Introduction
Breast cancer is one of the most frequently
diagnosed types of cancer and is a significant cause of mortality
amongst females each year (1). In
previous decades, a number of therapies have been developed to
treat various types of breast cancer. However, drug resistance
decreases the antitumor efficacy of treatments such as
chemotherapy, endocrine therapy and targeted cancer therapy, which
results in poor prognoses in certain patients. However, not all the
mechanisms underlying resistance have been revealed. In previous
years, increasing evidence has demonstrated that one important
process is involved in the resistance against breast cancer
therapies: autophagy (2,3).
Autophagy is a process that is responsible for the
degradation of long-lived or abnormal proteins, and organelles that
are damaged or incompetent (2–4). Under
normal conditions, autophagy within cells remains at a basal level,
but the process is upregulated during stressful conditions,
including starvation, nutritional or hormone insufficiency,
hypoxia, accumulation of metabolin and invasion by pathogens
(2,5,6). The
hallmarks of autophagy consist of the formation of autophagosomes,
which combine with lysosomes to form autophagolysosomes. The
enzymes inside the autophagolysosomes digest the contents enveloped
by the cysts and then release the fragments into the cytoplasm
(7,8).
In studies of the molecular-biological mechanisms of autophagy, a
series of genes termed autophagy-associated genes (ATG) were
identified. It has also been revealed that several proteins encoded
by these genes act concomitantly during autophagy and in processes
that occur prior to the onset of autophagy, including
autophagy-induced (ATG1, ATG13) (9–13)
formation of pro-autophagosomes (ATG6, ATG16, ATG17) (13–15) and
the formation of autophagosomes (ATG5, ATG7, ATG8, ATG12) (14,16–19).
Autophagy has been demonstrated to contribute to
numerous types of diseases: Neurodegenerative diseases, including
Alzheimer's disease (20),
myocardiosis (21), pathogenic
infections, including bacterial, viral and parasitic infections
(22,23), and tumors. Previous studies have
indicated that autophagy may exert two opposing effects within
tumor cells: Autophagy may induce the development of tumors through
the downregulation or deficiency of the autophagy-associated gene
BECLIN1, which induces the malignant transformation of
cells. Conversely, when exposed to chemotherapy or radiotherapy,
autophagy may decrease the rate of apoptosis amongst tumor cells
and, thus, may assist the pathogenesis of certain malignant
diseases (23–26). The exact mechanism underlying
autophagy-associated tumor regulation requires additional
investigation.
In addition to a possible intracellular signaling
mechanism, microRNAs (miR/miRNA) also serve an important role in
the regulation of autophagy. miRNAs cause the instability and/or
inhibition of translation of mRNAs, which leads to a decrease in
target gene expression (27). A
number of studies have indicated that, in stressful scenarios, a
small number of miRNAs are able to regulate autophagic activity via
a change in the expression level of certain specialized
autophagy-associated proteins (28).
In 2009, Zhu et al (28) first
proposed the association between miRNA and cellular autophagy and
stated that miR-30a may induce the downregulation of autophagy
within tumor cells by negatively affecting the translation of
BECLIN1. It was then observed that miR-181a blocks
starvation- and rapamycin-induced autophagy in cancer cell lines
through the regulation of the target protein ATG-5 (29). miR-376b was also revealed to regulate
autophagy via targeting of the BECLIN1 gene and ATG4C
(30). In addition to the
aforementioned miRNAs, over one hundred studies have demonstrated
evidence for the role of miRNAs in the modulation of autophagic
activity.
miRNA-96 is one member of the miR-183-96-182 cluster
(31–33). It was demonstrated to serve an
important role in the regulation of the biological behavior of
cancer cells (34–37). It is expressed at low levels within
breast cancer cells and exhibits the ability to affect the
translation of forkhead box protein O1 (FOXO1) and FOXO3 (34,38–41).
Additionally, evidence has revealed that FOXO1 regulates autophagy
(42–49). Primarily, the present study
hypothesized that connections exist between miR-96 and autophagy,
and that the likely target protein of this interaction is FOXO1.
Therefore, in the present study, it was demonstrated that miR-96-5p
blocked starvation-induced autophagy in breast cancer cell lines.
It was also determined that the key protein in this process, FOXO1,
is a direct autophagy-associated target of miR-96-5p.
Materials and methods
Cell culture
The human breast cancer cell lines MDA-MB-231,
MCF-7, BT-549, HS 578T, T47D, and ZR-75-1 and the MCF-10A normal
breast epithelial cell line were purchased from the American Type
Culture Collection (Manassas, VA, USA). These cells were cultured
in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
in a 5% CO2 humidified incubator at 37°C. To induce
autophagy, the cells were cultured under starvation conditions in
Earle's balanced salt solution (EBSS; Gibco; Thermo Fisher
Scientific, Inc.).
Reagents
Anti-light chain 3B (LC3B; cat. no. 3868; dilution,
1:1,000), FOXO1 (cat. no. 2880; dilution, 1:1,000), SQSTM1/p62
(cat. no. 5114; dilution, 1:1,000), GAPDH (cat. no. 2218 dilution,
1:1,000) antibodies and anti-rabbit IgG horseradish
peroxidase-linked secondary antibody (cat. no. 7074; dilution,
1:2,000) was purchased from were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA). The acetylated-FOXO1 antibody
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). The dimethyl sulfoxide vehicle control was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The DAPI- and
fluorescein isothiocyanate (FITC)-conjugated secondary antibodies
were also purchased from Sigma-Aldrich; Merck KGaA.
Plasmids and transient
transfection
LC3B-EGFP was purchased from Nanjing KeyGEN BioTech,
Co., Ltd. (Nanjing, China). The miR-96-5p mimic and the negative
control mimic were purchased from Guangzhou RiboBio Co., Ltd
(Guangzhou, China). The transient transfection of MCF-7 and
MDA-MB-231 cells was performed with either
Lipofectamine® RNA iMAX or Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA of the MCF-10A normal breast epithelial
cell line and the breast cancer cell lines (MCF-7, MDA-MB-231,
BT-549, HS 578T, T47D and ZR-75-1) were isolated with
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. cDNA was synthesized from
mRNA using the PrimeScript™ RT Master mix kit (catalog
no. RR036A; Takara Biotechnology, Co., Ltd., Dalian, China) and
from miRNA using the One Step PrimeScript miRNA cDNA Synthesis kit
(catalog no. D350; Takara Biotechnology, Co., Ltd.), according to
the manufacturer's instructions. The miRNAs were converted to cDNA
using TaqMan microRNA reverse transcription kit (Takara
Biotechnology, Co., Ltd.). The expression levels of miR-96-5p were
detected with a TaqMan microRNA kit-based quantitative PCR (Takara
Biotechnology, Co., Ltd.) and normalized to the expression of small
nuclear RNA, U6. The expression of FOXO1 was assessed by PCR
amplification using a TaqMan RNA kit (Takara Biotechnology, Co.,
Ltd.). PCR was performed with the PrimeScript RT Master Mix (Takara
Biotechnology, Co., Ltd.) in a Bio-Rad CFX96 real-time PCR machine
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The primer
sequences used for PCR were as follows: MiRNA-96-5p, forward,
5′-ACACTCCAGCTGGGTTTGGCACTAGCACATTT-3′; reverse,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGCAAAAA-3′; U6, forward,
5′-CTCGCTTCGGCAGCACA-3′; reverse, 5′-AACGCTTCACGAATTTGCGT-3′;
FOXO1, forward, 5′-GTTGCCCAACCAAAGCTTCC-3′; reverse,
5′-TCTCAGTTCCTGCTGTCAGACAATC-3′; GAPDH, forward,
5′-AGAAGGCTGGGGCTCATTTG-3′; reverse, 5′-AGGGGCCATCCACAGTCTTC-3′.
The cycling parameters for mRNA and miRNA were as follows: 95°C for
30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30
sec. The cycling parameters for mRNA were as follows: 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec.
All reactions were performed in duplicate, and the number of
independent experiments (n) was marked. Data analysis was performed
using the comparative ΔΔCq method with Bio-Rad Manager 2.1 software
(Bio-Rad Laboratories, Inc.) (50).
Western blot analysis
MCF-7 (~105) and MDA-MB-231
(~106) cells cultured in 6-well plates were lysed in
radioimmunoprecipitation assay lysis buffer (Cell Signaling
Technology, Inc.) supplemented with a protease inhibitor (Roche
Diagnostics, Basel, Switzerland). A total of 20 mg cell lysates
were resolved by 10% SDS-PAGE and then transferred to
polyvinylidene fluoride membranes. The membranes were blocked in 5%
non-fat dry milk in PBST for 2 h at 25°C and incubated overnight at
4°C with the respective primary antibodies (anti-LC3B antibody,
anti-SQSTM1/p62 antibody, anti-FOXO1 antibody, anti-GAPDH antibody,
acetylated-FOXO1 antibody) at a dilution of 1:1,000. The membranes
were washed by PBST for 3 times, and then incubated at room
temperature for 1 h with the appropriate HRP-conjugated secondary
antibodies as previously mentioned (Cell Signaling Technology,
Inc.) and were visualized with a Plus-enhanced chemiluminescence
reagent (PerkinElmer, Inc., Waltham, MA, USA), according to the
manufacturer's protocol.
Immunofluorescence
For the immunofluorescence experiments,
~105 MCF-7 and MDA-MB-231 cells were grown on glass
coverslips that were pre-coated with collagen and positioned in
24-well plates. The cells were incubated in a 5% CO2
humidified incubator at 37°C. The cells were transiently
transfected with the miRNA mimic negative control or 50 nM
miR-96-5p mimic for 24 h. The cells were then fixed in 4%
paraformaldehyde, which was followed by membrane permeabilization
with 0.2% Triton X-100. The cells were then blocked with 5% bovine
serum albumin and incubated with an anti-LC3B antibody overnight at
4°C. Subsequently, cells were incubated with a green fluorescent
protein (GFP)-tagged secondary antibody (Thermo Fisher Scientific,
Inc.) or with a green fluorescent secondary antibody without
GFP-tag (Thermo Fisher Scientific, Inc.) for 1 h at room
temperature, followed by incubation at room temperature with DAPI
(Roche Diagnostics) for 10 min. Images were captured by
fluorescence microscopy (Carl Zeiss Axio Observer Z1; Carl Zeiss,
Jena, Germany). The cells with >10 GFP-LC3 dots were identified
as positive cells, while all others were considered negative.
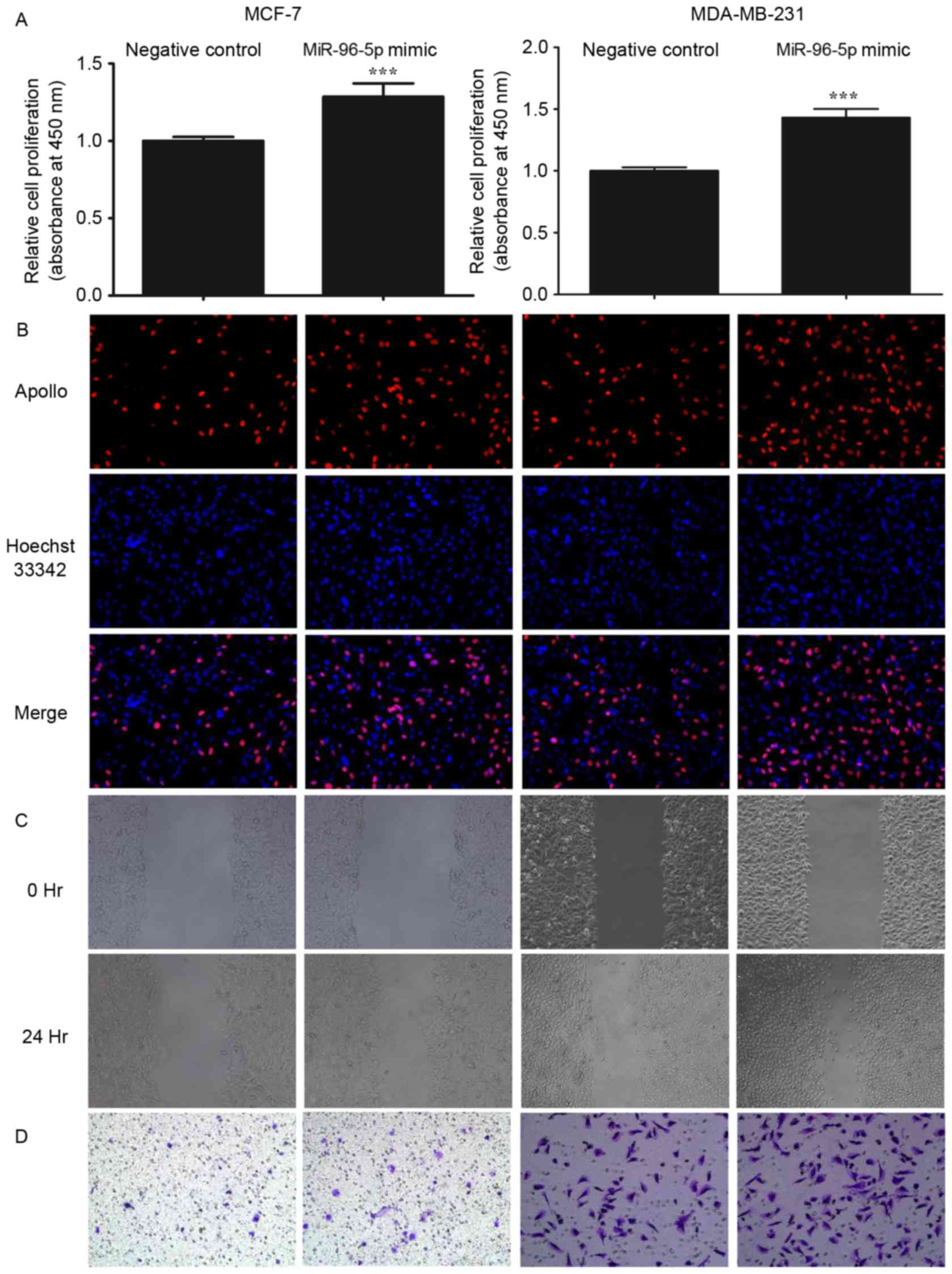
Cell proliferation assay
Cell proliferation was detected using a Cell
Counting kit-8 assay (CCK-8; Dojindo Molecular Technologies, Inc.,
Shanghai, China) and a 5-ethynyl-2′-deoxyuridine assay (EdU;
RiboBio Co., Ltd.). The cells were seeded into 96-well plates at a
density of 3×103 cells/well and allowed to attach
overnight. The cells that were transfected with the miRNA mimic or
with the negative control were then incubated with 1% FBS + DMEM
for 12 h subsequent to transfection at 37°C. For the CCK-8 assay,
10 µl CCK-8 solution was added to each well, and the cells were
allowed to incubate at 37°C for 1 h. The absorbance at 490 nm of
each well was read on a spectrophotometer. For the EdU assay, the
cells were incubated at 37°C with 50 µM EdU solution for 2 h and
then fixed in 4% paraformaldehyde for 15 min. The cells were
subsequently incubated at 37°C with 100 µl Apollo reagent (Thermo
Fisher Scientific, Inc.) for 30 min followed by 50 µl 1% Hoechst
33,342 for 30 min. Finally, the images were captured by
fluorescence microscopy. The images were analyzed using
Image-Pro® Plus software 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA). Three independent experiments were performed
in quadruplicate.
Cell migration and invasion assay
A scratch assay was performed to assess the
migratory potential of the cells. The cell layer was scratched with
a pipette tip when the cells reached ~95% confluence, and were then
incubated under serum starvation at 37°C. The medium was changed
every two days. Images were captured at ×40 magnification
immediately following the generation of the scratch, at 0, 12 and
24 h subsequent to the generation of the scratch. The cell invasion
assay was performed in 8 µm pore size and 6.5 mm diameter Transwell
plates (Corning Life Sciences, Tewksbury, MA, USA) that were
pre-coated with Matrigel® basement membrane matrix at a
concentration of 1 mg/ml (BD Biosciences, Franklin Lakes, NJ, USA),
according to the manufacturer's protocol. A total of
2×104 cells in 0.2 ml medium supplemented with 2% FBS
were seeded into the upper chamber, and 0.6 ml medium supplemented
with 10% FBS was placed in the lower chamber. The plates were
incubated at 37°C in an atmosphere of 5% CO2. After 48
h, the chambers were removed and a cotton swab was used to remove
the non-invading cells from the upper side of the chamber membrane.
The cells on the lower membrane were then fixed in methanol for 10
min and stained with 0.1% crystal violet in 20% methanol for 30 min
at room temperature.
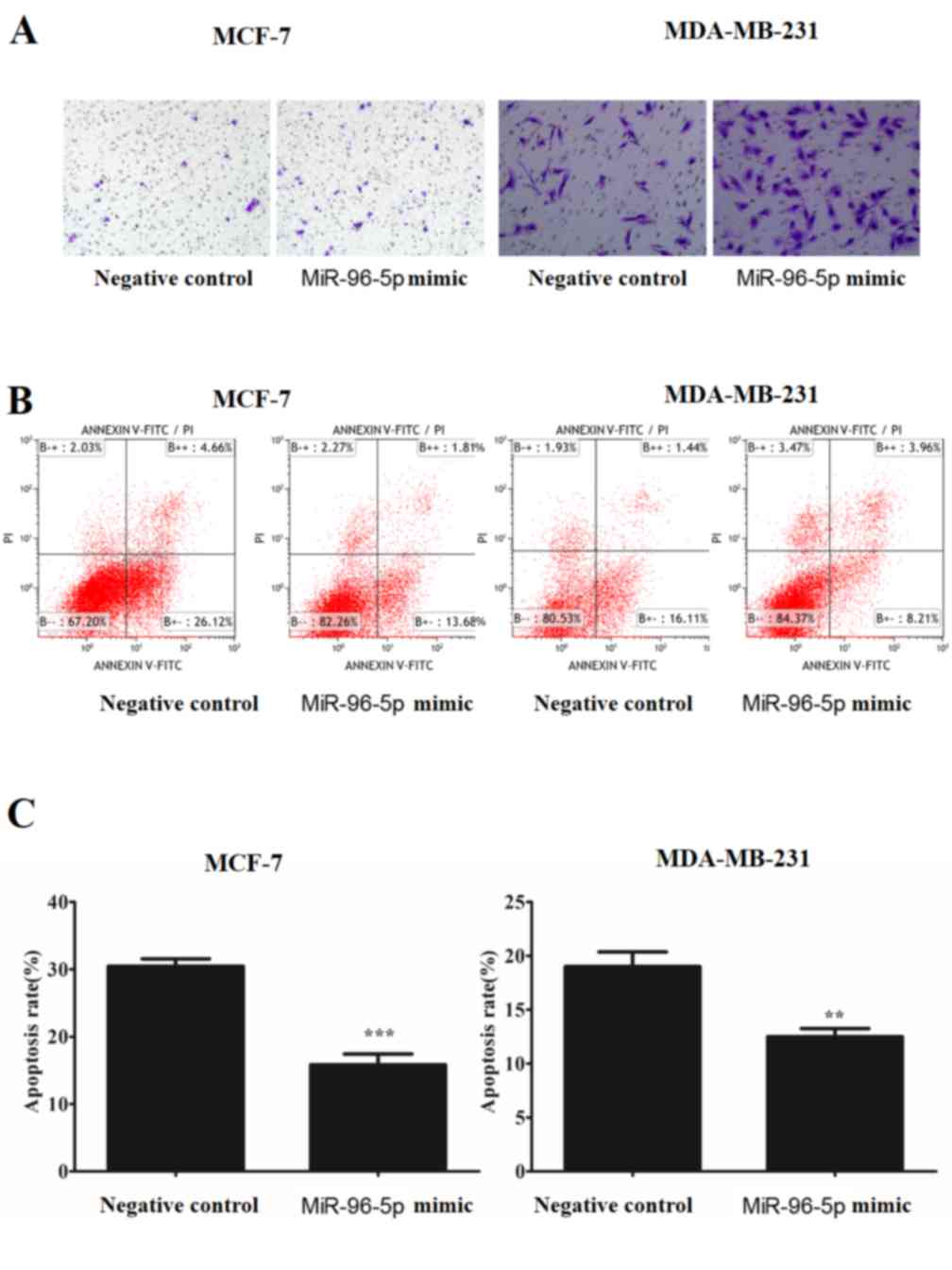
Cell apoptosis assay
Apoptosis was assessed using an Annexin V-FITC
apoptosis detection kit (Dojindo Molecular Technologies, Inc.)
according to the manufacturer's protocol. The cells were
transfected with the miRNA mimic or with the negative control for
12 h prior to incubation with serum-free DMEM for 48 h at 37°C. The
cells were harvested, resuspended in 500 µl binding solution from
the Annexin V-FITC apoptosis detection kit and incubated at room
temperature with 5 µl Annexin V and 5 µl propidium iodide (PI) for
10 min in the dark. Apoptosis was analyzed by flow cytometry using
a Beckman Coulter EPICS XL-MCL, and the results were analyzed by
Kaluza software (Beckman Coulter Inc., Brea, CA, USA).
Accession numbers of genes mentioned
in the present study
miRNA-96: Mus musculus, NCBI Gene ID: 407053;
FOXO1: Mus musculus, NCBI Gene ID: 2308.
Investigation of target genes
The target genes of miRNA were investigated using a
bioinformatics tool TargetScan (http://www.targetscan.org/).
Statistical analysis
The data are presented as histogram and gray
intensity analysis in the results. An unpaired Student's t-test or
one-way analysis of variation was used with GraphPad Prism version
5.0 (GraphPad Software Inc., La Jolla, CA, USA) to determine the
differences between the treatment groups and the control groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
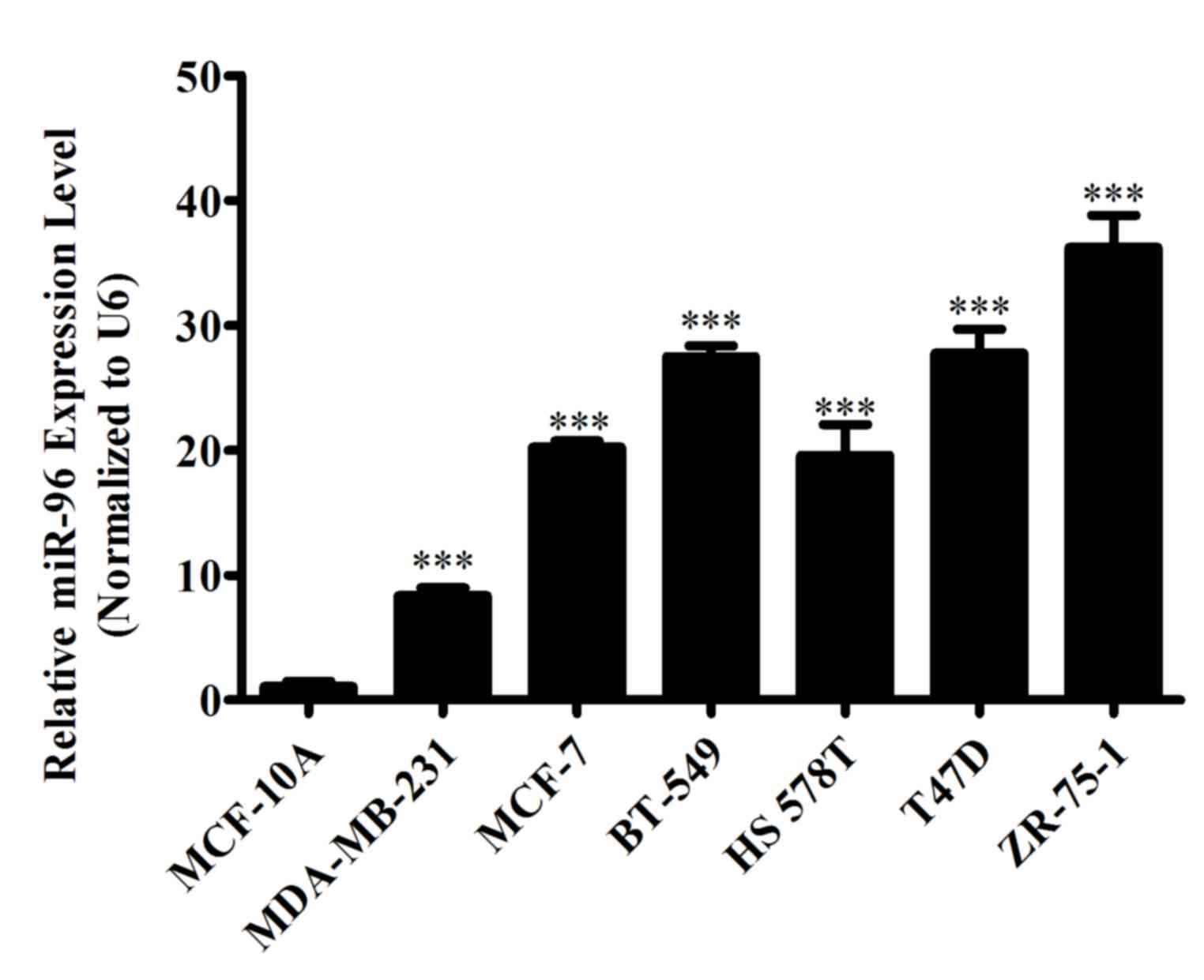
MiR-96-5p is overexpressed in breast
cancer cell lines
To evaluate the differential expression of miR-96-5p
in breast cancer cells and normal breast epithelial cells, RNA was
extracted from the normal breast epithelial MCF-10A cell line and
six distinct breast cancer cell lines, MCF-7, MDA-MB-231, BT-549,
HS 578T, T47D and ZR-75-1, at the proliferating time point. The
differential expression of miR-96-5p was then determined by
RT-qPCR. The results demonstrated that miR-96-5p is upregulated in
all breast cancer cell lines, as compared with in the normal breast
cell line (Fig. 1).
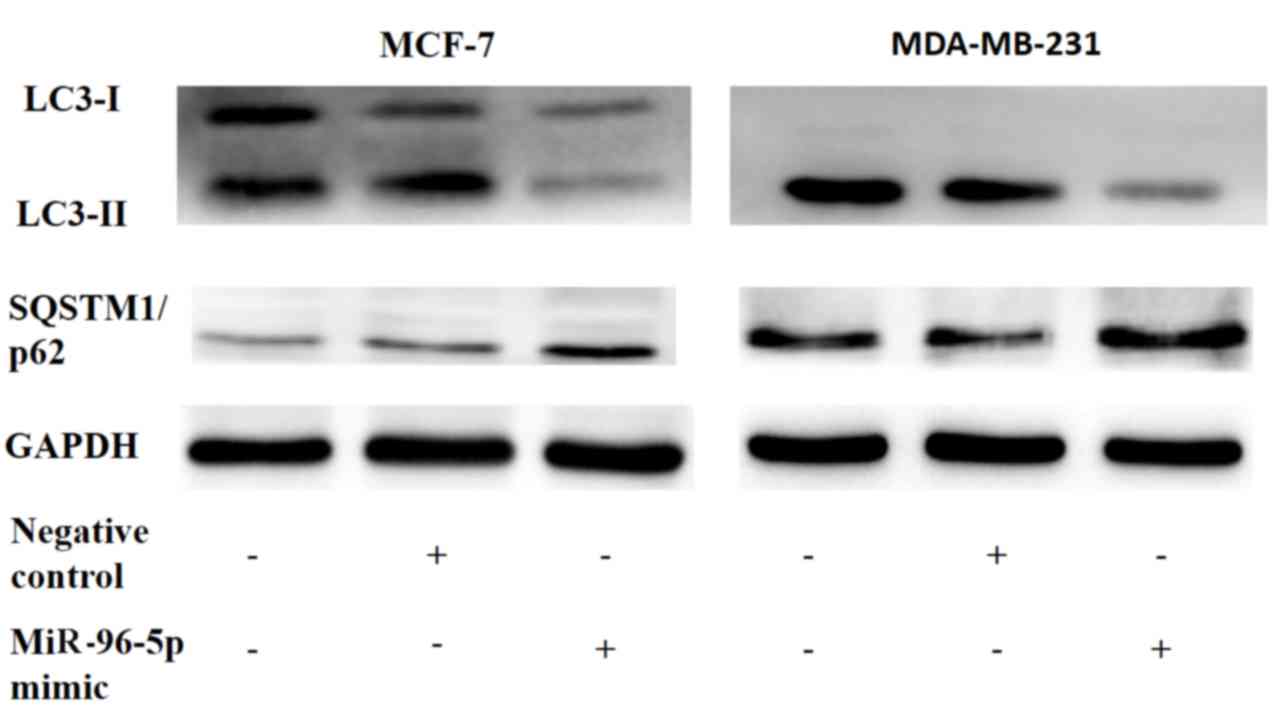
Overexpression of miR-96-5p suppresses
autophagy in MCF-7 and MDA-MB-231 breast cancer cells
Firstly, miR-96-5p was transiently overexpressed in
MCF-7 and MDA-MB-231 breast cancer cells, and the autophagic flux
was then estimated via western blot analysis (Fig. 2). This demonstrated that the elevation
of miR-96-5p in breast cancer cells may repress the conversion of
LC3-I to LC3-II, and that it may also block the degradation of
sequestosome-M1/tumor protein 62 (SQSTM1/p62). These data confirmed
an inhibitory effect of miR-96-5p on autophagy.
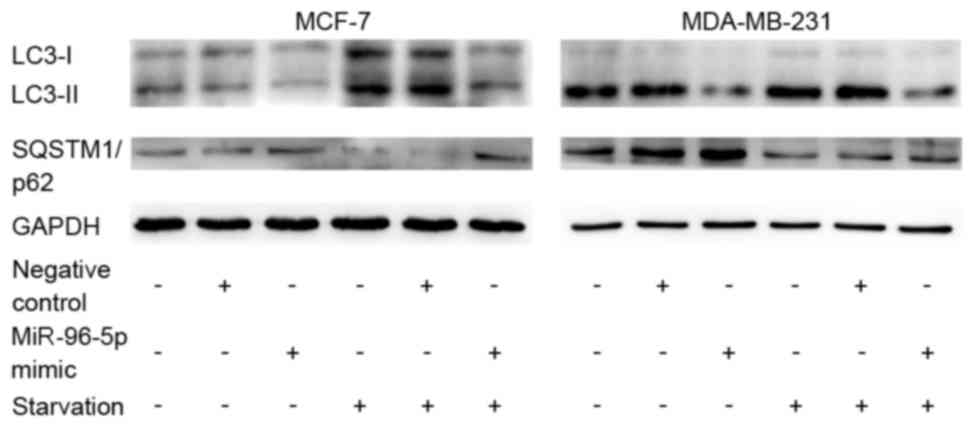
Overexpression of miR-96-5p suppresses
starvation-induced autophagy in MCF-7 and MDA-MB-231 breast cancer
cells
The present study investigated whether miR-96-5p may
regulate starvation-induced autophagy. A nutrient-starvation
environment was simulated via the application of EBSS for 4 h. As
illustrated in Fig. 3, starvation
significantly promoted autophagy (P=0.036), while the
overexpression of miR-96-5p markedly repressed the accumulation of
autophagic vacuoles in MCF-7 and MDA-MB-231 cells that were exposed
to starvation conditions according to the GFP-LC3 transfection
experiment and the immunofluorescence staining for LC3. In this
section, the cells with >10 GFP-LC3 dots were identified as
positive cells, while all others were considered negative.
Additionally, western blotting confirmed that the conversion of
LC3-I to LC3-II was inhibited whilst the degradation of SQSTM1/p62
was blocked in nutrient-starved-MCF-7 and MDA-MB-231 cells,
compared with control cells (Fig.
4).
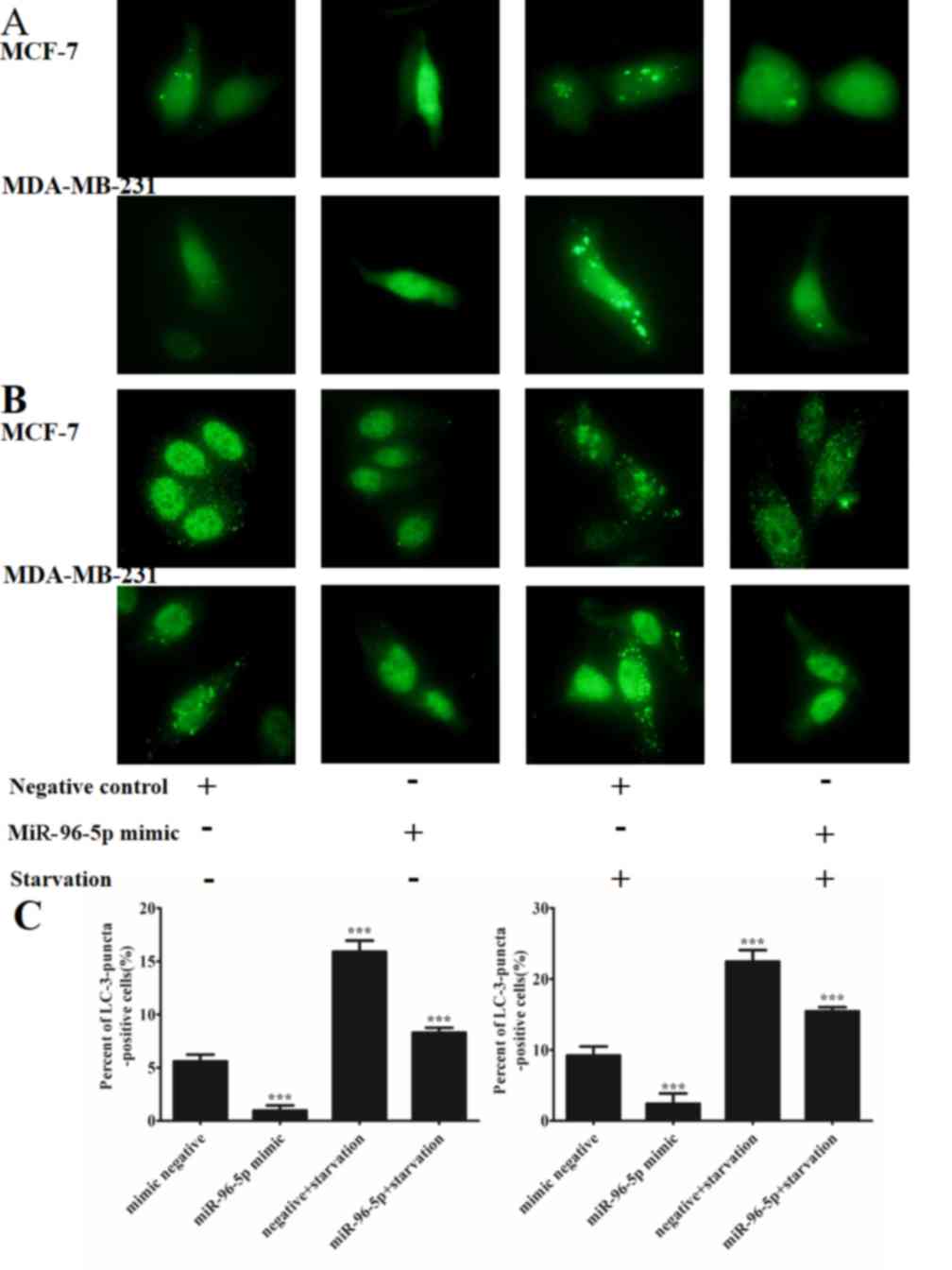 | Figure 3.The overexpression of miR-96-5p
resulted in decreased starvation-induced autophagic activity in
MCF-7 cells and MDA-MB-231 cells. (A) MCF-7 and MDA-MB-231 cells
were co-transfected with a miR-96-5p mimic or a control construct
with a GFP-LC3 plasmid. Autophagy was then assessed under nutrient
starvation conditions, Earle's balanced salt solution treatment for
4 h, by fluorescence microscopy at magnification, ×400. The cells
with >10 GFP-LC3 dots were identified as positive cells, while
all others were considered negative. This demonstrates that
miR-96-5p blocked GFP-LC3 dot formation in the setting of
starvation-induced autophagy in two different cell lines. (B) MCF-7
and MDA-MB-231 cells were transfected with a miR-96-5p mimic or a
control construct, and then endogenous LC3B was detected with an
LC3B antibody and a green Alexa Fluor 488-conjugated goat
anti-rabbit IgG. LC3 punctae were detected by fluorescence
microscopy at magnification, ×400, subsequent to exposure to
starvation conditions for 4 h. The cells with >10 LC3 punctae
were identified as positive cells, while all others were considered
negative. This suggests that miR-96-5p blocked LC3 puncta formation
in starvation-induced autophagy in two different cell lines
(***P<0.001 vs. mimic negative). miR, microRNA; GFP, green
fluorescent protein; LC, light chain. |
miR-96 post-transcriptionally
suppresses FOXO1 via interaction with its 3′UTR
To reveal the mechanism of the regulation of
autophagy by miR-96-5p, potential target genes were investigated
using bioinformatics tools, including TargetScan (http://www.targetscan.org/). Through this method,
FOXO1 was identified as a target gene of miR-96-5p. The interaction
between miR-96-5p and the FOXO1 3′ untranslated region is
illustrated in Fig. 5. To confirm
this prediction, RT-qPCR was performed in the normal breast
epithelial MCF-10A cell line, and in six breast cancer cell lines,
MCF-7, MDA-MB-231, BT-549, HS 578T, T47D and ZR-75-1. The results
demonstrated that FOXO1 mRNA levels were upregulated in MCF-10A
cells, but were downregulated in all of the breast cancer cell
lines tested (Fig. 5). This result
was in contrast with the expression of miR-96-5p in the same types
of cell lines.
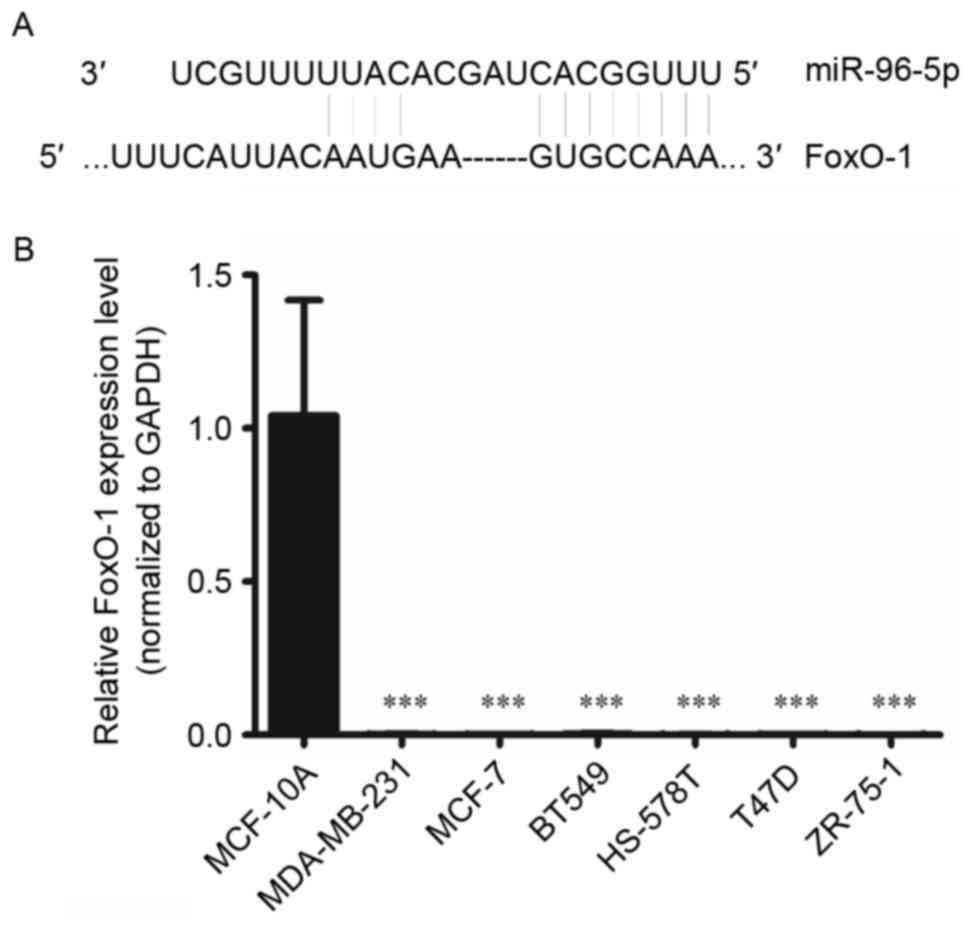 | Figure 5.(A) The miR-96-5p mature miRNA
sequence and miR-96-5p binding target sequence in the 3′
untranslated region of FOXO1 mRNA. (B) Expression levels of FOXO1
in MCF-10A normal breast epithelial cells and the breast cancer
cell lines MCF-7, MDA-MB-231, BT-549, HS 578T, T47D and ZR-75-1.
FOXO1 expression levels in cell lines were measured using reverse
transcription-quantitative polymerase chain reaction. FOXO1 was
downregulated in all the breast cancer cell lines tested, as
compared with in the non-malignant breast cell line (mean ±
standard deviation of five independent experiments; ***P<0.001
vs. MCF-10A). miR, microRNA; FOXO1, forkhead box O1; U, uracil; A,
adenosine; c, cytosine. |
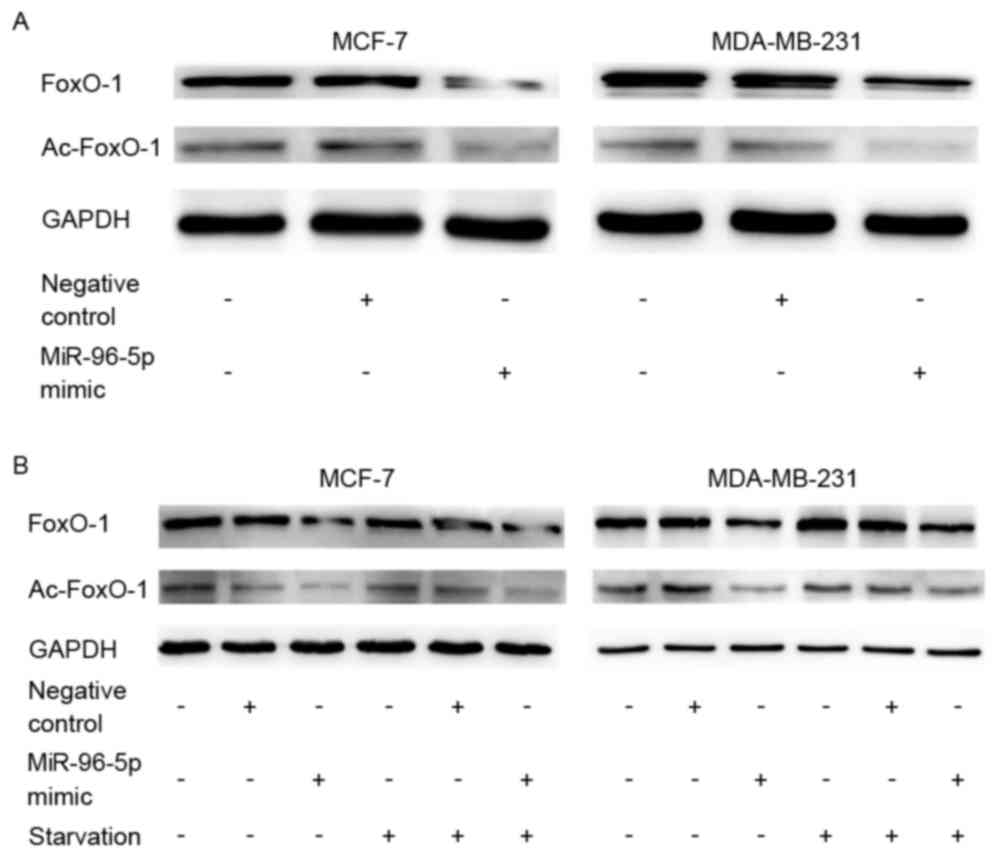
miR-96 may inhibit autophagy via
downregulation of the expression of FOXO1 and acetylated-FOXO1
A significant decrease was observed in FOXO1
(P=0.047) and acetylated-FOXO1 (P=0.024) protein expression levels
in miR-96-5p-overexpressing cells by western blot analysis
(Fig. 6). This demonstrated a
tendency towards an inverse expression pattern between miR-96-5p
and FOXO1. Additionally, the expression of FOXO1 and
acetylated-FOXO1 demonstrated a similar trend with autophagic
activities in MCF-7 and MDA-MB-231 cell lines that were cultured
under starvation conditions (Fig. 6).
Considering the previous reports, it is hypothesized that FOXO1 may
be a target of miR-96-5p and may inhibit autophagy.
miR-96-5p promotes proliferation,
migration and invasion, but suppresses apoptosis in MCF-7 and
MDA-MB-231 cells
To examine the effect of miR-96-5p on the malignant
biological processes in breast cancer cells, including cell
proliferation, migration and invasion, miR-96-5p was overexpressed
via the transfection of MCF-7 and MDA-MB-231 cells with a miR-96-5p
mimic or a control construct. CCK-8 and EdU assays were then
performed to evaluate the level of proliferation; scratch,
Transwell and Matrigel invasion assays were used to analyze
migration and invasion. As presented in Figs. 7 and 8,
it was demonstrated that the overexpression of miR-96-5p
significantly promoted cell proliferation, migration and invasion
(P<0.001). Additionally, the transfected cells were treated and
incubated without serum for 24–48 h and then stained with Annexin V
and PI; the apoptotic cells were subsequently quantified by flow
cytometry. The overexpression of miR-96-5p significantly inhibited
the rate of apoptosis of MCF-7 (P<0.001) and MDA-MB-231 cells
(P<0.005) (Fig. 8).
Discussion
miRNAs are a series of small RNAs that may affect
biological features through the regulation of the expression of
their target genes (51). In previous
studies, it was demonstrated that miR-96 is involved in several
types of cancer, including breast cancer, and contributes to cancer
progression (37,52–55).
Therefore, miRNAs are considered potential therapeutic targets in
cancer. In the present study, it was demonstrated that miR-96-5p is
overexpressed in breast cancer cell lines and that this
overexpression of miR-96-5p enhances the proliferation, migration
and invasiveness of these breast cancer cells. In addition, it was
revealed that the overexpression of miR-96 leads to the inhibition
of apoptosis. Therefore, it was confirmed that miR-96-5p may
promote the progression of breast cancer in a ‘traditional’
manner.
Additionally, autophagy is an important biological
feature that occurs in cells. Autophagy is another form of
programmed cell death that differs from apoptosis, in which cells
undergo self-digestion with increased autophagosome formation
(8,9,56).
Autophagy and apoptosis affect the rate cell death by parallel
pathways under certain conditions (57). However, previous studies have
demonstrated that autophagy serves as a pro-survival response to
several stressors, including starvation, hypoxia, radiation and
chemotherapy, particularly in tumor cells (25,53–55,58–61).
Autophagy may have antitumor or pro-tumor functions, and is thus
considered to be a ‘double-edged sword’ in the field of oncology
(25,53–55,58–61).
In breast cancer, autophagy may mitigate metabolic stress and
genomic damage in mammary tumorigenesis (5). Additionally, previous studies have
demonstrated that the upregulation of autophagy assists cancer
cells to survive common breast cancer therapies, including
tamoxifen, trastuzumab, anthracycline, taxane, temozolomide and
radiotherapy, amongst others, which may markedly affect the
prognosis of patients (60–70). Additionally, the inhibition of
autophagy increases drug sensitivity, such that chloroquine may
restore sensitivity to trastuzumab in HER2-positive breast cancer
(62,64).
The aforementioned data suggest that the combination
of autophagy inhibition and cancer therapy, particularly targeted
therapy based on miRNAs, may serve as an effective approach for the
treatment of tumors.
In the present study, miR-96-5p was introduced as an
autophagy-associated miRNA. The overexpression of miR-96-5p
inhibited GFP-LC3 dot formation, fluorescent LC3B accumulation, the
conversion of LC3-I to LC3-II and SQSTM1/p62 degradation. These are
markers of the initiation of standard and starvation-induced
autophagy, which is considered to be the classical and most
effective method of stimulating autophagy (71), in two breast cancer cell lines, MCF-7
and MDA-MB-231. Therefore, these data demonstrated that miR-96-5p
is a key miRNA that regulates autophagy in breast cancer cells.
The present study aimed to determine the association
between miR-96-5p and autophagy. According to the prediction
generated by TargetScan, a notable transcription factor, FOXO1, was
observed as one of the target genes of miR-96-5p. FOXO1 is a member
of the forkhead transcription factors of the O class family (FOXO)
that includes FOXO1 (FKHR), FOXO-3 (FKHRL1), FOXO-4 (AFX) and
FOXO-6. FOXO1 regulates the biological behavior of tumor cells,
including apoptosis, differentiation and oxidative stimulation
through the phosphoinositide 3-kinase and protein kinase B
signaling pathways (72,73). Guttilla et al (37) demonstrated that miR-96 was
overexpressed in breast cancer cell lines, and that it
downregulated the expression of FOXO1. Guo et al (74) and Haflidadóttir et al (75) suggested that miR-96 regulates
FOXO1-mediated cell apoptosis and proliferation in prostate cancer.
The association between FOXO1 and autophagy in multiple types of
cancer, including bladder transitional cell carcinoma, prostate
cancer and endometrial carcinoma, was identified in recent years
(42–44,47,48,66,76).
Hariharan et al (47)
demonstrated that the deacetylation of FOXO1 by Sirtuin 1 serves a
key role in the mediation of starvation-induced autophagy in
cardiac myocytes. In a series of articles by Zhao et al
(44,48,77), it
was noted that cytosolic FOXO1 combined with ATG5 is essential for
cancer cell autophagy, and may be suppressed by unspliced X-box
protein-1 and induced by FOXO3 in varying conditions.
In the present study, it was determined that the
expression of FOXO1 mRNA was lower in the breast cancer cell lines,
as compared with in the normal breast epithelial cell line, but
that the expression of miR-96-5p was high in the breast cancer cell
lines. The overexpression of miR-96-5p significantly inhibited the
expression of FOXO1 and acetylated-FOXO1; this effect was also
observed under starvation-induced conditions together with the
inhibition of autophagy activation in the two breast cancer cell
lines MDA-MB 231 and MCF-7.
Considering the results of previous studies, it is
reasonable to predict that miR-96-5p may regulate the activation of
autophagy in breast cancer cells through the target protein, FOXO1.
During the preparation of this manuscript, Ma et al
(78) published an article that
hypothesized a biphasic regulation of autophagy by miR-96 in
prostate cancer cells in hypoxic conditions. These data suggests
that there may be an expression level threshold of miR-96 in
certain situations, including hypoxia, and on either side of this
threshold the miR-96 may have the opposite effect. Therefore,
additional studies are required to explore the mechanisms
underlying the regulation of miR-96 and FOXO1-associated autophagy
in breast cancer.
In conclusion, the present study demonstrated that
miR-96-5p is overexpressed in breast cancer cells. The upregulation
of miR-96-5p enhances proliferation, migration and invasion, but
inhibits apoptosis. Notably, the overexpression of miR-96-5p may
suppress standard and starvation-induced autophagy in breast cancer
cells, which is most likely to be regulated by FOXO1. Therefore,
miR-96-5p is an important molecule in the study of chemotherapeutic
sensitivity and tolerance, and it may become a potential
therapeutic target in patients with breast cancer.
References
|
1
|
Libson S and Lippman M: A review of
clinical aspects of breast cancer. Int Rev Psychiatry. 26:4–15.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
White E and Dipaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kamada Y, Sekito T and Ohsumi Y: Autophagy
in yeast: A TOR-mediated response to nutrient starvation. Curr Top
Microbiol Immunol. 279:73–84. 2004.PubMed/NCBI
|
|
4
|
Krustev LP: Cell autophagy of the liver in
starvation and undernutrition. Bibl Nutr Dieta. 145–154.
1976.PubMed/NCBI
|
|
5
|
Karantza-Wadsworth V, Patel S, Kravchuk O,
Chen G, Mathew R, Jin S and White E: Autophagy mitigates metabolic
stress and genome damage in mammary tumorigenesis. Genes Dev.
21:1621–1635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chera S, Buzgariu W, Ghila L and Galliot
B: Autophagy in hydra: A response to starvation and stress in early
animal evolution. Biochim Biophys Acta. 1793:1432–1443. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N, Ohsumi Y and Yoshimori T:
Autophagosome formation in mammalian cells. Cell Struct Funct.
27:421–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Duve C and Wattiaux R: Functions of
lysosomes. Annu Rev Physiol. 28:435–492. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Noda T, Suzuki K and Ohsumi Y: Yeast
autophagosomes: De novo formation of a membrane structure. Trends
Cell Biol. 12:231–235. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kabeya Y, Kamada Y, Baba M, Takikawa H,
Sasaki M and Ohsumi Y: Atg17 functions in cooperation with Atg1 and
Atg13 in yeast autophagy. Mol Biol Cell. 16:2544–2553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reggiori F, Tucker KA, Stromhaug PE and
Klionsky DJ: The Atg1-Atg13 complex regulates Atg9 and Atg23
retrieval transport from the pre-autophagosomal structure. Dev
Cell. 6:79–90. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kabeya Y, Kamada Y, Baba M, Takikawa H,
Sasaki M and Ohsumi Y: Atg17 functions in cooperation with Atg1 and
Atg13 in yeast autophagy. Mol Biol Cell. 16:2544–2553. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matsushita M, Suzuki NN, Obara K, Fujioka
Y, Ohsumi Y and Inagaki F: Structure of Atg5. Atg16, a complex
essential for autophagy. J Biol Chem. 282:6763–6772. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsushita M, Suzuki NN, Fujioka Y, Ohsumi
Y and Inagaki F: Expression, purification and crystallization of
the Atg5-Atg16 complex essential for autophagy. Acta Crystallogr
Sect F Struct Biol Cryst Commun. 62:1021–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Codogno P and Meijer AJ: Atg5: More than
an autophagy factor. Nat Cell Biol. 8:1045–1047. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujioka Y, Noda NN, Fujii K, Yoshimoto K,
Ohsumi Y and Inagaki F: In vitro reconstitution of plant Atg8 and
Atg12 conjugation systems essential for autophagy. J Biol Chem.
283:1921–1928. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishino I: Autophagic vacuolar myopathies.
Curr Neurol Neurosci Rep. 3:64–69. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rubinsztein DC, Difiglia M, Heintz N,
Nixon RA, Qin ZH, Ravikumar B, Stefanis L and Tolkovsky A:
Autophagy and its possible roles in nervous system diseases, damage
and repair. Autophagy. 1:11–22. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miller S and Krijnse-Locker J:
Modification of intracellular membrane structures for virus
replication. Nat Rev Microbiol. 6:363–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakagawa I, Amano A, Mizushima N, Yamamoto
A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et
al: Autophagy defends cells against invading group A streptococcus.
Science. 306:1037–1040. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kubisch J, Türei D, Földvári-Nagy L, Dunai
ZA, Zsákai L, Varga M, Vellai T, Csermely P and Korcsmáros T:
Complex regulation of autophagy in cancer - Integrated approaches
to discover the networks that hold a double-edged sword. Semin
Cancer Biol. 23:252–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lorin S, Hamaï A, Mehrpour M and Codogno
P: Autophagy regulation and its role in cancer. Semin Cancer Biol.
23:361–379. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu B, Wen X and Cheng Y: Survival or
death: Disequilibrating the oncogenic and tumor suppressive
autophagy in cancer. Cell Death Dis. 4:e8922013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X,
Liu CG and Yang JM: Regulation of autophagy by a beclin 1-targeted
microRNA, miR-30a, in cancer cells. Autophagy. 5:816–823. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tekirdag KA, Korkmaz G, Ozturk DG, Agami R
and Gozuacik D: MIR181A regulates starvation- and rapamycin-induced
autophagy through targeting of ATG5. Autophagy. 9:374–385. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Korkmaz G, le Sage C, Tekirdag KA, Agami R
and Gozuacik D: MiR-376b controls starvation and mTOR
inhibition-related autophagy by targeting ATG4C and BECN1.
Autophagy. 8:165–176. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sacheli R, Nguyen L, Borgs L, Vandenbosch
R, Bodson M, Lefebvre P and Malgrange B: Expression patterns of
miR-96, miR-182 and miR-183 in the development inner ear. Gene Expr
Patterns. 9:364–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhu W, Liu X, He J, Chen D, Hunag Y and
Zhang YK: Overexpression of members of the microRNA-183 family is a
risk factor for lung cancer: A case control study. BMC Cancer.
11:3932011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mihelich BL, Khramtsova EA, Arva N,
Vaishnav A, Johnson DN, Giangreco AA, Martens-Uzunova E, Bagasra O,
Kajdacsy-Balla A and Nonn L: MiR-183-96-182 cluster is
overexpressed in prostate tissue and regulates zinc homeostasis in
prostate cells. J Biol Chem. 286:44503–44511. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fendler A, Jung M, Stephan C, Erbersdobler
A, Jung K and Yousef GM: The antiapoptotic function of miR-96 in
prostate cancer by inhibition of FOXO1. PLoS One. 8:e808072013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W,
Liu J, Yu J and Chen J: MiRNA-96 suppresses KRAS and functions as a
tumor suppressor gene in pancreatic cancer. Cancer Res.
70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin H, Dai T, Xiong H, Zhao X, Chen X, Yu
C, Li J, Wang X and Song L: Unregulated miR-96 induces cell
proliferation in human breast cancer by downregulating
transcriptional factor FOXO3a. PLoS One. 5:e157972010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96 and miR-182 in breast cancer
cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li J, Li P, Chen T, Gao G, Chen X, Du Y,
Zhang R, Yang R, Zhao W, Dun S, et al: Expression of microRNA-96
and its potential functions by targeting FOXO3 in non-small cell
lung cancer. Tumour Biol. 36:685–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kundu ST, Byers LA, Peng DH, Roybal JD,
Diao L, Wang J, Tong P, Creighton CJ and Gibbons DL: The miR-200
family and the miR-183~96~182 cluster target Foxf2 to inhibit
invasion and metastasis in lung cancers. Oncogene. 35:173–186.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu JJ, Wu YX, Zhao FJ and Xia SJ: MiR-96
promotes cell proliferation and clonogenicity by down-regulating of
FOXO1 in prostate cancer cells. Med Oncol. 31:9102014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Haflidadóttir BS, Larne O, Martin M,
Persson M, Edsjö A, Bjartell A and Ceder Y: Upregulation of miR-96
enhances cellular proliferation of prostate cancer cells through
FOXO1. PLoS One. 8:e724002013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou J, Liao W, Yang J, Ma K, Li X, Wang
Y, Wang D, Wang L, Zhang Y, Yin Y, et al: FOXO3 induces
FOXO1-dependent autophagy by activating the AKT1 signaling pathway.
Autophagy. 8:1712–1723. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vidal RL and Hetz C: Unspliced XBP1
controls autophagy through FoxO1. Cell Res. 23:463–464. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao Y, Li X, Cai MY, Ma K, Yang J, Zhou
J, Fu W, Wei FZ, Wang L, Xie D and Zhu WG: XBP-1u suppresses
autophagy by promoting the degradation of FoxO1 in cancer cells.
Cell Res. 23:491–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiao D, Bommareddy A, Kim SH, Sehrawat A,
Hahm ER and Singh SV: Benzyl isothiocyanate causes FoxO1-mediated
autophagic death in human breast cancer cells. PLoS One.
7:e325972012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
van der Vos KE, Eliasson P,
Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen
IJ, Mauthe M, Zellmer S, Pals C, et al: Modulation of glutamine
metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat
Cell Biol. 14:829–837. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hariharan N, Maejima Y, Nakae J, Paik J,
Depinho RA and Sadoshima J: Deacetylation of FoxO by Sirt1 plays an
essential role in mediating starvation-induced autophagy in cardiac
myocytes. Circ Res. 107:1470–1482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao Y, Yang J, Liao W, Liu X, Zhang H,
Wang S, Wang D, Feng J, Yu L and Zhu WG: Cytosolic FoxO1 is
essential for the induction of autophagy and tumour suppressor
activity. Nat Cell Biol. 12:665–675. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi
J, Liu Z and Cao W: Hepatic autophagy is suppressed in the presence
of insulin resistance and hyperinsulinemia: inhibition of
FoxO1-dependent expression of key autophagy genes by insulin. J
Biol Chem. 284:31484–31492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Krützfeldt J and Stoffel M: MicroRNAs: A
new class of regulatory genes affecting metabolism. Cell Metab.
4:9–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu XM, Qian JC, Deng ZL, Cai Z, Tang T,
Wang P, Zhang KH and Cai JP: Expression of miR-21, miR-31, miR-96
and miR-135b is correlated with the clinical parameters of
colorectal cancer. Oncol Lett. 4:339–345. 2012.PubMed/NCBI
|
|
53
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kang C and Avery L: To be or not to be,
the level of autophagy is the question: Dual roles of autophagy in
the survival response to starvation. Autophagy. 4:82–84. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Münz C: Autophagy in cellular
transformation, survival and communication with the tumor
microenvironment. Semin Cancer Biol. 23:299–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Macintosh RL and Ryan KM: Autophagy in
tumour cell death. Semin Cancer Biol. 23:344–351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Milani M, Rzymski T, Mellor HR, Pike L,
Bottini A, Generali D and Harris AL: The role of ATF4 stabilization
and autophagy in resistance of breast cancer cells treated with
bortezomib. Cancer Res. 69:4415–4423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Notte A, Ninane N, Arnould T and Michiels
C: Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast
cancer cells: Role of autophagy and JNK activation. Cell Death Dis.
4:e6382013. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jain K, Paranandi KS, Sridharan S and Basu
A: Autophagy in breast cancer and its implications for therapy. Am
J Cancer Res. 3:251–265. 2013.PubMed/NCBI
|
|
63
|
Xiao D, Bommareddy A, Kim SH, Sehrawat A,
Hahm ER and Singh SV: Benzyl isothiocyanate causes FoxO1-mediated
autophagic death in human breast cancer cells. PLoS One.
7:e325972012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cufí S, Vazquez-Martin A,
Oliveras-Ferraros C, Corominas-Faja B, Cuyàs E, López-Bonet E,
Martin-Castillo B, Joven J and Menendez JA: The anti-malarial
chloroquine overcomes primary resistance and restores sensitivity
to trastuzumab in HER2-positive breast cancer. Sci Rep. 3:24692013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cook KL, Shajahan AN and Clarke R:
Autophagy and endocrine resistance in breast cancer. Expert Rev
Anticancer Ther. 11:1283–1294. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xue LY, Chiu SM and Oleinick NL: Atg7
deficiency increases resistance of MCF-7 human breast cancer cells
to photodynamic therapy. Autophagy. 6:248–255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Debnath J: The multifaceted roles of
autophagy in tumors-implications for breast cancer. J Mammary Gland
Biol Neoplasia. 16:173–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Di X, Shiu RP, Newsham IF and Gewirtz DA:
Apoptosis, autophagy, accelerated senescence and reactive oxygen in
the response of human breast tumor cells to adriamycin. Biochem
Pharmacol. 77:1139–1150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gewirtz DA, Hilliker ML and Wilson EN:
Promotion of autophagy as a mechanism for radiation sensitization
of breast tumor cells. Radiother Oncol. 92:323–328. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhang Y, Gan B, Liu D and Paik JH: FoxO
family members in cancer. Cancer Biol Ther. 12:253–259. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kousteni S: FoxO1: A molecule for all
seasons. J Bone Miner Res. 26:912–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Guo Y, Liu H, Zhang H, Shang C and Song Y:
MiR-96 regulates FOXO1-mediated cell apoptosis in bladder cancer.
Oncol Lett. 4:561–565. 2012.PubMed/NCBI
|
|
75
|
Haflidadóttir BS, Larne O, Martin M,
Persson M, Edsjö A, Bjartell A and Ceder Y: Upregulation of miR-96
enhances cellular proliferation of prostate cancer cells through
FOXO1. PLoS One. 8:e724002013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Chen C, Xu T, Zhou J, Yan Y, Li W, Yu H,
Hu G, Ding X, Chen J and Lu Y: High cytoplasmic FOXO1 and pFOXO1
expression in astrocytomas are associated with worse surgical
outcome. PLoS One. 8:e692602013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhao Y, Wang Y and Zhu WG: Applications of
post-translational modifications of FoxO family proteins in
biological functions. J Mol Cell Biol. 3:276–282. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ma Y, Yang HZ, Dong BJ, Zou HB, Zhou Y,
Kong XM and Huang YR: Biphasic regulation of autophagy by miR-96 in
prostate cancer cells under hypoxia. Oncotarget. 5:9169–9182. 2014.
View Article : Google Scholar : PubMed/NCBI
|