Introduction
Choroidal melanoma is the most common primary
intraocular tumor in adults (1,2). The mean
incidence of choroidal melanoma in the USA is ~6 novel
cases/106 people every year (3). Currently, there are several treatment
protocols for primary choroidal melanoma, including surgery,
radiation therapy, thermotherapy and external beam proton therapy
(4–7).
The 5-year relative survival rate of choroidal melanoma is ~80%
when the tumors are confined to the eyes (3,8). However,
patients with choroidal melanoma have a high risk of developing
metastasis (typically to the liver), which is usually fatal with a
median survival time of 6–9 months subsequent to the detection of
liver metastasis (3,8). Therefore, development of novel effective
therapies for choroidal melanoma is required urgently.
Cepharanthine(CEP; 6′,12′-dimethoxy-2,2′-dimethyl-
6,7-[methylenebis(oxy)]oxyacanthan) is a biscoclaurine alkaloid
extracted from the roots of the plant Stephania cepharantha
Hayata, which has been broadly used in Japan for chemoprevention
and treatment of numerous diseases by virtue of its
anti-inflammatory and immunomodulatory activities (9,10). CEP has
been reported to exert antitumor effects in numerous cancers by
inhibiting cancer cell proliferation (11), cell cycle progression (12), tumor invasion (13), generating reactive oxygen species
(ROS) (11,14), inducing cell apoptosis (11,14,15),
regulating cell survival signaling pathways (16,17) and
increasing the competence of the host (18,19). It
was also identified to potentiate the anticancer effects of other
chemotherapeutic agents (20–22), and was able to circumvent the
multidrug resistance of doxorubicin, vincristine and other
anticancer agents (21,23–26).
However, little is known about the effect and molecular mechanism
of CEP on choroidal melanoma.
The present study investigated the effects of CEP on
choroidal melanoma cell proliferation and survival, and on a
choroidal melanoma xenograft tumor. In addition, the potential
underlying molecular mechanisms for CEP-induced choroidal melanoma
cell apoptosis were explored.
Materials and methods
Reagents
CEP, propidium iodide (PI) and crystal violet dye
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
CellTiter 96® AQueous One Solution reagent (MTS) was
purchased from Promega Corporation (Madison, WI, USA).
N-acetyl-L-cysteine (NAC) and SP600125 were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA).
Cells and cell culture
The human choroidal melanoma MEL15-1 cell line was
derived from a primary choroidal melanoma patient (age 60 years;
female) with metastatic outcome. The human subject studies were
approved by the ethical standards committee of Jilin University
(Jilin, China). Written informed consent for isolating cancer cells
was obtained from the patient involved in the present study. Cells
were maintained in minimum essential medium supplemented with 4 mM
L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, 1% sodium
pyruvate, 1% nonessential amino acids and 10% fetal bovine serum
(all Corning Incorporated, Corning, NY, USA) at 37°C in a 5%
CO2 atmosphere.
Cell viability assay
MEL15-1 cells (1×105 cells/well)
maintained in minimum essential medium (Cellgro; Corning
Incorporated) supplemented with 4 mM L-glutamine, 100 U/ml
penicillin, 100 µg/ml streptomycin, 1% sodium pyruvate, 1%
nonessential amino acids and 10% fetal bovine serum at 37°C, and
were placed in 96-well plates overnight. CEP at various
concentrations (0, 20, 40, 60 and 80 µM), NAC, SP600125 or
identical volumes of control [dimethylsulfoxide (DMSO)] was added
to the appropriate wells. The cells were treated for 24 or 48 h
prior to the addition of 20 µl CellTiter 96 Aqueous One Solution
reagent. Following incubation for 4 h at 37°C, the number of cells
in each well was determined by measuring the optical densities at
490 nm. The results were expressed as the percentages of the
control cultures.
Cell death assay
A cell death assay was performed using the Cell
Death Detection ELISAPLUS kit (Roche Applied Science, Penzberg,
Germany), according to the manufacturer's protocol. This
photometric enzyme immunoassay was used for the quantitative in
vitro determination of cytoplasmic histone-associated DNA
fragments (mono- and oligonucleosomes) following induced cell
death. MEL15-1 cells were treated with indicated concentrations (0,
40 and 60 µM) of CEP at 37°C for 24 h. The cell lysates were then
placed into a streptavidin-coated microplate and incubated with a
mixture of biotin-conjugated anti-histone and horseradish
peroxidase (HRP)-conjugated anti-DNA (provided in the kit) at
15–25°C for 2 h. The amount of peroxidase retained in the
immunocomplex was photometrically determined with 2,2′-azinobis
(3-ethylbenzothiazoline-6-sulfonic acid)-diammonium salt as the
substrate. Absorbance was measured at 405 nm. The intensity of
absorbance at 405 nm was proportional to the amount of cell
death.
Colony formation assay
Colony formation of MEL15-1 cells following drug
treatment was performed as follows. Briefly, cells were seeded onto
6-well plates (1,000 cells/well), incubated at 37°C overnight and
exposed to CEP at different concentrations (0, 40 and 60 µM) for 48
h. The cells were then incubated at 37°C in fresh medium for
another 12–14 days. Subsequent to washing with PBS, the cells were
then fixed with 10% neutral buffered formalin for 5 min, and
stained with 0.05% crystal violet solution for 30 min.
Cell cycle analysis
Cells (1×106/well) maintained in complete
medium at 37°C were placed in 6-well plates overnight. CEP (40 and
60 µM), or 2 µl of vehicle control (DMSO), was added to the
appropriate wells. The control and treated cells were cultured for
an additional 24 h at 37°C, and the cells were stained with PI.
Living cells were gated for cell cycle analysis. Cell cycle data
were acquired using the Cell Lab Quanta™ SC system (Beckman
Coulter, Inc., Brea, CA, USA) and analyzed using FlowJo software
(version 9.9.5; Tree Star, Inc., Ashland, OR, USA).
Western blot analysis
Whole cell lysates were prepared by using
radioimmunoprecipitation assay buffer (1% NP-40, 0.5% sodium
deoxycholate and 0.1% SDS in PBS) supplemented with 100X protease
inhibitor cocktail (Roche Applied Science) and phosphatase
inhibitor cocktail (Cell Signaling Technology, Inc., Danvers, MA,
USA). The mitochondrial and cytosolic fractions were separated
using the digitonin method (27).
Briefly, 5×106 cells (200 µl) were permeabilized on ice
with 21.33 µg digitonin (Sigma-Aldrich; Merck KGaA) in 80 µl buffer
containing 75 mM NaCl, 8 mM Na2HPO4, 1 mM
NaH2PO4 and 250 mM sucrose, supplemented with
100X protease inhibitor cocktail and phosphatase inhibitors.
Following incubation for 30 sec in ice cold buffer, cells were
centrifuged at 4°C for 2 min at 13,000 × g. The supernatant was
removed as the cytosolic fraction, and the remaining pellet was
resuspended in the same volume of buffer not containing digitonin.
A total of 20 µl of Laemmli sample buffer (Sigma-Aldrich; Merck
KGaA) supplemented with 10% dithiothreitol and 10% SDS was added to
all samples. Protein samples were analyzed by SDS-PAGE (12% gels)
and blotted onto the polyvinylidene fluoride membrane (GE
Healthcare Life Sciences, Chalfont, UK). Primary antibodies used
were specific to: B-cell lymphoma 2 (Bcl-2; catalog no. 2872),
Bcl-2-associated X protein (Bax; catalog no. 2774), caspase-3
(catalog no. 9662) cleaved caspase-3 (catalog no. 9661), caspase-9
(catalog no. 9502), cleaved caspase-9 (catalog no. 9505), cleaved
poly (ADP-ribose) polymerase(PARP) (catalog no. 9542),
phosphorylated c-Jun N-terminal kinase (JNK;
Thr183/Tyr185) (catalog no. 9251) (1:1,000;
all from Cell Signaling Technology, Inc., Danvers, MA, USA),
cytochrome c (cyt c) (1:2,000; catalog no. ab13575), JNK1 (1:2,000;
catalog no. ab110724;), JNK2 (1:2,000; catalog no. ab76125) (all
from Abcam, Cambridge, UK) and β-actin (1:8,000; catalog no.
SAB5500001; Sigma-Aldrich; Merck KGaA). The HRP-conjugated
anti-mouse (1:4,000; catalog no. sc-2371) and anti-rabbit secondary
antibodies (1:4,000; catalog no. sc-2357) were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Signals were detected
using the Immun-Star HRP peroxide luminol/enhancer kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Quantification of the
western blots was performed using ImageJ software (National
Institutes of Health, Bethesda, MD, USA).
Luminescence assay/detection of
ROS
The detection of ROS was performed using the
ROS-Glo™ assay (Promega Corporation) according to the
manufacturer's protocol. Cells treated with CEP in combination with
or without NAC for 24 h were incubated with ROS-Glo™
H2O2 substrate for 6 h at 37°C. The ROS-Glo™
detection solution was then added and the plate was incubated for
20 min at room temperature. The luminescence was measured using a
SpectraMax L microplate reader (Molecular Devices, LCC, Sunnyvale,
CA, USA). The intensity of luminescence was proportional to the
amount of ROS.
Nude mice and tumor inoculations
The protocol was approved by the Committee on the
Ethics of Animal Experiments of Jilin University. Mice were
sacrificed by CO2 asphyxiation followed by cervical
dislocation when they became moribund and when they reached defined
study end points. The present study was performed in accordance
with the recommendations from the Institutional Animal Care and Use
Committee of Jilin University. A total of 24 male athymic nude mice
(Charles River Laboratories, Inc., Wilmington, MA, USA) purchased
at 4 weeks of age with ~20 g weight were kept under sterile
conditions in a pathogen-free environment. The mice were provided
with sterile water and food ad libitum. All manipulations
were carried out aseptically inside a laminar flow hood. The
xenograft model was established in the nude mice using cells.
Briefly, tumor cells (1×106) were suspended in 0.1 ml
serum-free medium and injected into the subcutaneous tissue of
6-week-old nude mice using a 27-gauge needle. Tumors were allowed
to grow for 10 days prior to treatment. The mice were then divided
into 4 groups, each of 6 mice with similar mean tumor volumes
(between 100 and 150 mm3).
In vivo treatment protocol
When solid tumors grew to 100–150 mm3,
mice were treated with vehicle control or CEP (25 mg/kg) for 4
weeks (5 times/week) via peritumoral injection. The tumors were
measured every 3 days and the relative tumor volumes were
calculated. At 27 days, mice were sacrificed by CO2
asphyxiation followed by cervical dislocation.
Statistical analysis
All quantitative data are presented as the mean ±
standard deviation. Statistical tests were performed with the SPSS
software package (version 19.0; IBM SPSS, Armonk, NY, USA).
Differences between two groups were tested using paired Student's
t-tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
CEP inhibits MEL15-1 cell
proliferation and induces MEL15-1 cell death
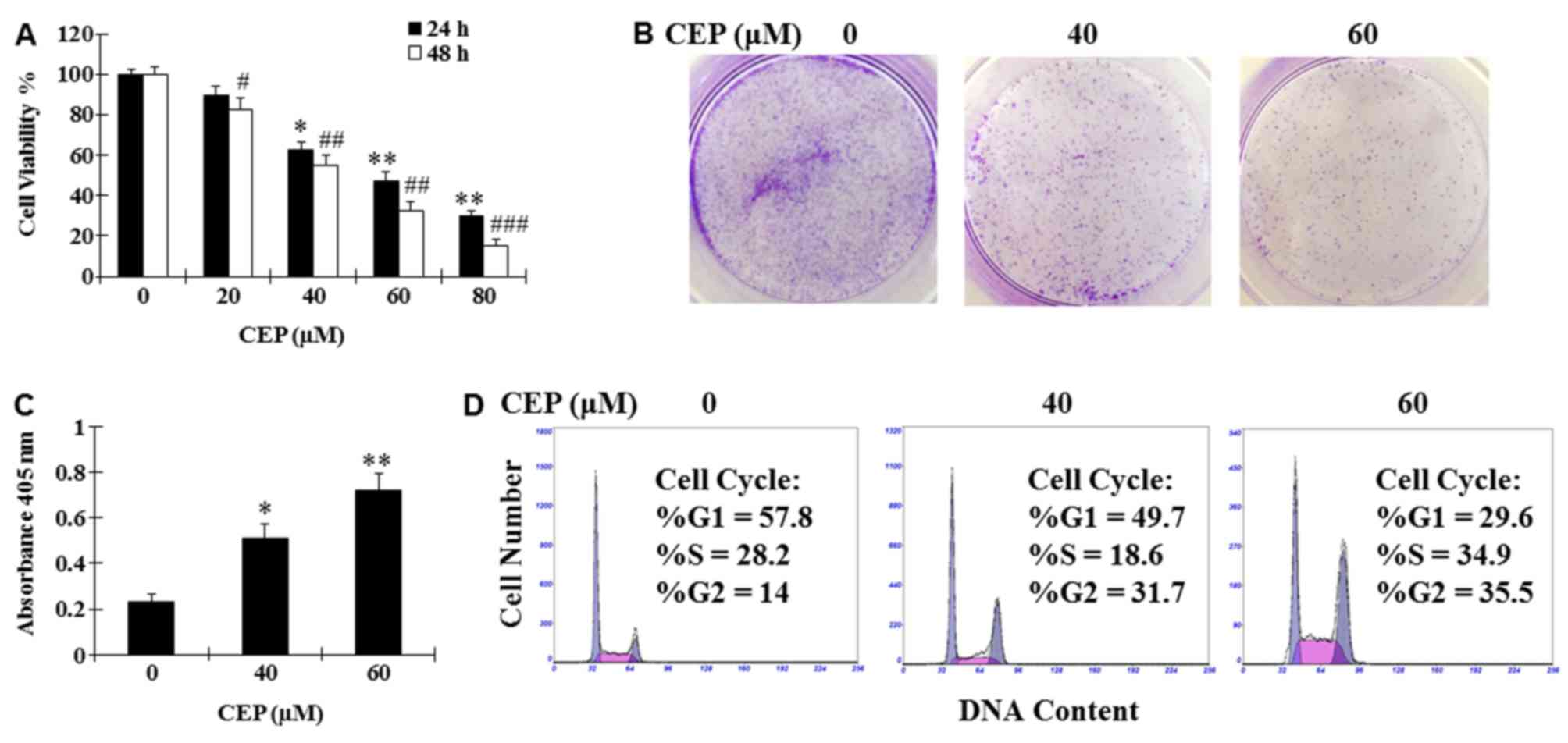
A large body of evidence demonstrated that CEP
effectively inhibits various cancer cell lines (11,12,14–17).
To test whether CEP may also inhibit MEL15-1 cell proliferation,
MEL15-1 cells were treated with CEP at various concentrations (0,
20, 40, 60 and 80 µM) for 24 or 48 h, and then subjected to an MTS
assay. Results demonstrated that CEP was able to dose-dependently
inhibit cell proliferation with a half-maximal inhibitory
concentration of 40–60 µM (Fig. 1A).
Treatment with CEP (40 and 60 µM) for 24 h was also revealed to
markedly suppress colony formation and induce cell death of MEL15-1
cells compared with the vehicle control (Fig. 1B and C). To investigate how CEP
affects MEL15-1 cell proliferation, cell cycle analysis was
performed following MEL15-1 cell exposure to 24 h treatment with
CEP (40 and 60 µM). Results demonstrated that treatment with CEP at
40 and 60 µM induced G2 arrest (31.7 and 35.5%, respectively)
compared with the vehicle control (14.0%) (Fig. 1D).
CEP induces apoptosis in MEL15-1 cells
via the caspase signaling pathway
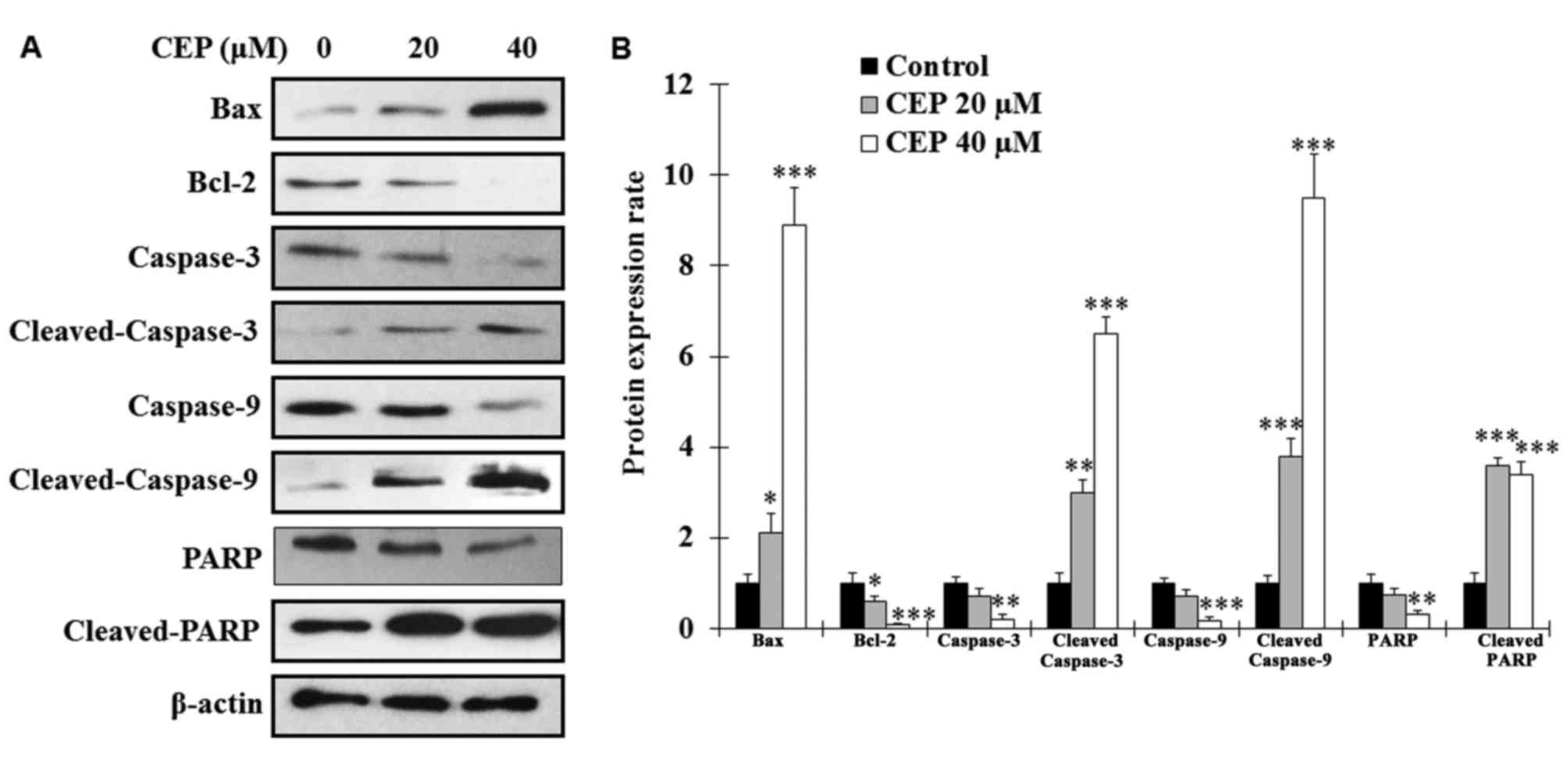
To analyze whether CEP induces MEL15-1 cell
cytotoxicity by triggering the apoptotic process, the expression
levels of the pro-apoptotic protein Bax and the anti-apoptotic
protein Bcl-2 were examined. The results revealed that, following
CEP treatment for 24 h, the expression levels of Bax in MEL15-1
cells were markedly increased, whereas the levels of Bcl-2 were
significantly decreased (Fig. 2).
Since enhanced Bax/Bcl-2 ratio is known as an activator of
caspase-3 (28,29), the effects of CEP on
caspase-associated proteins were analyzed. The results revealed
that procaspase-3, procaspase-9 and PARP were decreased
dose-dependently in MEL15-1 cells treated with CEP (Fig. 2). By contrast, increased levels of
active (cleaved) caspase-3, caspase-9 and PARP were observed in
cells treated with CEP (Fig. 2)
compared with control cells.
CEP induces cellular ROS production
and cyt c release from mitochondria
Previous studies have demonstrated that CEP inhibits
cancer cell proliferation by inducing ROS production (11,14).
Therefore, in the present study, it was hypothesized that CEP
induces MEL15-1 cells via affecting cellular ROS production. ROS
production was measured in MEL15-1 cells following 24 h treatment
with CEP (0–60 µM) in the presence or the absence of 5 mM NAC (a
commonly used antioxidant). Results demonstrated that treatment
with CEP significantly induced cellular ROS production compared
with the vehicle control, whereas concurrent treatment with NAC
attenuated this effect (Fig. 3A).
 | Figure 3.CEP induces cellular ROS production
and cyt c release from mitochondria. (A) Detection of ROS
production of MEL15-1 cells treated with between 0 and 60 µM CEP in
combination with or without 5 mM NAC for 24 h. Data represent the
mean ± SD from three separate experiments. (B) Western blot
analysis of cyt c from MEL15-1 cells treated with 40 µM CEP at
various time points, and quantification of cyt c expression rate.
(C) Western blot analysis of cyt c from MEL15-1 cells treated with
40 µM CEP with or without 5 mM NAC at 4 h, and quantification of
cyt c expression rate. Protein expression rate in control groups
was normalized to 1. β-Actin served as a loading control. Data are
presented as the mean ± SD from three separate experiments.
**P<0.01, ***P<0.001 vs. control; #P<0.05,
##P<0.01 vs. control. SD, standard deviation; cyt c,
cyctochrome c; ROS, reactive oxygen species; CEP, cepharanthine;
NAC, N-acetyl-L-cysteine; RLU, relative light units. |
Considering the aforementioned observation and the
evidence that ROS production is associated with mitochondria
function, it was investigated whether CEP-induced cell death
resulted from the effect of CEP on mitochondria function. It was
revealed that cyt c was released from mitochondria in MEL15-1 cells
following between 2 and 4 h treatment with 40 µM CEP, whereas
concurrent treatment with 5 mM NAC prevented this event (Fig. 3B and C). These results indicated that
CEP triggers MEL15-1 cell apoptosis through the mitochondrial
signaling pathway by inducing ROS production.
Proliferation inhibitory effects of
CEP on MEL15-1 cells are mediated by activation of the JNK
signaling pathway
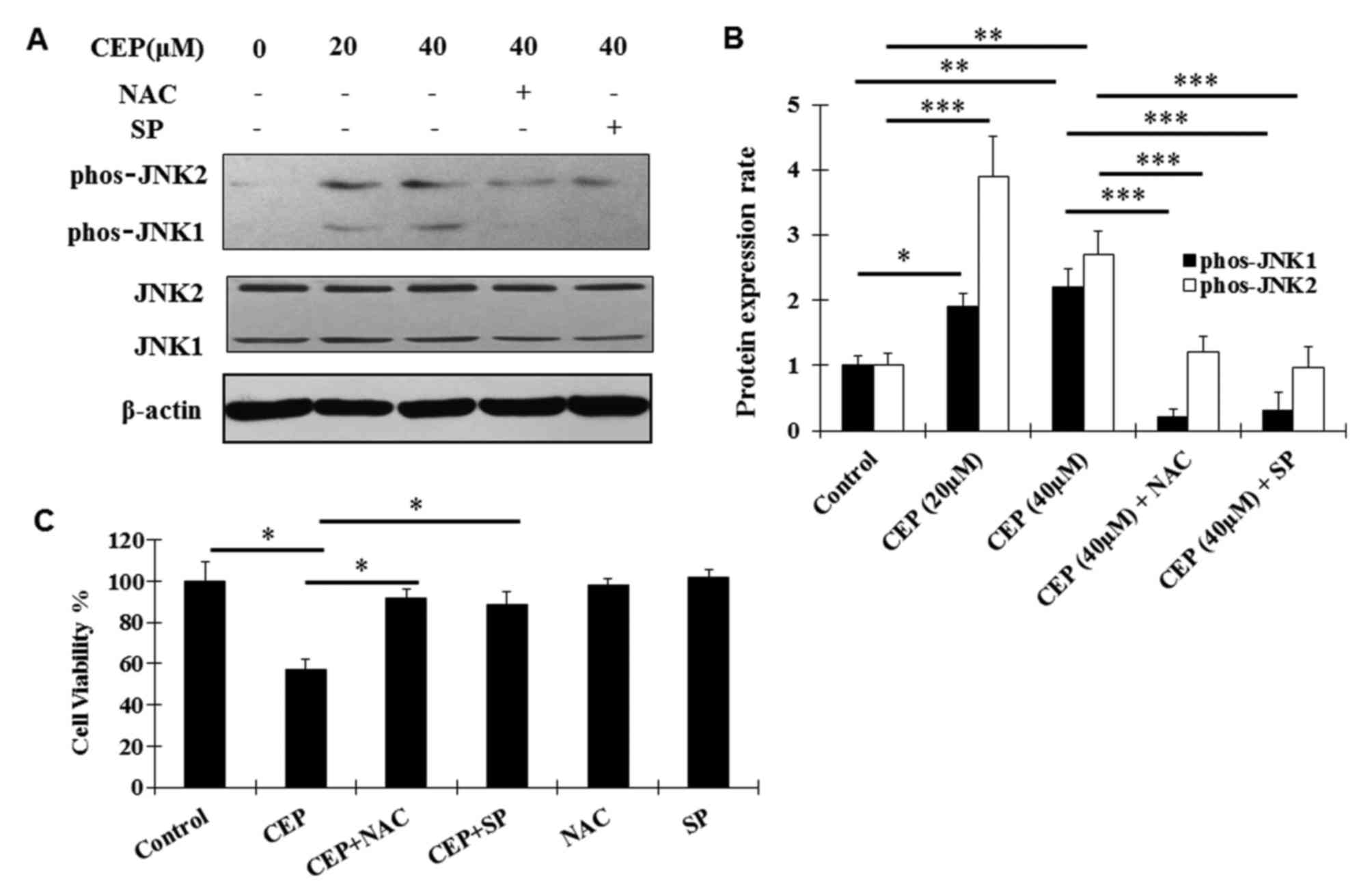
The JNK signaling pathway is known to be a
pro-apoptotic signaling pathway and has been identified to be
implicated in CEP-induced apoptosis in other tumor cells (14,30).
Aiming at dissection of the molecular mechanism of the antitumor
effects of CEP in MEL, the regulation of CEP on the JNK signaling
pathway was examined using western blot analysis. Activation
(phosphorylation) of JNK1 and JNK2 was observed following CEP
treatment (Fig. 4A and B). Such
activation was abolished by the co-treatment with CEP and SP600125,
a novel and selective inhibitor of JNK (31) (Fig. 4A and
B). In addition, the ROS scavenger NAC significantly decreased
the CEP-induced phosphorylation of JNK1/2 (Fig. 4A and B), indicating that the
activation of the JNK signaling pathway is dependent on ROS
production.
To further analyze whether CEP exerts the inhibitory
effects on MEL15-1 cells, dependent on ROS production and the JNK
signaling pathway, MEL15-1 cells were treated with CEP in
combination with or without the ROS scavenger NAC and the JNK
inhibitor SP600125. The results demonstrated that NAC and SP600125
reversed the CEP-induced inhibition on MEL15-1 cell viability
(Fig. 4C).
Inhibitory effects of CEP on tumor
growth
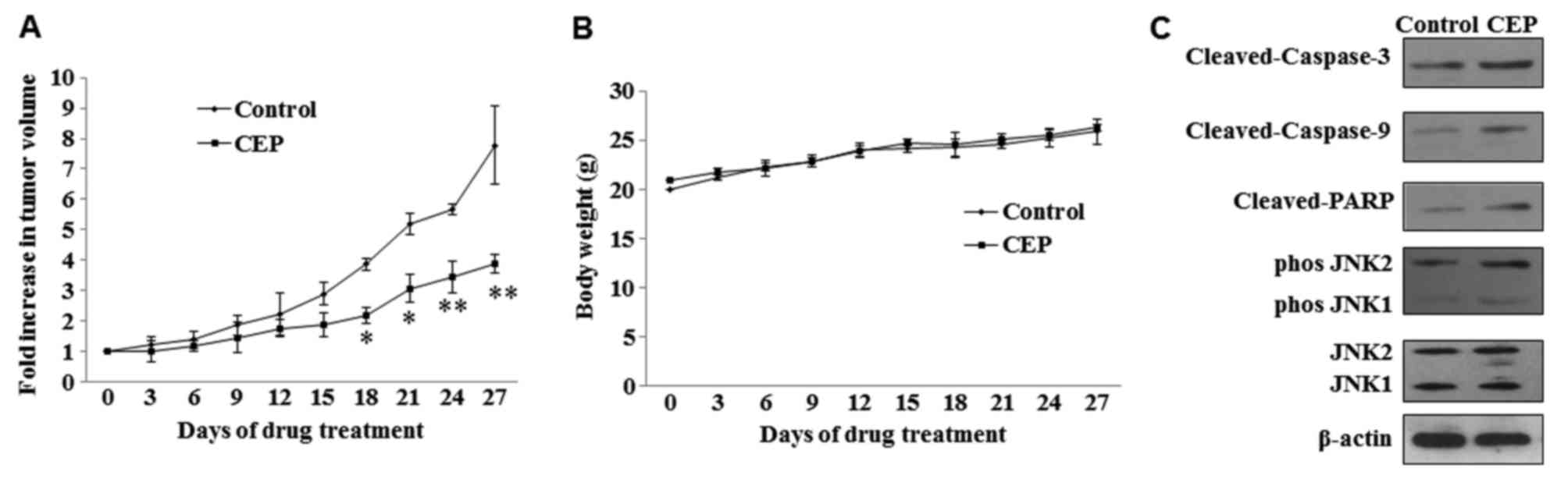
Mice were treated with 25 mg/kg doses of CEP for 4
weeks (5 times/week) and tumor growth was measured during the
treatment period. Statistically significant growth inhibition was
seen with CEP treatment when compared with the control (Fig. 5A). No significant loss in body weight
was observed in mice treated with CEP (Fig. 5B). To examine whether CEP induces
MEL15-1 cell apoptosis and regulates the JNK signaling pathway
in vivo, the expression of proteins in the caspase signaling
pathway and JNK signaling pathway in tumor tissues treated with or
without CEP was analyzed. Consistent with the in vitro
results of the present study, increased levels of active (cleaved)
caspase-3, caspase-9 and PARP were observed in tumors treated with
CEP compared with those treated with vehicle control. Activation
(phosphorylation) of JNK1 and JNK2 was also observed upon CEP
treatment (Fig. 5C).
Discussion
The results of the present study demonstrated that
CEP, a natural alkaloid extracted from the roots of S.
cepharantha Hayata, potently suppresses the proliferation of
choroidal melanoma cells, induces cell cycle arrest and activates
cellular apoptotic proteins, including Bax, caspase and PARP. The
results of the present study also revealed that CEP induced the
production of ROS and led to cyt c release, whereas the ROS
scavenger NAC was able to attenuate the situation. In addition,
consistent with a previous study (14), CEP was also revealed to activate JNK1
and JNK2 (members of the mitogen-activated protein kinase pathway),
which may serve an important role in CEP induced apoptosis.
In the present study, it was demonstrated that CEP
induces MEL15-1 cell apoptosis in a dose-dependent manner. The
Bcl-2 protein family is known to control the commitment of cells to
apoptosis (32). The increased
expression level of Bax and a reduced level of Bcl-2, as well as
increased levels of cleaved caspase-3 and caspase-9, induced by CEP
treatment in MEL15-1 cells indicated that CEP-induced apoptosis
occurs via the activation of caspase-3 and caspase-9 and the
mitochondrial pathways. In addition, consistent with previous
studies of CEP in other cancer cells (11,14), the
results demonstrated that the production of ROS increased in
CEP-treated MEL15-1 cells. The excessive production of ROS is
likely due to stimuli that act on the Bcl-2 family protein,
subsequently altering the mitochondrial membrane integrity. This
was substantiated by the observation that CEP induced the release
of cyt c from mitochondria.
JNK activation has been revealed to lead to
apoptosis and correlate positively with oxidative stress-induced
apoptosis (33–35). The activation of JNK was also revealed
to be implicated in CEP-triggered apoptosis in human hepatocellular
carcinoma cells (14). In the present
study, it was observed that ROS and JNK activation are positively
associated with the anticancer effects of CEP in MEL15-1 cells.
Notably, the results of the present study revealed that treatment
with the ROS scavenger NAC may abolish the activation of JNK in
MEL15-1 cells and the inhibitory effects of CEP on cell viability,
indicating that CEP-induced JNK activation is ROS-dependent. Other
functional consequences for CEP-induced ROS, as well as JNK
activation, are not yet established and require additional
investigation.
CEP, as a natural alkaloid extract, has been
demonstrated to effectively inhibit the proliferation of various
cancer cell lines (9–11,14–17).
Notably, it was also revealed to possess anti-metastasis activity
against different types of cancer cells (13,36). CEP
was demonstrated to suppress invasion and migration of
cholangiocarcinoma cells, possibly by suppressing intercellular
adhesion molecule-1 and matrix metalloproteinase-2 (13). Intraperitoneal injection of CEP was
reported to inhibit the metastasis of Lewis lung carcinoma in a
mouse model (37), and CEP inhibited
metastasis of human colon cancer cells in a mouse model (36). In addition, the combination of CEP and
other chemotherapeutic agents, including 5-fluorouracil, or
recombinant human interferon-β or -γ, were demonstrated to be even
more effective to prevent metastasis (36,37). In
the present study, the inhibitory effects of CEP on primary
choroidal melanoma tumor growth were observed. Since metastasis
remains a life-threatening risk for choroidal melanoma (38), future studies will focus on the effect
of CEP (alone or in combination with other agents) on metastasis of
choroidal melanoma using a metastatic choroidal melanoma model to
develop a safe and effective regimen for choroidal melanoma
therapy.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant nos. 81201188 and 81472344) and
the International Cooperation Project of Jilin Provincial Science
and Technology Department (grant no. 20150414031GH).
References
|
1
|
Laver NV, McLaughlin ME and Duker JS:
Ocular melanoma. Arch Pathol Lab Med. 134:1778–1784.
2010.PubMed/NCBI
|
|
2
|
Singh AD, Turell ME and Topham AK: Uveal
melanoma: Trends in incidence, treatment, and survival.
Ophthalmology. 118:1881–1885. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh P and Singh A: Choroidal melanoma.
Oman J Ophthalmol. 5:3–9. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Finger PT: Radiation therapy for choroidal
melanoma. Surv Ophthalmol. 42:215–232. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Finger PT: Transpupillary thermotherapy in
choroidal melanoma. Ophthalmology. 104:731–732. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oosterhuis JA, Journée-de Korver HG and
Keunen JE: Transpupillary thermotherapy: Results in 50 patients
with choroidal melanoma. Arch Ophthalmol. 116:157–162. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wilson MW and Hungerford JL: Comparison of
episcleral plaque and proton beam radiation therapy for the
treatment of choroidal melanoma. Ophthalmology. 106:1579–1587.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gragoudas ES, Egan KM, Seddon JM, Glynn
RJ, Walsh SM, Finn SM, Munzenrider JE and Spar MD: Survival of
patients with metastases from uveal melanoma. Ophthalmology.
98:383–390. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rogosnitzky M and Danks R: Therapeutic
potential of the biscoclaurine alkaloid, cepharanthine, for a range
of clinical conditions. Pharmacol Rep. 63:337–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gulcin I, Elias R, Gepdiremen A, Chea A
and Topal F: Antioxidant activity of bisbenzylisoquinoline
alkaloids from Stephania rotunda: Cepharanthine and fangchinoline.
J Enzyme Inhib Med Chem. 25:44–53. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Furusawa S, Wu J, Fujimura T, Nakano S,
Nemoto S, Takayanagi M, Sasaki K and Takayanagi Y: Cepharanthine
inhibits proliferation of cancer cells by inducing apoptosis.
Methods Find Exp Clin Pharmacol. 20:87–97. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kikukawa Y, Okuno Y, Tatetsu H, Nakamura
M, Harada N, Ueno S, Kamizaki Y, Mitsuya H and Hata H: Induction of
cell cycle arrest and apoptosis in myeloma cells by cepharanthine,
a biscoclaurine alkaloid. Int J Oncol. 33:807–814. 2008.PubMed/NCBI
|
|
13
|
Uthaisar K, Seubwai W, Srikoon P,
Vaeteewoottacharn K, Sawanyawisuth K, Okada S and Wongkham S:
Cepharanthine suppresses metastatic potential of human
cholangiocarcinoma cell lines. Asian Pac J Cancer Prev. 13:(Suppl).
S149–S154. 2012.
|
|
14
|
Biswas KK, Tancharoen S, Sarker KP,
Kawahara K, Hashiguchi T and Maruyama I: Cepharanthine triggers
apoptosis in a human hepatocellular carcinoma cell line (HuH-7)
through the activation of JNK1/2 and the downregulation of Akt.
FEBS Lett. 580:703–710. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu J, Suzuki H, Zhou YW, Liu W, Yoshihara
M, Kato M, Akhand AA, Hayakawa A, Takeuchi K, Hossain K, et al:
Cepharanthine activates caspases and induces apoptosis in Jurkat
and K562 human leukemia cell lines. J Cell Biochem. 82:200–214.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Z, Huang C, Yang YL, Ding Y, Ou-Yang
HQ, Zhang YY and Xu M: Inhibition of the STAT3 signaling pathway is
involved in the antitumor activity of cepharanthine in SaOS2 cells.
Acta Pharmacol Sin. 33:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seubwai W, Vaeteewoottacharn K, Hiyoshi M,
Suzu S, Puapairoj A, Wongkham C, Okada S and Wongkham S:
Cepharanthine exerts antitumor activity on cholangiocarcinoma by
inhibiting NF-kappaB. Cancer Sci. 101:1590–1595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morioka S, Ono M, Tanaka N and Orita K:
Synergistic activation of rat alveolar macrophages by cepharanthine
and OK-432. Gan To Kagaku Ryoho. 12:1470–1475. 1985.(In Japanese).
PubMed/NCBI
|
|
19
|
Furusawa S and Wu J: The effects of
biscoclaurine alkaloid cepharanthine on mammalian cells:
Implications for cancer, shock, and inflammatory diseases. Life
Sci. 80:1073–1079. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ita M, Halicka HD, Tanaka T, Kurose A,
Ardelt B, Shogen K and Darzynkiewicz Z: Remarkable enhancement of
cytotoxicity of onconase and cepharanthine when used in combination
on various tumor cell lines. Cancer Biol Ther. 7:1104–1108. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kato T and Suzumura Y: Potentiation of
antitumor activity of vincristine by the biscoclaurine alkaloid
cepharanthine. J Natl Cancer Inst. 79:527–532. 1987.PubMed/NCBI
|
|
22
|
Harada K, Ferdous T, Itashiki Y, Takii M,
Mano T, Mori Y and Ueyama Y: Effects of cepharanthine alone and in
combination with fluoropyrimidine anticancer agent, S-1, on tumor
growth of human oral squamous cell carcinoma xenografts in nude
mice. Anticancer Res. 29:1263–1270. 2009.PubMed/NCBI
|
|
23
|
Kisara S, Furusawa S, Murata R, Ogata M,
Hikichi N, Takayanagi Y and Sasaki K: Combined effects of
buthionine sulfoximine and cepharanthine on cytotoxic activity of
doxorubicin to multidrug-resistant cells. Oncol Res. 7:191–200.
1995.PubMed/NCBI
|
|
24
|
Zhou Y, Hopper-Borge E, Shen T, Huang XC,
Shi Z, Kuang YH, Furukawa T, Akiyama S, Peng XX, Ashby CR Jr, et
al: Cepharanthine is a potent reversal agent for MRP7
(ABCC10)-mediated multidrug resistance. Biochem Pharmacol.
77:993–1001. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiraishi N, Akiyama S, Nakagawa M,
Kobayashi M and Kuwano M: Effect of bisbenzylisoquinoline
(biscoclaurine) alkaloids on multidrug resistance in KB human
cancer cells. Cancer Res. 47:2413–2416. 1987.PubMed/NCBI
|
|
26
|
Hotta T, Tanimura H, Yamaue H, Iwahashi M,
Tani M, Tsunoda T, Noguchi K, Mizobata S and Terasawa H:
Synergistic effects of tamoxifen and cepharanthine for
circumventing the multidrug resistance. Cancer Lett. 107:117–123.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Single B, Leist M and Nicotera P:
Simultaneous release of adenylate kinase and cytochrome c in cell
death. Cell Death Differ. 5:1001–1003. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Salakou S, Kardamakis D, Tsamandas AC,
Zolota V, Apostolakis E, Tzelepi V, Papathanasopoulos P, Bonikos
DS, Papapetropoulos T, Petsas T and Dougenis D: Increased Bax/Bcl-2
ratio up-regulates caspase-3 and increases apoptosis in the thymus
of patients with myasthenia gravis. In Vivo. 21:123–132.
2007.PubMed/NCBI
|
|
29
|
Yang B, Johnson TS, Thomas GL, Watson PF,
Wagner B, Furness PN and El Nahas AM: A shift in the Bax/Bcl-2
balance may activate caspase-3 and modulate apoptosis in
experimental glomerulonephritis. Kidney Int. 62:1301–1313. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen HM and Liu ZG: JNK signaling pathway
is a key modulator in cell death mediated by reactive oxygen and
nitrogen species. Free Radic Biol Med. 40:928–939. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakano H, Nakajima A, Sakon-Komazawa S,
Piao JH, Xue X and Okumura K: Reactive oxygen species mediate
crosstalk between NF-kappaB and JNK. Cell Death Differ. 13:730–737.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How Can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ono M, Tanaka N and Orita K: Positive
interactions between human interferon and cepharanthin against
human cancer cells in vitro and in vivo. Cancer Chemother
Pharmacol. 35:10–16. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ito H, Ito H, Amano H and Noda H:
Inhibitory effect of a biscoclaurine alkaloid, cepharanthin, on
lung metastasis of Lewis lung carcinoma. Jpn J Pharmacol.
56:195–202. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rietschel P, Panageas KS, Hanlon C, Patel
A, Abramson DH and Chapman PB: Variates of survival in metastatic
uveal melanoma. J Clin Oncol. 23:8076–8080. 2005. View Article : Google Scholar : PubMed/NCBI
|