Introduction
Nasopharyngeal carcinoma (NPC) is a common cancer of
the head and neck that is prevalent in Southern China and Southeast
Asia (1). Radiotherapy is the primary
treatment for this disease, with chemotherapy usually administered
in combination with radiotherapy to patients with locally advanced,
metastatic or recurrent disease (1,2).
Cisplatin-based chemotherapy is the standard first-line regimen for
advanced NPC and achieves good responses (1). The major obstacle to the efficacy of
cisplatin is drug resistance, the underlying molecular mechanisms
of which remain obscure (3).
Therefore, dissecting the complicated mechanisms involved in
cisplatin resistance in NPC and identifying valuable targets to
reverse this resistance, as well as developing novel strategies for
improving the clinical efficacy of cisplatin, is of great
value.
Autophagy is an evolutionarily conserved degradation
and recycling process (4–6). Double-membrane autophagosomes sequester
cytoplasmic components, such as damaged proteins and organelles,
and fuse with lysosomes to form autolysosomes, in which cytoplasmic
components are degraded for recycling (4–6).
Microtubule-associated protein 1 light chain 3β (LC3B) is a
mammalian homologue of the protein autophagy-related gene 8 (Atg8)
in yeast. LC3B exists in two forms: LC3B-I and LC3B-II. LC3B-I is a
non-lipidated form that can be modified to the lipidated form,
LC3B-II, which is closely correlated with the number of
autophagosomes (7). Autophagy
proceeds at a low basal level in cells to maintain cellular
homeostasis, but is activated in response to different forms of
metabolic stress, including nutrient starvation, growth factor
depletion and hypoxia (4–6). Tumour cells can utilize autophagy to
evade death induced by chemotherapies, with the inhibition of
autophagy enhancing drug cytotoxicity (8–10).
Nevertheless, certain evidence suggests that autophagy is not
cytoprotective, but is necessary for the anticancer effect of drugs
(11–13). This ‘double-edged sword’
characteristic of autophagy necessitates the identification of its
precise role in chemotherapy under specific tumour settings.
The epithelial-mesenchymal transition (EMT) is a
process by which epithelial cells transform into a mesenchymal
phenotype with increased mobility (14). It is well known that the EMT
facilitates a potential mechanism of metastasis, by which
epithelial cancer cells detach from adjacent cells and invade the
surrounding stroma (15). Moreover,
compelling evidence has indicated that the EMT of tumour cells also
contributes to the chemoresistance to drugs, including cisplatin
(16,17). Previously, autophagy induction was
shown to impair migration and invasion by reversing the EMT in
glioblastoma cells, indicating the presence of a potential link
between autophagy and the EMT (18).
However, this link is controversial and the effect of autophagy on
the EMT process in NPC has not been investigated. The present study
therefore aimed to clarify the role of autophagy and EMT in
response to cisplatin treatment in NPC and to identify the
underlying impact of autophagy on the EMT process.
Materials and methods
Reagents
Cisplatin and CQ were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Recombinant human transforming
growth factor β1 (TGFβ1) was purchased from Peprotech Inc. (Rocky
Hill, NJ, USA). The polyclonal mouse anti-glyceraldehyde
3-phosphate dehydrogenase (GAPDH) antibody (catalogue no. AG019),
secondary antibodies HRP-labeled goat anti-rabbit IgG (catalogue
no. A0208) and HRP-labelled goat anti-mouse IgG (catalogue no.
A0216), cell lysis buffer for Western and IP, BeyoECL Plus,
4′,6-diamidino-2-phenylindole (DAPI) and Cell Counting Kit-8
(CCK-8) were purchased from Beyotime Institute of Biotechnology
(Shanghai, China). The monoclonal rabbit antibodies against LC3B
(catalogue no. 3868), E-cadherin (catalogue no. 3195) and vimentin
(catalogue no. 5741) were purchased from Cell Signalling Technology
(Danvers, MA, USA). Fluorescein isothiocyanate (FITC)-conjugated
secondary antibody goat anti-rabbit IgG (catalogue no. 70-GAR001)
was purchased from MultiSciences Biotech Co., Ltd. (Hangzhou,
China). LC3B small interfering RNA (siRNA; catalogue no. sc-43390)
and control siRNA (catalogue no. sc-37007) were purchased from
Santa Cruz (Dallas, TX, USA). The riboFECT™ Transfection kit was
purchase from Guangzhou RiboBio Co., Ltd. (Guangzhou, China).
TRIzol Reagent was purchased from Invitrogen (Thermo Fisher
Scientific Inc., Waltham, MA, USA). The SuperQuickRT MasterMix kit
and the UltraSYBR Mixture kit were purchased from Beijing ComWin
Biotech Co., Ltd. (Beijing, China).
Cell culture
The poorly-differentiated NPC 6–10B and 5–8F cell
lines were purchased from the Cell Centre of Central South
University (Changsha, China). The cells were cultured in RPMI-1640
medium (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA)
supplemented with 10% foetal bovine serum (FBS), 100 U/ml
penicillin and 100 µg/ml streptomycin (all Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in a humidified atmosphere with 5%
CO2. All experiments were conducted on cells in the
logarithmic growth stage.
Transmission electron microscopy
(TEM)
The 6–10B cells were incubated with 2 µg/ml
cisplatin or vehicle for 24 h, and then collected and centrifuged
at 1,000 × g for 10 min at 4°C. The cells incubated with vehicle
were used as the control group. The cells were fixed with 2.5%
glutaraldehyde for 24 h and 2% osmic acid for 2 h. The fixed
samples were dehydrated with increasing concentrations (50–70%) of
acetone. Next, the samples were infiltrated in the mixed liquor of
epoxy resin and acetone for 24 h at 37°C and finally embedded in
epoxy resin for 24 h at 60°C. Ultrathin sections were prepared with
an ultramicrotome, and then stained with uranyl acetate and lead
citrate. The sections were observed under a transmission electron
microscope (Hitachi HT-7700; Hitachi, Ltd., Tokyo, Japan).
Cell viability assay
Cell viability was measured using the CCK-8 assay
according to the manufacturer's instructions. In brief, the 6–10B
and 5–8F cells were cultured at 3×103/well in triplicate
in 96-well plates and allowed to attach for 12 h. The complete
RPMI-1640 medium was then removed and replaced with fresh medium
containing drugs. The 6–10B and 5–8F cells were treated with
cisplatin (0.31–10 µg/ml) and CQ (10 µM) or cisplatin and vehicle
for 48 h at 37°C. For the TGFβ1 treatment, the 6–10B and 5–8F cells
were pre-incubated with TGFβ1 (10 ng/ml) or vehicle for 24 h at
37°C, followed by the addition of cisplatin (0.31–10 µg/ml) for 48
h at 37°C. Subsequently, the absorbance was determined at a
wavelength of 450 nm using a microplate reader (BioTek Instruments,
Inc., Winooski, VT, USA). Cell viability was calculated by dividing
the absorbance values of the treated cells by that of the control
cells.
Immunofluorescence staining
The 6–10B and 5–8F cells were seeded at a
concentration of 1×105 cells/well in 12-well plates and
allowed to attach for 12 h at 37°C. The 6–10B and 5–8F cells were
treated with TGFβ1 (10 ng/ml) or vehicle for 48 h at 37°C. For the
CQ treatment, the 6–10B and 5–8F cells were pre-incubated with
TGFβ1 (10 ng/ml) for 24 h and followed by the addition of CQ (10
µM) or vehicle for 48 h at 37°C. Subsequently, the cells were fixed
with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton
X-100 for 15 min and then blocked with 1% bovine serum albumin
(BSA) for 30 min. The cells were incubated with primary antibody
(vimentin, 1:200) diluted with 1% BSA at 37°C for 1 h, followed by
FITC-conjugated secondary antibody (1:200) at 37°C for 1 h. The
cells were then counterstained with DAPI and examined using a
fluorescence microscope (Leica Microscopes GmbH, Wetzlar,
Germany).
Western blotting
The 6–10B and 5–8F cells were washed three times
with PBS and then lysed with cell lysis buffer for 30 min on ice.
The lysates were centrifuged at 12,000 × g for 10 min at 4°C. The
supernatants were collected for western blot analyses. Total
protein (50 µg) was separated by 12% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). After blocked with 5% non-fat milk for 1 h at room
temperature, the blotted membranes were incubated with the
corresponding primary antibodies overnight at 4°C, followed by the
relevant secondary antibodies for 1 h at room temperature. The
primary antibodies included monoclonal rabbit anti-LC3B (1:800),
E-cadherin (1:800), vimentin (1:800) and polyclonal mouse
anti-GAPDH (1:1,000). The blots were detected using BeyoECL Plus
reagent and exposed in a ChemiDoc MP imaging system (Bio-Rad
Laboratories, Inc.).
siRNA transfection
The 6–10B cells were transfected with LC3B siRNA or
control siRNA using the riboFECT™ Transfection kit
according to the manufacturer's protocol. The transfected cells
were collected after a 72-h incubation and the efficacy of the
siRNA knockdown was determined by western blot assay.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from the 6–10B cells was isolated using
TRIzol reagent according to the manufacturer's protocol. Total RNA
(1 µg) was reverse-transcribed using a SuperQuickRT MasterMix kit.
qPCR was performed using an UltraSYBR Mixture kit in a Bio-Rad IQ5™
Multicolor Real-Time PCR detection system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The cycling conditions used were as
follows: 95°C for 10 min, 95°C for 15 sec and 62.5°C for 60 sec for
45 cycles. qPCR quantification was conducted using the
2−ΔΔCt method (19). The
following primer sequences were used: E-cadherin forward,
5′-TGCCCAGAAAATGAAAAAGG-3′ and reverse, 5′-GTGTATGTGGCAATGCGTTC-3′;
Vimentin forward, 5′-GAGAACTTTGCCGTTGAAGC-3′ and reverse,
5′-TCCAGCAGCTTCCTGTAGGT-3′; Snail forward,
5′-GCGAGCTGCAGGACTCTAAT-3′ and reverse, 5′-GGACAGAGTCCCAGATGAGC-3′;
Slug forward, 5′-TTCGGACCCACACATTACCT-3′ and reverse,
5′-TGACCTGTCTGCAAATGCTC-3′; and β-actin forward,
5′-CTCTTCCAGCCTTCCTTCCT-3′ and reverse,
5′-AGCACTGTGTTGGCGTACAG-3′.
Statistical analysis
All experiments were performed at least three times.
Statistical analysis was performed using SPSS 13.0 software (SPSS,
Inc., Chicago, IL, USA). Quantitative data were expressed as the
mean ± standard deviation. Comparison between two groups was
performed using a two-sided, unpaired Student's t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Cisplatin induces autophagy, the
inhibition of which enhances the cytotoxicity of cisplatin in NPC
cells
To clarify the effects of cisplatin on autophagy in
NPC cells, TEM assays were initially performed to visualize
autophagic vacuoles, which are canonical morphological alterations
of autophagy and indicate the occurrence of autophagy (7). After a 24-h incubation with cisplatin,
the 6–10B cells contained more autophagic vacuoles than the control
cells, including early autophagic vacuoles and late degradative
autophagic vacuoles (Fig. 1A). Next,
the expression of LC3B-II in the cells under cisplatin treatment
was assessed by western blot analysis. Cisplatin incubation
increased the level of LC3B-II in a dose- and time-dependent manner
in the 5–8F and 6–10B cells (Fig.
1B). Blocking the late stage of autophagy by the addition of CQ
further increased the expression of LC3B-II in cisplatin-treated
NPC cells (Fig. 1C), suggesting that
cisplatin induces autophagosome accumulation. Finally, the CCK-8
assay revealed that the half-maximal inhibitory concentration
(IC50) value decreased in the CQ group compared with the
control group (6–10B cells: 2.17±0.01 vs. 2.67±0.13, P<0.01;
5–8F cells: 1.01±0.08 vs. 1.35±0.03, P<0.01) (Fig. 1D). Collectively, these data suggested
that cisplatin induces autophagy and that inhibiting autophagy
augments the sensitivity of NPC cells to cisplatin.
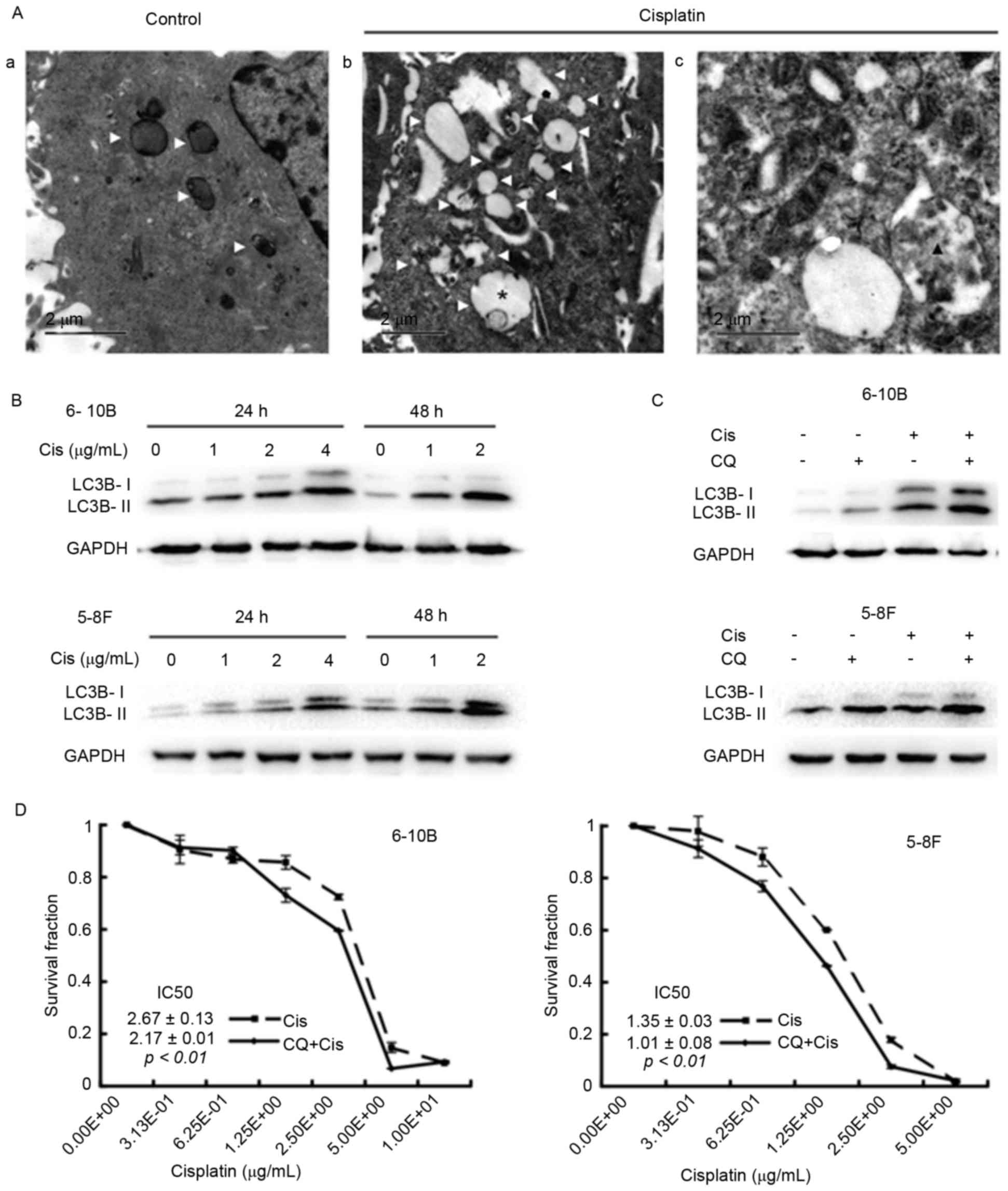 | Figure 1.Cisplatin induces autophagy, while
the inhibition of autophagy by CQ elevates the cytotoxicity of
cisplatin in 6–10B and 5–8F nasopharyngeal carcinoma cells. (A) The
6–10B cells were incubated with (Aa) vehicle or with (Ab and Ac) 2
µg/ml cisplatin for 24 h and then subjected to transmission
electron microscopy. White triangles denote an autophagic vacuole,
the asterisk denotes an early autophagic vacuole and the black
triangle denotes a degradative autophagic vacuole. (B) The 6–10bB
and 5–8F cells were treated with 0, 1, 2 or 4 µg/ml cisplatin for
24 or 48 h. Western blot analysis revealed a dose- and
time-dependent increase in LC3B-II expression in the
cisplatin-treated 6–10B and 5–8F cells. (C) The 6–10B and 5–8F
cells were incubated with 2 µg/ml cisplatin and/or 10 µM CQ for 24
h. Western blot analysis showed that cisplatin enhanced the
expression of LC3B-II, and the combination of cisplatin and CQ
resulted in more LC3B-II expression. (D) The 6–10B and 5–8F cells
were treated with increasing concentrations of cisplatin (0.31–10
µg/ml) in the presence or absence of 10 µM CQ for 48 h. The CCK-8
assay revealed that the IC50 value decreased in the CQ
group compared with the blank group (6–10B cells: 2.17±0.01 vs.
2.67±0.13, P<0.01; 5–8F cells: 1.01±0.08 vs. 1.35±0.03,
P<0.01). Cis, cisplatin; CQ, chloroquine; LC3B,
microtubule-associated protein 1 light chain 3B; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; IC50,
half-maximal inhibitory concentration. |
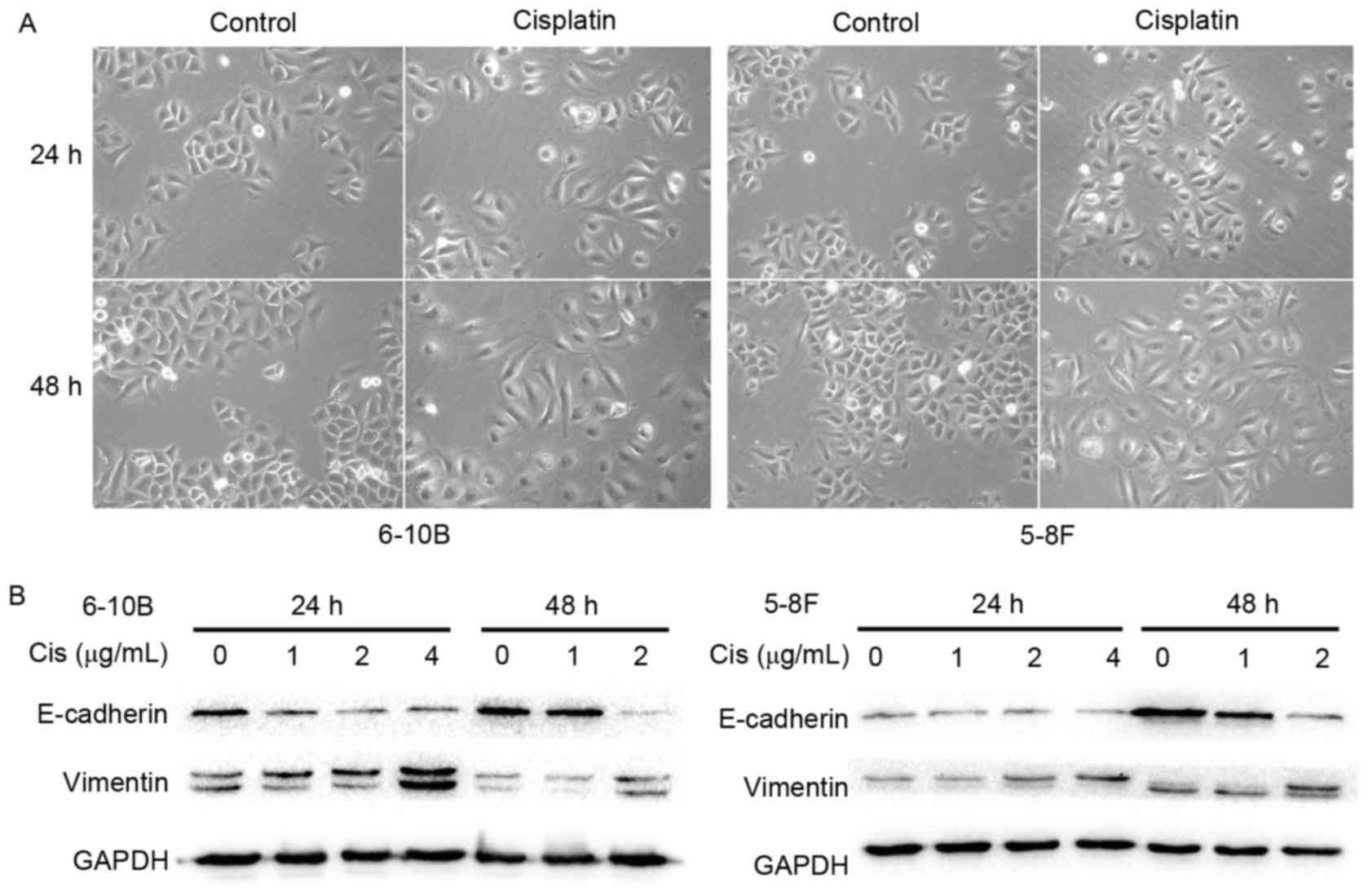
Cisplatin induces the EMT phenotype in
NPC cells
To clarify the impact of cisplatin on the EMT
process, the morphological changes of NPC cells under cisplatin
treatment were first observed using an inverted phase-contrast
microscope. After 24 or 48 h of treatment with 2 µg/ml cisplatin,
the 6–10B and 5–8F cells gradually exhibited a spindle-like
morphology with pseudopodia and reduced cell-cell contacts
(Fig. 2A). These observations
suggested that the NPC cells underwent a characteristic EMT
morphological change under stimulation with cisplatin. To further
elucidate this observation, the expression of EMT-related
biomolecules was measured by western blot analysis. A marked
decrease in the epithelial marker E-cadherin was observed together
with an increase in the mesenchymal marker vimentin in the 6–10B
and 5–8F cells following incubation with cisplatin (Fig. 2B). These results indicated that
cisplatin elicits the EMT phenotype in NPC cells.
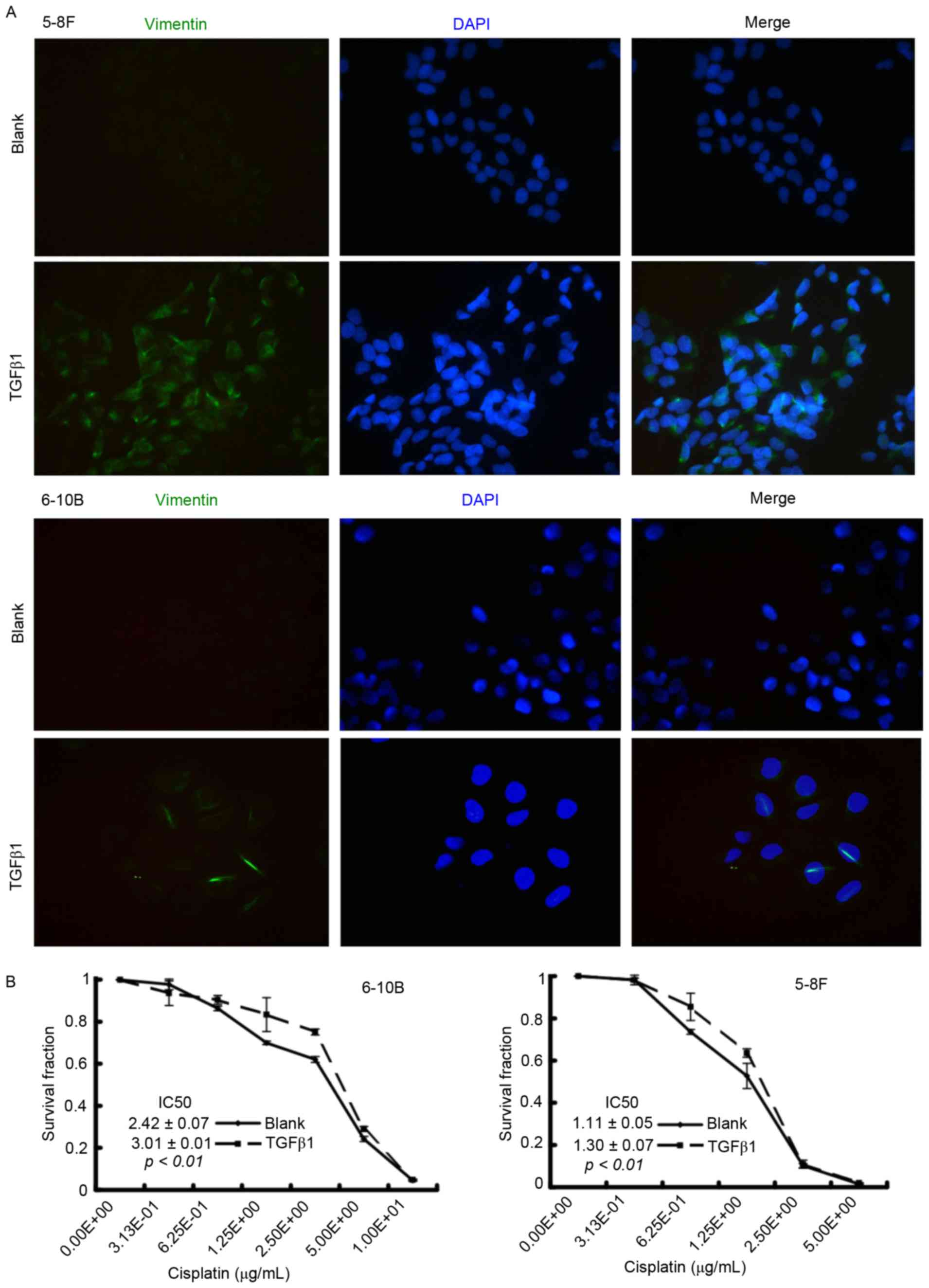
TGFβ1 induces EMT and promotes
cisplatin resistance in NPC cells
To clarify the role of EMT in cisplatin sensitivity,
the NPC cells were firstly treated with TGFβ1, which is a critical
inducer of EMT (15), to activate the
EMT process. After a 48-h incubation, immunofluorescence microscopy
showed that TGFβ1-treated NPC cells underwent an EMT-like
alteration, characterized by the upregulation of the mesenchymal
marker vimentin (Fig. 3A). Results of
the CCK-8 assay demonstrated that the IC50 value
increased in the TGFβ1 group compared with the blank group (6–10B:
3.01±0.01 vs. 2.42±0.07, P<0.01; 5–8F: 1.30±0.07 vs. 1.11±0.05,
P<0.01) (Fig. 3B). These results
indicated that TGFβ1-induced EMT is associated with cisplatin
resistance in NPC cells.
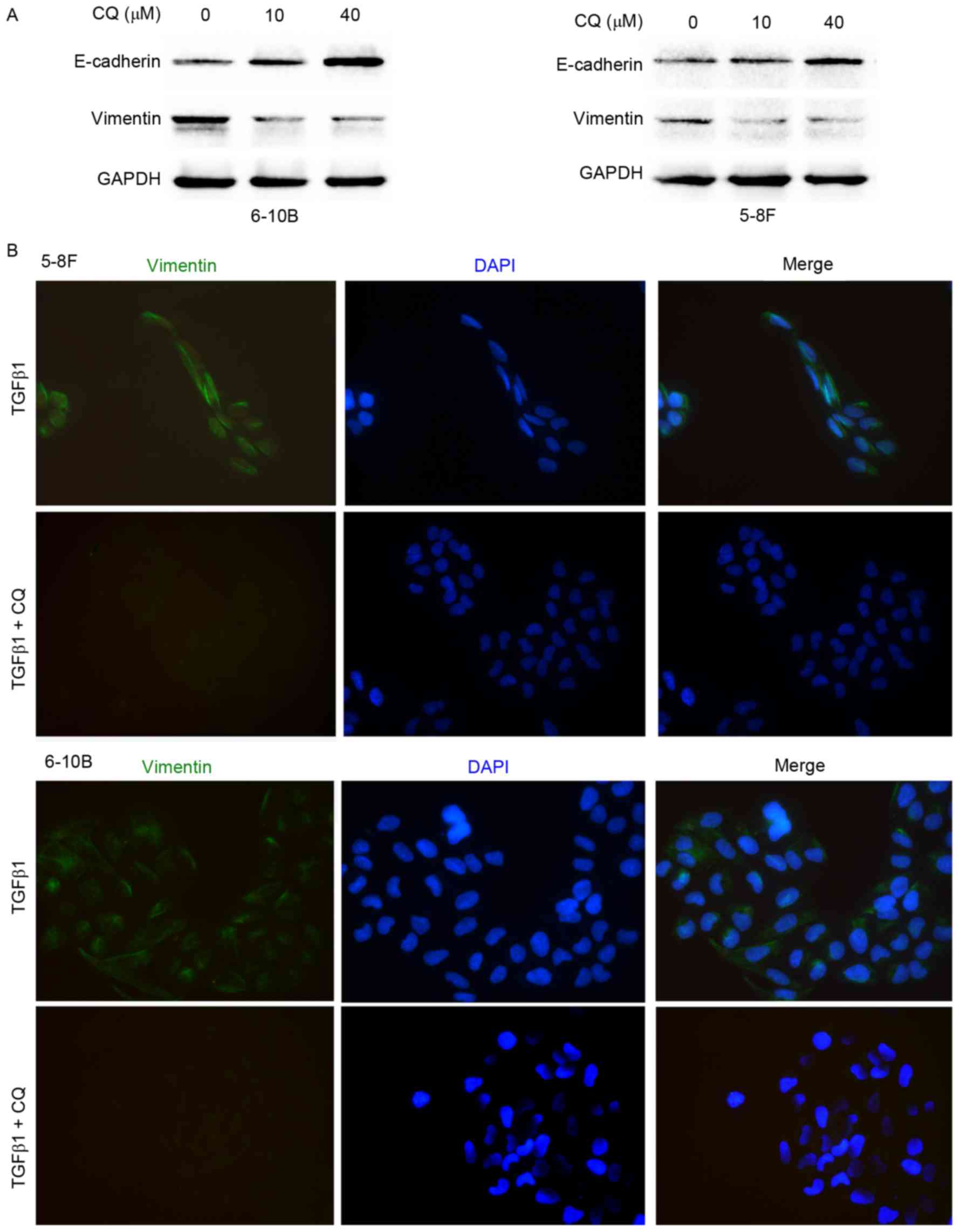
Autophagy inhibition impairs the EMT
process in NPC cells
To elucidate the effect of autophagy on the EMT
process, western blot analysis and immunofluorescence microscopy
were performed. As shown in Fig. 4A,
CQ downregulated the expression of vimentin and upregulated the
expression of E-cadherin in the 5–8F and 6–10B cells. Moreover,
immunofluorescence microscopy showed that CQ inhibited the
TGFβ1-induced increase in vimentin expression, suggesting that CQ
reverses the mesenchymal phenotype activated by TGFβ1 in 5–8F and
6–10B cells (Fig. 4B). To clarify the
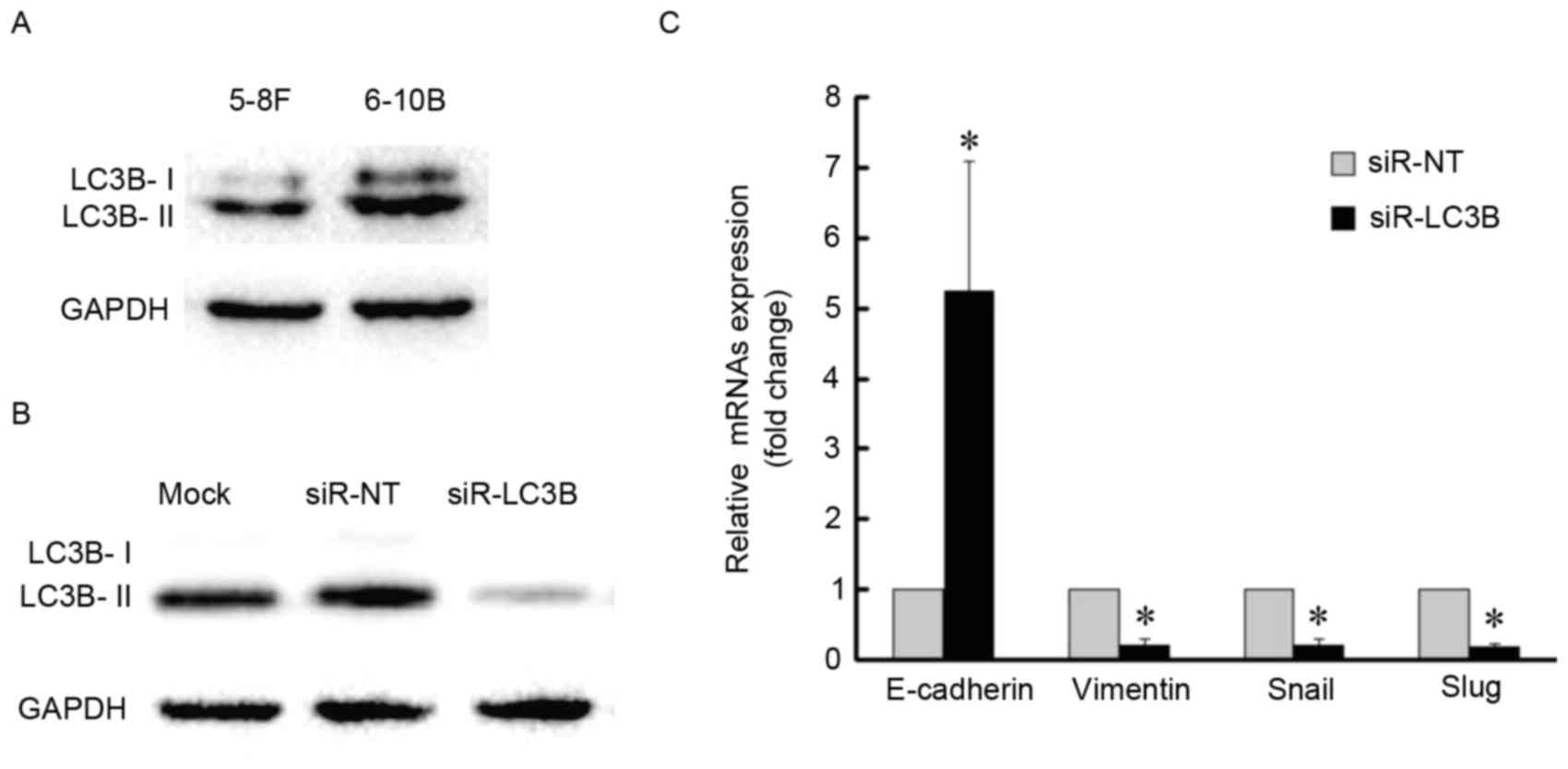
association between autophagy and EMT further, basal levels of LC3B
protein were detected in the two NPC cell lines and 6–10B cells
demonstrated a higher level of autophagy with a higher expression
level of LC3B-II (Fig. 5A). The 6–10B
cells were then selected for LC3B-knockdown by siRNA (Fig. 5B and C). LC3B-knockdown increased the
amount of E-cadherin mRNA, whereas the amount of vimentin, Snail
and Slug mRNA was decreased compared with the control (Fig. 5C). Together, these data indicated that
blockage of autophagy impairs the EMT process.
Discussion
Cisplatin is used in the clinical management of
various solid tumours, including NPC (1). Although good responses have been
observed, chemoresistance and cytotoxicity limit the use of
cisplatin in clinical management. Several mechanisms have been
described as being involved in the development of cisplatin
resistance, including alterations in drug transport, the DNA repair
apparatus and apoptotic signalling pathways (3,20).
Nevertheless, the molecular mechanism of cisplatin resistance
remains poorly understood. Previously, autophagy and EMT, two
interrelated biological processes, have been proposed to
participate in chemoresistance. In the present study, it was found
that cisplatin elicited cytoprotective autophagy and EMT in NPC
cells. Inhibition of autophagy enhanced sensitivity to cisplatin,
whereas activating the EMT process promoted resistance to cisplatin
in NPC cells. Moreover, inhibition of autophagy impaired the EMT
process in NPC cells.
EMT always characterizes advanced neoplastic
lesions. As such, EMT has been proposed to promote cancer
metastasis, the cancer stem-cell phenotype, immune evasion, and
radiotherapy and drug resistance (14,21,22).
Tumour cells able to survive chemotherapy treatment exhibit an EMT
phenotype. In NPC, paclitaxel- and cisplatin-resistant NPC cells
transform into a mesenchymal phenotype (23,24). In
the present study, NPC cells underwent characteristic morphological
and molecular transformations, such as EMT, upon cisplatin
treatment. Furthermore, TGFβ1, a well-known inducer of EMT, induced
the EMT phenotype and promoted cisplatin resistance in the NPC
cells, supporting the theory that the EMT process establishes a
resistance phenotype to cisplatin treatment in NPC.
Autophagy affects drug sensitivity in tumour
treatment, and can serve as either a mechanism of drug resistance
or a mechanism of killing tumour cells (25). Chloroquine is a well-established
inhibitor of autophagy that blocks the late stage of autophagy by
preventing lysosomal degradation (7).
The present study demonstrates that treatment with cisplatin
induced autophagy in the NPC cells. Autophagy inhibition by CQ
enhanced sensitivity to cisplatin in NPC cells. This result was
consistent with that of another study, in which combining the
autophagy inhibitor 3-methyladenine with cisplatin resulted in
enhanced cell death in NPC cells in vitro (26). Use of CQ is much safer in patients
than 3-methyladenine and has been adopted to inhibit autophagy in
clinical trials for cancer treatment (25).
Autophagy and the EMT process share a number of
common features. The two processes can, for example, be activated
by adverse stresses, including starvation and hypoxia (27,28). The
well-documented EMT inducer TGF-β can activate autophagy (29,30),
whereas the inhibition of mammalian target of rapamycin, an
important mediator of autophagy, can trigger the EMT process
(31,32). The connection between autophagy and
EMT has gradually emerged in the past few years. In breast cancer,
autophagy activation is involved in the degradation of the Snail
and Twist transcription factors, which are significant inducers of
the EMT process (33). Induction of
autophagy has been shown to impair migration and invasion in
glioblastoma, triggering a molecular switch from a mesenchymal
phenotype to an epithelial-like one (18). These lines of evidence have confirmed
that autophagy can inhibit the EMT process in tumours. Conversely,
however, Li et al reported that autophagy induces EMT and
enhances the invasion capability of hepatocellular carcinoma
(34). Blockade of autophagy by
either gene knockdown or CQ treatment promoted the reversal of the
mesenchymal phenotype and decreased the cancer stem cell phenotype
in breast cancer (35). In the
present study, under treatment with cisplatin, the surviving NPC
cells established a defensive phenotype, with autophagy and EMT
activated. Blockage of autophagy by CQ reversed the mesenchymal
phenotype in the NPC cells, a novel finding of this study.
Moreover, knockdown of LC3B increased the expression of E-cadherin
mRNA, whereas expression of vimentin, Snail and Slug mRNA was
decreased in the NPC cells. Snail and Slug are transcription
factors that repress the expression of E-cadherin and induce EMT
(15). Autophagy may therefore
regulate the expression of EMT-related transcription factors,
switching the EMT phenotype in NPC.
In summary, the present study indicated that
cisplatin activates autophagy and EMT in NPC cells and that the two
processes contribute to the resistance to cisplatin. Moreover,
inhibition of autophagy promotes the reversal of the EMT process.
Blockage of autophagy, therefore, represents an encouraging
strategy to reverse drug-induced adaptive responses (i.e.,
autophagy and EMT) and to overcome chemoresistance in NPC
management.
Acknowledgements
Grants were provided by the National Natural Science
Foundation of China (nos. 81372426, 81172558 and No. 81202128) and
the Natural Science Foundation of Hunan Province (nos. 14JJ2018 and
2015JJ3137).
Glossary
Abbreviations
Abbreviations:
|
NPC
|
nasopharyngeal carcinoma
|
|
EMT
|
epithelial-mesenchymal transition
|
|
CQ
|
chloroquine
|
|
TGFβ1
|
transforming growth factor β1
|
|
LC3B
|
microtubule-associated protein 1 light
chain 3β
|
|
TEM
|
transmission electron microscopy
|
References
|
1
|
Chan AT: Nasopharyngeal carcinoma. Ann
Oncol. 21 Suppl 7:i308–i312. 2010. View Article : Google Scholar
|
|
2
|
Chua DT, Sham JS and Au GK: A phase II
study of docetaxel and cisplatin as first-line chemotherapy in
patients with metastatic nasopharyngeal carcinoma. Oral Oncol.
41:589–595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galluzzi L, Vitale I, Michels J, Brenner
C, Szabadkai G, Harel-Bellan A, Castedo M and Kroemer G: Systems
biology of cisplatin resistance: Past, present and future. Cell
Death Dis. 5:e12572014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. Embo J. 7:856–880. 2015.
View Article : Google Scholar
|
|
5
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun K, Deng W, Zhang S, Cai N, Jiao S,
Song J and Wei L: Paradoxical roles of autophagy in different
stages of tumorigenesis: Protector for normal or cancer cells. Cell
Biosci. 3:352013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
He J, Yu JJ, Xu Q, Wang L, Zheng JZ, Liu
LZ and Jiang BH: Downregulation of ATG14 by EGR1-MIR152 sensitizes
ovarian cancer cells to cisplatin-induced apoptosis by inhibiting
cyto-protective autophagy. Autophagy. 11:373–384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hashimoto D, Bläuer M, Hirota M, Ikonen
NH, Sand J and Laukkarinen J: Autophagy is needed for the growth of
pancreatic adenocarcinoma and has a cytoprotective effect against
anticancer drugs. Eur J Cancer. 50:1382–1390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cufi S, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E,
Martin-Castillo B, Joven J and Menendez JA: The anti-malarial
chloroquine overcomes primary resistance and restores sensitivity
to trastuzumab in HER2-positive breast cancer. Sci Rep. 3:24692013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fulda S and Kögel D: Cell death by
autophagy: Emerging molecular mechanisms and implications for
cancer therapy. Oncogene. 34:5105–5113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chen CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Salazar M, Carracedo A, Salanueva IJ,
Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C,
Torres S, García S, et al: Cannabinoid action induces
autophagy-mediated cell death through stimulation of ER stress in
human glioma cells. J Clin Invest. 119:1359–1372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dave B, Mittal V, Tan NM and Chang JC:
Epithelial-mesenchymal transition, cancer stem cells and treatment
resistance. Breast Cancer Res. 14:2022012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Micalizzi DS and Ford HL:
Epithelial-mesenchymal transition in development and cancer. Future
Oncol. 5:1129–1143. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu X, Shen H, Yin X, Long L, Xie C, Liu
Y, Hui L, Lin X, Fang Y, Cao Y, et al: miR-186 regulation of Twist1
and ovarian cancer sensitivity to cisplatin. Oncogene. 35:323–332.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Catalano M, D'Alessandro G, Lepore F,
Corazzari M, Caldarola S, Valacca C, Faienza F, Esposito V,
Limatola C, Cecconi F and Di Bartolomeo S: Autophagy induction
impairs migration and invasion by reversing EMT in glioblastoma
cells. Mol Oncol. 9:1612–1625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nantajit D, Lin D and Li JJ: The network
of epithelial-mesenchymal transition: Potential new targets for
tumor resistance. J Cancer Res Clin Oncol. 141:1697–1713. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akalay I, Janji B, Hasmim M, Noman MZ,
André F, de Cremoux P, Bertheau P, Badoual C, Vielh P, Larsen AK,
et al: Epithelial-to-mesenchymal transition and autophagy induction
in breast carcinoma promote escape from T-cell-mediated lysis.
Cancer Res. 73:2418–2427. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou Z, Zhang L, Xie B, Wang X, Yang X,
Ding N, Zhang J, Liu Q, Tan G, Feng D and Sun LQ: FOXC2 promotes
chemoresistance in nasopharyngeal carcinomas via induction of
epithelial mesenchymal transition. Cancer Lett. 363:137–145. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang P, Liu H, Xia F, Zhang QW, Zhang YY,
Zhao Q, Chao ZH, Jiang ZW and Jiang CC: Epithelial-mesenchymal
transition is necessary for acquired resistance to cisplatin and
increases the metastatic potential of nasopharyngeal carcinoma
cells. Int J Mol Med. 33:151–159. 2014.PubMed/NCBI
|
|
25
|
Thorburn A, Thamm DH and Gustafson DL:
Autophagy and cancer therapy. Mol Pharmacol. 85:830–838. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song L, Liu H, Ma L, Zhang X, Jiang Z and
Jiang C: Inhibition of autophagy by 3-MA enhances endoplasmic
reticulum stress-induced apoptosis in human nasopharyngeal
carcinoma cells. Oncol Lett. 6:1031–1038. 2013.PubMed/NCBI
|
|
27
|
Talbot LJ, Bhattacharya SD and Kuo PC:
Epithelial-mesenchymal transition, the tumor microenvironment and
metastatic behavior of epithelial malignancies. Int J Biochem Mol
Biol. 3:117–136. 2012.PubMed/NCBI
|
|
28
|
Klymkowsky MW and Savagner P:
Epithelial-mesenchymal transition: A cancer researcher's conceptual
friend and foe. Am J Pathol. 174:1588–1593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kiyono K, Suzuki HI, Matsuyama H,
Morishita Y, Komuro A, Kano MR, Sugimoto K and Miyazono K:
Autophagy is activated by TGF-beta and potentiates
TGF-beta-mediated growth inhibition in human hepatocellular
carcinoma cells. Cancer Res. 69:8844–8852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzuki HI, Kiyono K and Miyazono K:
Regulation of autophagy by transforming growth factor-β (TGF-β)
signaling. Autophagy. 6:645–647. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mikaelian I, Malek M, Gadet R, Viallet J,
Garcia A, Girard-Gagnepain A, Hesling C, Gillet G, Gonzalo P,
Rimokh R and Billaud M: Genetic and pharmacologic inhibition of
mTORC1 promotes EMT by a TGF-β-independent mechanism. Cancer Res.
73:6621–6631. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang L, Graham PH, Hao J, Ni J, Bucci J,
Cozzi PJ, Kearsley JH and Li Y: Acquisition of
epithelial-mesenchymal transition and cancer stem cell phenotypes
is associated with activation of the PI3K/Akt/mTOR pathway in
prostate cancer radioresistance. Cell Death Dis. 4:e8752013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H,
Yan J, Lv X, Chen X and Hu ZW: DEDD interacts with PI3KC3 to
activate autophagy and attenuate epithelial-mesenchymal transition
in human breast cancer. Cancer Res. 72:3238–3250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo
Y, Song Z, Zheng Q and Xiong J: Autophagy promotes hepatocellular
carcinoma cell invasion through activation of
epithelial-mesenchymal transition. Carcinogenesis. 34:1343–1351.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cufi S, Vazquez-Martin A,
Oliveras-Ferraros C, Martin-Castillo B, Vellon L and Menendez JA:
Autophagy positively regulates the CD44(+) CD24(−/low) breast
cancer stem-like phenotype. Cell Cycle. 10:3871–3885. 2011.
View Article : Google Scholar : PubMed/NCBI
|