinic acetylcholine
receptor downstream p38 MAPK signaling pathway. Oncol Rep.
35:205–210. 2016.PubMed/NCBI
|
|
15
|
Peng C, Jia X, Xiong Y, Yin J, Li N, Deng
Y, Luo K, Zhang Q, Wang C, Zhang Z, et al: The
14-3-3s/GSK3β/β-catenin/ZEB1 regulatory loop modulates
chemo-sensitivity in human tongue cancer. Oncotarget.
6:20177–20189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan G, Zou R, Chen Z, Fan B, Wang Z, Wang
Y, Yin X, Zhang D, Tong L, Yang F, et al: Silencing RhoA inhibits
migration and invasion through Wnt/β-catenin pathway and growth
through cell cycle regulation in human tongue cancer. Acta Biochim
Biophys Sin (Shanghai). 46:682–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng L, Li N, Guo F, Jian XC, Jiang CH,
Yin P, Min AJ and Huang L: Twist-related protein 1 enhances oral
tongue squamous cell carcinoma cell invasion through β-catenin
signaling. Mol Med Rep. 11:2255–2261. 2015.PubMed/NCBI
|
|
18
|
Zhang A, He S, Sun X, Ding L, Bao X and
Wang N: Wnt5a promotes migration of human osteosarcoma cells by
triggering a phosphatidylinositol-3 kinase/Akt signals. Cancer Cell
Int. 14:152014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Z, Bi Q and Wang Y: Hydrogen sulfide
accelerates cell cycle progression in oral squamous cell carcinoma
cell lines. Oral Dis. 21:156–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seidensaal K, Nollert A, Feige AH, Muller
M, Fleming T, Gunkel N, Zaoui K, Grabe N, Weichert W, Weber KJ, et
al: Impaired aldehyde dehydrogenase 1 subfamily member 2A-dependent
retinoic acid signaling is related with a mesenchymal-like
phenotype and an unfavorable prognosis of head and neck squamous
cell carcinoma. Mol Cancer. 14:2042015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang CM, Chang PY, Tu MG, Lu CC, Kuo SC,
Amagaya S, Lee CY, Jao HY, Chen MY and Yang JS: Epigallocatechin
gallate sensitizes CAL-27 human oral squamous cell carcinoma cells
to the anti-metastatic effects of gefitinib (Iressa) via
synergistic suppression of epidermal growth factor receptor and
matrix metalloproteinase-2. Oncol Rep. 28:1799–1807.
2012.PubMed/NCBI
|
|
22
|
Psyrri A, Kotoula V, Fountzilas E,
Alexopoulou Z, Bobos M, Televantou D, Karayannopoulou G, Krikelis
D, Markou K, Karasmanis I, et al: Prognostic significance of the
Wnt pathway in squamous cell laryngeal cancer. Oral Oncol.
50:298–305. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maruyama K, Ochiai A, Akimoto S, Nakamura
S, Baba S, Moriya Y and Hirohashi S: Cytoplasmic beta-catenin
accumulation as a predictor of hematogenous metastasis in human
colorectal cancer. Oncology. 59:302–309. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Whang YM, Jo U, Sung JS, Ju HJ, Kim HK,
Park KH, Lee JW, Koh IS and Kim YH: Wnt5a is associated with
cigarette smoke-related lung carcinogenesis via protein kinase C.
PLoS One. 8:e530122013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu G, Sengupta PK, Jamal B, Yang HY,
Bouchie MP, Lindner V, Varelas X and Kukuruzinska MA:
N-glycosylation induces the CTHRC1 protein and drives oral cancer
cell migration. J Biol Chem. 288:20217–20227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schwam ZG and Judson BL: Improved
prognosis for patients with oral cavity squamous cell carcinoma:
Analysis of the National Cancer Database 1998–2006. Oral Oncol.
52:45–51. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu C and Chang Y: Enhancement of cancer
stem-like and epithelial-mesenchymal transdifferentiation property
in oral epithelial cells with long-term nicotine exposure: Reversal
by targeting SNAIL. Toxicol Appl Pharmacol. 266:459–469. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiao J, Zhao X, Liang Y, Tang D and Pan C:
FGF1-FGFR1 axis promotes tongue squamous cell carcinoma (TSCC)
metastasis through epithelial-mesenchymal transition (EMT). Biochem
Biophys Res Commun. 466:327–332. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pasquier J, Abu-Kaoud N, Al Thani H and
Rafii A: Epithelial to mesenchymal transition in a clinical
perspective. J Oncol. 2015:7921822015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhu R, Jiang XY, Song XL, Zhang W, Chen S
and Liu LK: Expression of E-cadherin and beta-catenin in oral
squamous cell carcinomas of tongue: Correlation with the
clinicopathologic features and patient prognosis. Zhonghua Kou
Qiang Yi Xue Za Zhi. 45:295–298. 2010.(In Chinese). PubMed/NCBI
|
|
31
|
Chow V, Yuen AP, Lam KY, Tsao GS, Ho WK
and Wei WI: A comparative study of the clinicopathological
significance of E-cadherin and catenins (alpha, beta, gamma)
expression in the surgical management of oral tongue carcinoma. J
Cancer Res Clin Oncol. 127:59–63. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chaw SY, Majeed Abdul A, Dalley AJ, Chan
A, Stein S and Farah CS: Epithelial to mesenchymal transition (EMT)
biomarkers-E-cadherin, beta-catenin, APC and Vimentin-in oral
squamous cell carcinogenesis and transformation. Oral Oncol.
48:997–1006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Walker A, Frei R and Lawson KR: The
cytoplasmic domain of N-cadherin modulates MMP9 induction in oral
squamous carcinoma cells. Int J Oncol. 45:1699–1706.
2014.PubMed/NCBI
|
|
34
|
Jiang Q, Wei MD, Wang KW, Lan YX, Zhu N
and Wang Y: Nicotine contributes to the neural stem cells fate
against toxicity of microglial-derived factors induced by Aβ via
the Wnt/β-catenin pathway. Int J Neurosci. 126:257–268. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gong WY, Wu JF, Liu BJ, Zhang HY, Cao YX,
Sun J, Lv YB, Wu X and Dong JC: Flavonoid components in Scutellaria
baicalensis inhibit nicotine-induced proliferation, metastasis and
lung cancer-associated inflammation in vitro. Int J Oncol.
44:1561–1570. 2014.PubMed/NCBI
|
|
36
|
Shojima K, Sato A, Hanaki H, Tsujimoto I,
Nakamura M, Hattori K, Sato Y, Dohi K, Hirata M, Yamamoto H and
Kikuchi A: Wnt5a promotes cancer cell invasion and proliferation by
receptor-mediated endocytosis-dependent and -independent
mechanisms, respectively. Sci Rep. 5:80422015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kobayashi M, Shibuya Y, Takeuchi J, Murata
M, Suzuki H, Yokoo S, Umeda M, Minami Y and Komori T: Ror2
expression in squamous cell carcinoma and epithelial dysplasia of
the oral cavity. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.
107:398–406. 2009. View Article : Google Scholar : PubMed/NCBI
|