Introduction
Despite numerous therapeutic advances, epithelial
ovarian cancer remains the most lethal type of gynecologic
malignancy, as the 5-year survival rate is <25% for patients who
are diagnosed with stage III–IV disease (1). Cytoreductive surgery followed by the
administration of systemic chemotherapy remains an important
treatment for advanced stage ovarian cancer. A combination of
platinum- and taxane-based chemotherapy is recommended as the
therapy subsequent to surgery for at least 70% of patients with
ovarian cancer. However, the initial response to chemotherapy is
not permanent, and the tumors become resistant (2). Novel treatment strategies are therefore
required.
The targeting of histone deacetylase (HDAC) activity
using pharmacological small molecule HDAC inhibitors (HDACi) has
become a notable therapeutic strategy (3). HDACi cause changes in the acetylation
status of chromatin and other non-histone proteins, resulting in
changes in gene expression, the induction of apoptosis, cell cycle
arrest and the inhibition of angiogenesis and metastasis (4). Currently, 7 structurally distinct
classes of HDACi are known, and inhibitors of 4 different classes
are now in clinical development (5).
Vorinostat, a broad spectrum HDACi, which inhibits zinc-dependent
HDAC (Class I, II and IV), was the first HDACi approved by the
United States Food and Drug Administration (FDA) and has been
demonstrated to be successful in the treatment of refractory
cutaneous T cell lymphoma (6).
Trichostatin A (TSA), 1 of the most extensively studied HDACi, has
been shown to inhibit cell proliferation and induce apoptosis in
ovarian cancer cells in preclinical studies. This raises the
possibility that TSA may serve a role in the treatment of ovarian
cancer (4,7).
Proteasome inhibitors are a novel group of
therapeutic agents that are designed to restrict the degradation of
proteins through the inhibition of proteasome activity. The
inhibitors constrain cancer progression through the recovery of key
protein functions in the regulation of apoptosis, cell cycle
progression and angiogenesis (8). The
anti-tumor effect of PS-341 (bortezomib/Velcade), the first
FDA-approved proteasome inhibitor for the treatment of multiple
myeloma and mantle cell lymphoma, has been extended to numerous
other types of malignancy through the preferential induction of
toxicity and cell death in tumor cells (9). In addition, the inhibitor is being
evaluated for the treatment of solid tumors, including ovarian
cancer (10). PS-341 exhibits a wide
range of molecular effects, including the stabilization of cell
cycle regulation proteins, the inhibition of nuclear factor-κB
activation, the induction of apoptosis and the counterbalance of
B-cell lymphoma 2 resistance and angiogenesis (11). Despite being the most widely used
proteasome inhibitor, and the first to have been identified, the
efficacy of PS-341 is limited when it is used as a single agent.
The present study considers whether the anticancer effect of PS-341
is enhanced when combined with other therapeutic agents, such as
TSA.
A growing number of studies in the present field
provide evidence to support the hypothesis that the combined
treatment of proteasome inhibitors and HDACi in malignancies
exhibits a synergistic effect, including in the treatment of human
multiple myeloma, recurrent glioblastoma, and renal, pancreatic and
hepatocellular cancer (12–15). A previous study demonstrated that TSA
significantly improved PS-341-mediated inhibition of head and neck
squamous cell carcinoma both in vitro and in vivo
(16). The present study examined the
efficacy of the combination of HDAC inhibition by TSA, and
proteasome inhibition by PS-341, in ovarian cancer cell lines in
vitro with respect to their potential synergistic effect on
levels of apoptosis and cell cycle arrest. The present study also
sought to investigate the molecular mechanism of taxane resistance
and the synergistic activity of TSA and PS-341 by the assessment of
downstream effector pathways, to provide supporting data for a
mechanism-based regimen for the treatment of ovarian cancer,
particularly for taxane-resistant ovarian cancer.
Materials and methods
Cell lines and cell culture
The taxane-sensitive ovarian cancer A2780 cell line
was obtained from the American Type Culture Collection (Manassas,
VA, USA) and cultured in RPMI-1640 medium supplemented with 10%
fetal bovine serum (FBS; both from Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) (17). The
taxane resistant variant ovarian cancer A2780T cell line was
donated from the Biological Sciences Collegiate Division of
Huazhong University of Science and Technology (Wuhan, China), and
was cultured in RPMI-1640 medium supplemented with 10% FBS and 80
nM taxane (Bristol-Myers Squibb Caribbean Company, New York, NY,
USA) at 37°C with 5% CO2. For time-course study of
cyclin B1 expression, cells were trypsinized and transferred to a
new 6-well plate. After 24 h of attachment, the culture medium was
replaced for fresh RPIM-1640 supplemented with 10% FBS, containing
DMSO or 1 µM taxane. Cells were harvested at the indicated time
points and analyzed by western blot analysis.
Chemicals and antibodies
TSA was purchased from Sigma-Aldrich (Merck
Millipore, St. Louis, MO, USA), and the specific proteasome
inhibitor PS-341 was purchased from Millenium Pharmaceuticals
(Takeda Pharmaceutical Company, Ltd., Cambridge, MA, USA). The
stock solutions of TSA and PS-341 were reconstituted in
dimethylsulfoxide (DMSO) at a concentration of 1 mM, stored at
−20°C and diluted into the complete cell culture medium prior to
use. Taxane was purchased from Bristol-Myers Squibb Caribbean
Company (New York, NY, USA). Propidium iodide and DMSO were
purchased from Sigma-Aldrich (Merck Millipore). Antibodies were
obtained from the following commercial sources: Anti-cyclin A (cat.
no., 611268; dilution, 1:500), anti-cyclin B1 (cat. no., 554179;
dilution, 1:500), anti-cyclin D (cat. no., 554181; dilution,
1:500), anti-CDK1 (cat. no., 610037; dilution, 1:1,000), anti-CDK2
(cat. no., 610146; dilution, 1:1,000), anti-Rb (cat. no., 554145;
dilution, 1:500) and anti-MAD2 (cat. no., 610679; dilution,
1:1,000) were obtained from BD Pharmingen (San Diego, CA, USA);
anti-BUB1 (cat. no., sc-28257; dilution, 1:500),
anti-phosphorylated-histone H3 (H3P; Ser10; cat. no., sc-8656-R;
dilution, 1:1000) and anti-β-actin (cat. no., sc-7210; dilution,
1:1,000) were obtained from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA).
Flow cytometry
All experiments were performed in triplicate. A
total of 1×106 cells were plated in 6-well plates.
Subsequent to 24 h treatment with the chemotherapy drugs (1 µM
taxane, 500 nM TSA and 40 nM PS-341, individually or in
combination) or DMSO as control, the cells were washed twice with
PBS, stained with 5 µl Annexin V- Phycoerythrin (PE) and 5 µl
7-amino-actinomycin D (5 µg/ml) in 1X binding buffer following the
protocol of the manufacturer (Nanjing Keygen Biotech, Co., Ltd,
Nanjing, China) and analyzed using fluorescence-activated cell
sorting (FACS) analysis. With regard to cell cycle detection, the
cells were harvested and washed twice prior to being fixed in
ice-cold 70% ethanol and stored at −20°C overnight. The cells were
then washed once in PBS and resuspended in a solution of 5 mg/ml
propidium iodide and 0.5 mg/ml RNase A (Thermo Fisher Scientific,
Inc.) in PBS for 30 min in the dark prior to being sorted by
FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA). The results
were analyzed with Cell Quest version 3.3 software (BD
Biosciences).
Protein extraction and western blot
analysis
Subsequent to incubation, the cells were harvested
and lysed in an ice-cold SDS lysis buffer (Beyotime Institute of
Technology, Shanghai, China) for 30 min. The lysates were
centrifuged at 12,000 × g at 4°C for 15 min, and the supernatant
was denatured in SDS sample buffer at 100°C for 8 min, then stored
at −20°C. Equal amounts of protein were separated by
Tricine-SDS-PAGE and transferred to nitrocellulose membranes
(18). The membrane was blocked in
TBS with Tween 20 with 5% non-fat milk for 1 h at 37°C, then
incubated with primary antibodies targeting genes investigated in
the experiment (e.g., cyclin A, B1 and D) at 4°C overnight followed
by incubation with the appropriate secondary antibodies against the
species of the primary antibody [alkaline phosphatase labeled goat
anti-mouse IgG (cat. no., A0258; dilution, 1:1,000), alkaline
phosphatase labeled goat anti-rabbit IgG (cat. no., A0239;
dilution, 1:1,000; Beyotime Institute of Technology)] at 37°C for 1
h. The proteins were subsequently visualized with the BCIP/NBT
Alkaline Phosphatase Color Development kit following the
manufacturer's protocol (cat. no., C3206; Beyotime Institute of
Technology) which included a nitro-blue tetrazolium
chloride/5-bromo-4-chloro-3′-indolyphosphate p-toluidine
salt/buffer. β-actin was used as an internal reference.
Overexpression and knockdown of cyclin
B1 by transfection
The full-length coding sequence of the human cyclin
B1 plasmid with a green fluorescent protein (GFP) tag
(chloroxylenol/cyclin B1-GFP; lot no., 26061) was purchased from
Addgene, Inc. (Cambridge, MA, USA). In total, 1.5×105
ovarian cancer cells were plated per well in a 6-well plate on day
0. On day 1, the A2780 cells were transfected with 2 µg plasmid in
Opti-minimal essential medium (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) using Lipofectamine 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). On day 2, the cells
were treated with taxane alone or combined TSA and PS341 for an
additional 24 h. The small interfering RNA (siRNA) corresponding to
positions 776–796 of cyclin B1 (5′-CUCGUACAGCCUUGGGAGACAtt-3′) and
the control siRNA targeting GFP were ordered from Invitrogen
(Thermo Fisher Scientific, Inc.). The A2780T cells were transfected
with siRNA using Lipofectamine 2000 according to the protocol of
the manufacturer (Invitrogen; Thermo Fisher Scientific, Inc.). A
total of 24 h subsequent to transfection, the cells were treated
with taxane or combined TSA and PS341 for an additional 24 h.
Immunofluorescence assay
The confluent cells grown on glass cover-slips were
rinsed once with PBS, fixed in 4% paraformaldehyde for 10 min and
permeabilized with 0.2% Triton-X in PBS for 5 min at room
temperature. The cells were incubated with the primary antibody
phospho-histone H3 (H3P; Ser10) at a dilution of 1:100 or PBS
overnight at 4°C, followed by incubation with the fluorescein
isothiocyanate-conjugated secondary antibody at 37°C for 1 h (cat.
no., A0562; dilution, 1:200, Beyotime Institute of Technology). All
controls were incubated with PBS. The nuclei were stained with DAPI
(Millipore Sigma, St. Louis, Missouri, USA). All immunofluorescence
images were captured with a Nikon Eclipse 80i Microscope (Nikon
Corporation, Tokyo, Japan). With respect to drugs treatment, the
cells culture medium were replaced for fresh containing 1 µM taxane
alone, 500 nM TSA combined with 40 nM PS-341, or equal volume
percentage of DMSO as control. Each treatment lasted 24 h and was
stopped by rinsing the cells at 3 time points with cold PBS.
Colony formation assay
Cell proliferation was determined by colony
formation assay according to a published protocol (17). The cells were seeded at 200 cells per
well in 6-well plates and treated with DMSO as control, taxane
alone or TSA combined with PS-341 for 3 h. Subsequent to 10–14
days, colonies were fixed in 4% paraformaldehyde for 20 min and
stained with 0.5% crystal violet in distilled water for 15 min at
room temperature. The cells were then rinsed with tap water and
allowed to air dry. The number of colonies formed was counted with
a charge-coupled device camera at ×4 magnification on a DM-IRB
inverted microscope (Leitz, Wetzlar, Germany).
Statistical analysis
The data are presented as the mean ± standard
deviation of at least 3 separate experiments. Comparisons between 2
groups were analyzed using a Student's t-test. P<0.05 was
considered to indicate a statistically significant difference in
all experiments.
Results
TSA interacts synergistically with
PS-341 to induce G2/M arrest in taxane-resistant ovarian
cancer cells
In order to explore the combined effect of TSA and
PS-341 on the taxane-resistant A2780T cell line, the present study
treated the taxane sensitive and resistant cell lines with combined
500 nM TSA and 40 nM PS-341, or with each as a single agent.
Furthermore, the present study compared the
synergistic effect of TSA and PS-341 with the effect induced by
taxane alone in ovarian cancer cells. The results of the flow
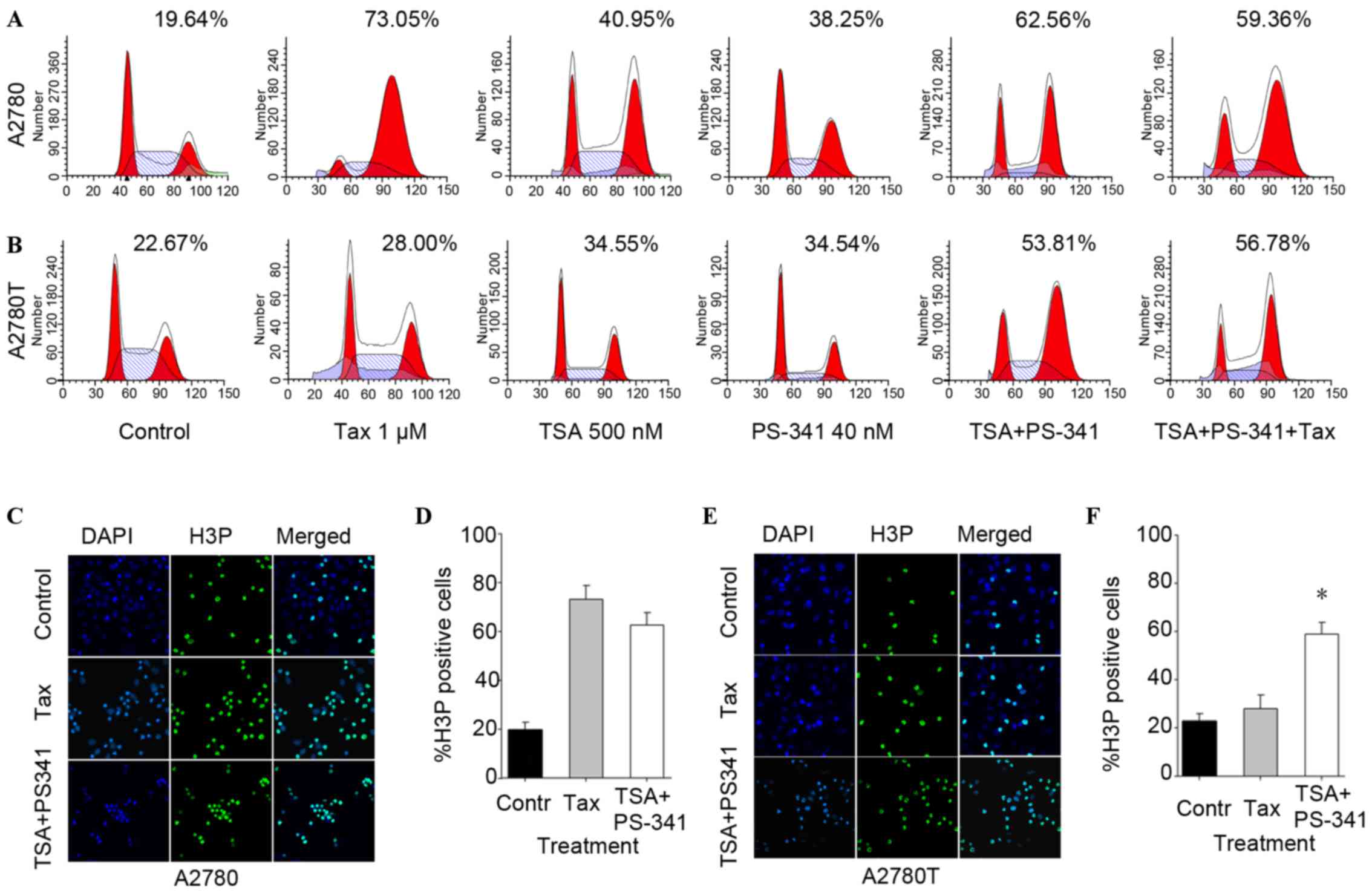
cytometric analysis showed that treatment with TSA and PS-341
induced G2/M arrest in the 2 ovarian cancer cell lines
while taxane treatment induced G2/M arrest in A2780 but
not A2780T cells (Fig. 1A and B).
Compared with the percentage cell cycle arrest induced by TSA or
PS-341 alone, a greater percentage of A2780 and A2780T cells
arrested in the G2/M phase was observed subsequent to
the cells being treated with combined TSA and PS-341.
The expression of the mitotic phase-specific protein
H3P (Ser10) was examined by immunofluorescence staining subsequent
to the aforementioned treatment. A similar pattern of change was
observed in A2780 and A2780T cells subsequent to TSA and PS-341
treatment (figures not shown). Consistent with the cell cycle
arrest results, the combination of TSA and PS341 increased H3P
(Ser10) expression in A2780T cells compared with taxane, which was
not observed in A2780 cells (Fig.
1C-F). The increase of H3P (Ser10) indicated that the
combination of TSA and PS-341 demonstrated synergistic effects in
the induction of G2/M arrest, particularly mitotic phase
arrest in taxane-resistant ovarian cancer cells.
Combination of TSA and PS-341 resulted
in enhanced expression of cyclin B1 in taxane-sensitive and
taxane-resistant cell lines
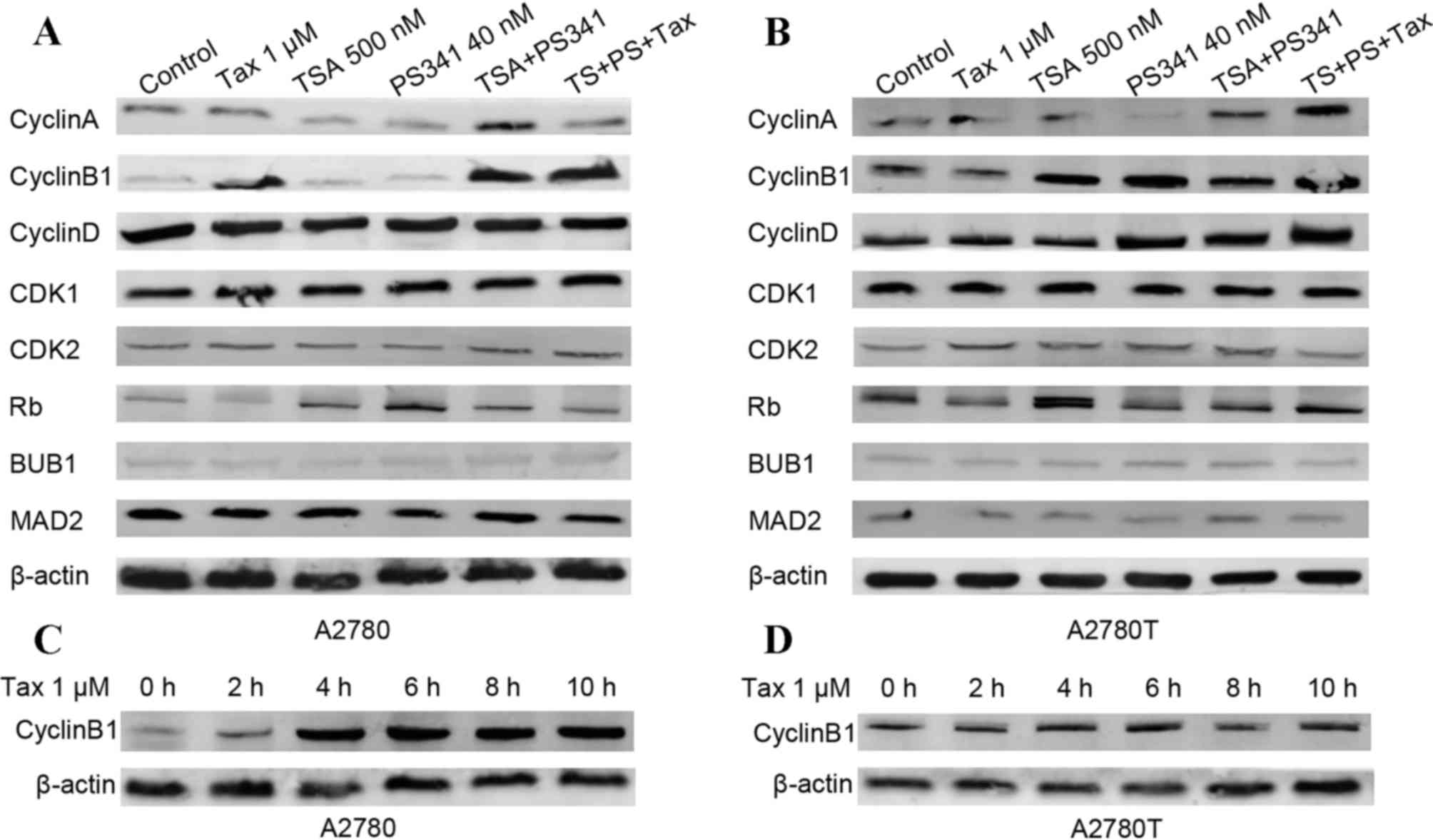
Western blot analysis was subsequently used to
assess the effect of the combination of TSA and PS-341 on the
expression of various cell cycle-associated proteins in
taxane-sensitive and taxane-resistant cell lines. As shown in
Fig. 2A, the treatment of cells with
taxane alone and the combination of TSA and PS-341 induced a marked
increase in the expression of cyclin B1 protein in the A2780 cells.
However, the upregulation of cyclin B1 expression was only detected
subsequent to treatment with TSA and PS-341 in A2780T cells, not
subsequent to taxane treatment alone, as shown in Fig. 2B. The difference in cyclin B1
expression in response to taxane treatment indicated that cyclin B1
may be involved in taxane resistance. Consistent with the previous
result, the expression of cyclin B1 in A2780 cells increased in a
time-dependent manner subsequent to treatment (Fig. 2C), yet no change of cyclin B1 protein
was observed in the A2780T cells (Fig.
2D). In addition, the basal expression level of cyclin B1 in
the A2780T cells was higher compared with the level in the A2780
cells. Collectively, the findings of the western blot analysis
indicated that the induction of G2/M arrest by taxane in
A2780 cells primarily depends on the increased expression of cyclin
B1.
Overexpression of cyclin B1 resisted
the effect of taxane in A2780 cells but did not affect the
synergistic effect of TSA and PS-341
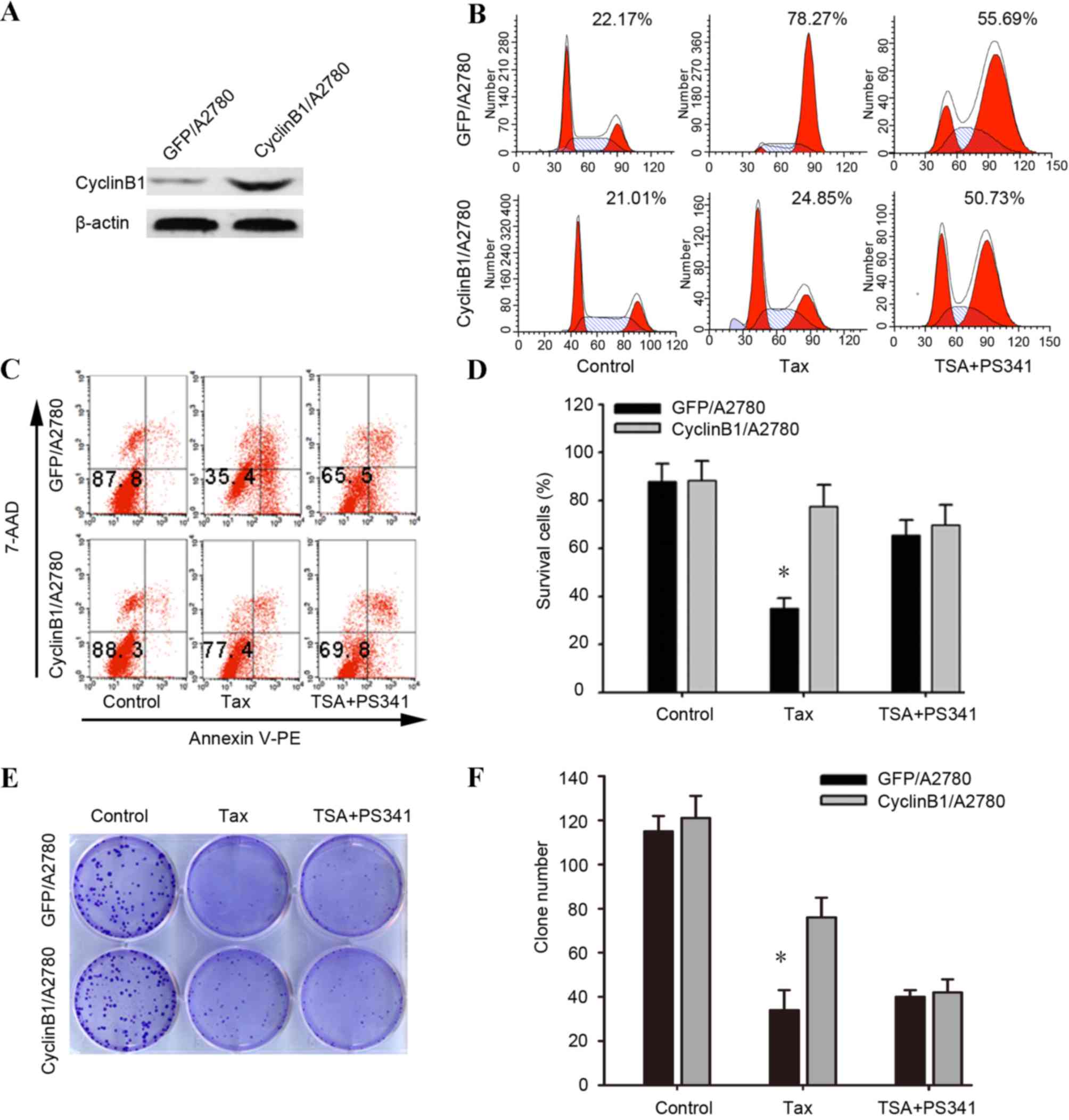
In order to investigate the biological consequence
of the high basal expression of cyclin B1 and the upregulation of
cyclin B1 in response to taxane or to the coadministration of TSA
and PS-341, the present study overexpressed cyclin B1 in the
taxane-sensitive A2780 cell line via plasmid transfection. Firstly,
the result the of western blot analysis confirmed that plasmid
transfection increased the level of cyclin B1 protein expression
(Fig. 3A). The transfected cells were
then treated with 1 µM taxane or the combination of 500 nM TSA and
40 nM PS-341 for 24 h.
The percentage cyclin B1-overexpressing cells in
G2/M arrest subsequent to treatment with taxane was
detected to be 24.85% by flow cytometry, while that of the control
GFP-transfected cells was 78.27%. No significant difference was
observed between the 2 groups of transfected cells subsequent to
treatment with a combination of 500 nM TSA and 40 nM PS-341
(Fig. 3B) (P=0.378). The flow
cytometry analysis with respect to apoptosis revealed that cyclin
B1-transfected cells resulted in 77.4±2.6% Annexin V-PE and
7-amino-actinomycin (AA) D-negative cells subsequent to treatment
of taxane. In contrast, the control GFP-transfected cells, treated
with taxane, resulted in 35.4±1.4% annexin V-PE and 7-AAD-negative
cells (Fig. 3C-D). Similar results
were found in the colony formation assay (Fig. 3E-F). The aforementioned results
indicated that the overexpression of cyclin B1 in A2780 cells
enabled the cells to override the G2 DNA damage
checkpoint, thus reducing the anticancer effect of taxane. The
effect of combined TSA and PS-341 was not affected by the
upregulation of cyclin B1.
Knockdown of cyclin B1 expression
reversed the resistance to taxane in A2780T cells and enhanced the
synergistic effect induced by TSA and PS-341
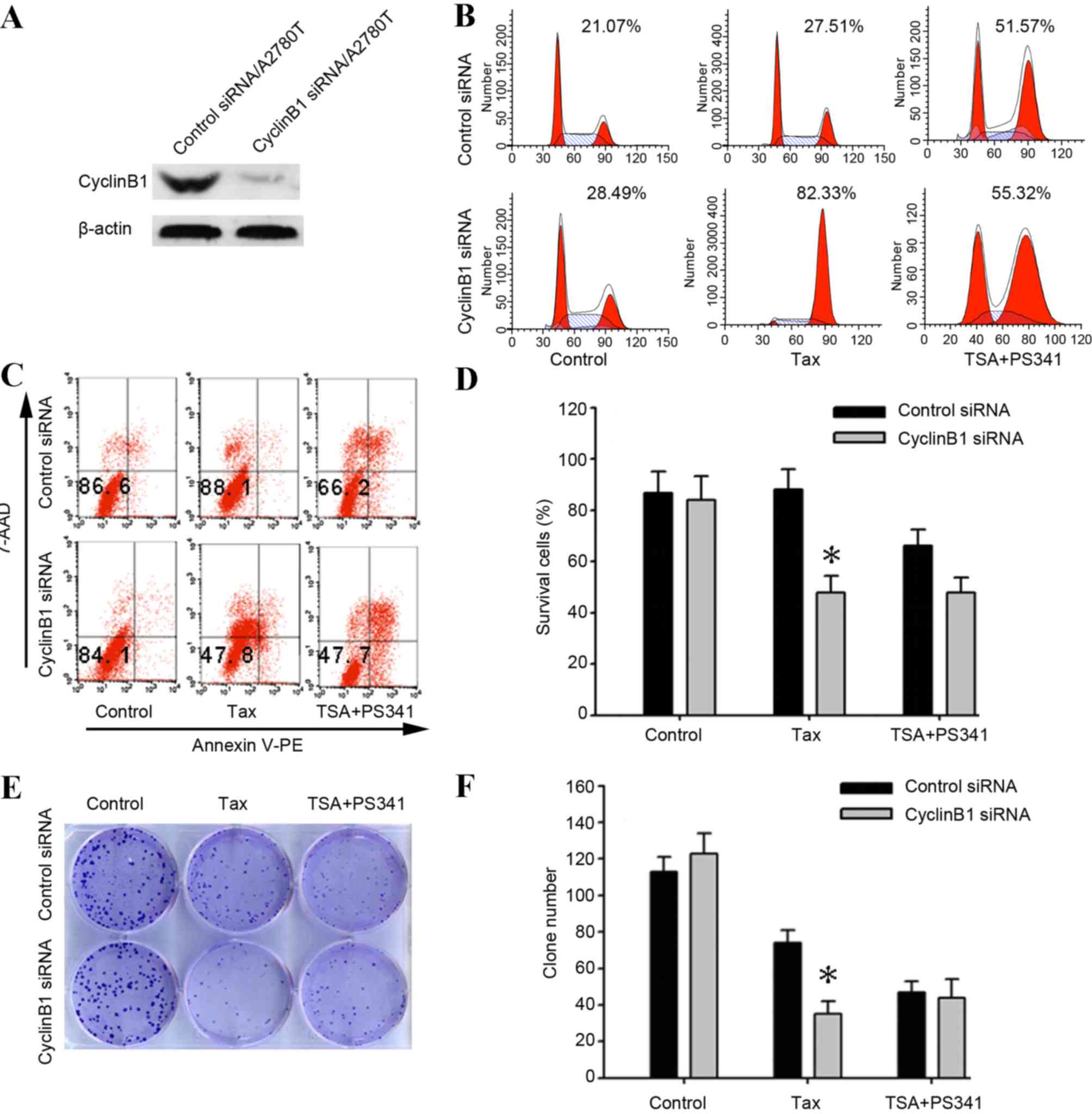
To additionally explore the role of cyclin B1 in
taxane resistance, the present study designed synthetic siRNA in
order to silence cyclin B1 expression. The results of western blot
analysis confirmed that the cyclin B1 siRNA suppressed the cyclin
B1 protein expression in taxane-resistant A2780T cells (Fig. 4A). The flow cytometry results revealed
that 24 h exposure of the cyclin B1-siRNA transfection A2780T cells
to 1 µM taxane resulted in 82.33±2.8% cells arrested in the
G2/M phase and 47.8±2.2% Annexin V-PE and 7-AAD-negative
cells. By contrast, the aforementioned treatment of control cells
resulted in 27.51±2.6% cells arrested in the G2/M phase
and 88.1±2.1% Annexin V-PE and 7-AAD-negative cells (Fig. 4B-D). For proliferation investigation,
the siRNA transfected cells were subsequently treated with 1 µM
taxane or a combination of 500 nM TSA and 40 nM PS-341 for 3 h. The
surviving fraction of cells were detected by crystal violet
staining. It was revealed that the clone number was significantly
reduced subsequent to treatment with taxane in the cyclin B1
siRNA-transfected cells compared with the negative scrambled
siRNA-transfected cells (P=0.011). By contrast, similar clone
numbers were observed in the negative scrambled siRNA- and cyclin
B1 siRNA-transfected cells subsequent to treatment with TSA plus
PS-341 (P=0.133; Fig. 4E and F).
Discussion
Encouraging in vitro and in vivo
results have resulted in several clinical trials with HDAC or
proteasome inhibitors alone. Although the efficacy of the
inhibitors is limited when used as single agent therapies, previous
studies have shown that the coadministration of a HDAC and
proteasome inhibitor may exert greater therapeutic efficacy in
numerous tumor cell lines and patients with lymphoma (19,20).
Additionally, it was reported that the combined molecular targeting
of HDAC and proteasomes synergistically induced apoptosis in
non-small cell lung cancer (21).
Another study demonstrated that PS-341 increased paclitaxel
sensitivity in ovarian cancer cells (22). Thus, the present authors hypothesized
that the HDACi TSA and the proteasome inhibitor PS-341 in
combination may overcome taxane resistance and induce cell cycle
arrest and apoptosis in taxane-resistant ovarian cancer cells.
Indeed, the present results indicated that the combination of TSA
and PS-341 resulted in the synergistic effect of G2/M
arrest in the taxane-sensitive and the taxane-resistant ovarian
cancer cell lines (Fig. 1).
Taxane blocks the progression of the cell cycle
through interference with the assembly and dynamics of microtubule
spindles, thus preventing their attachment to kinetochores
(23). The therapeutic efficacy of
taxane depends on whether or not it can induce persistent cell
cycle arrest and apoptosis. The molecular mechanism that underlies
the regulation of the cell cycle and apoptosis, and that determines
susceptibility to taxane in tumor cells, is not presently known. In
addition, the molecular targets by which TSA and PS-341 induce
G2/M arrest and inhibit tumor cell proliferation is not
fully understood.
Uncontrolled cell proliferation, which is associated
with the loss of proper cell cycle control, is a prominent feature
of ovarian cancer. The cell cycle is controlled by a highly
conserved family of cyclin-dependent kinases (Cdks) and their
regulatory subunits, known as cyclins. A number of studies have
reported that several Cdks and cyclins are involved in the
prognosis of various types of cancer (24–26). The
present study found that the most marked difference between the
response of A2780 and A2780T cells to taxane was the protein
expression level of cyclin B1 (Fig.
2). The present study also found that the baseline expression
level of cyclin B1 cells was higher in A2780T compared with A2780
cells that exhibit more sensitivity to taxane treatment. Cyclin B1
is indispensable for the transition between the G2 phase
and mitosis, and the upregulation of the protein is closely
associated with a poor prognosis in various types of cancer
(27,28). Additionally, the overexpression of
cyclin B1 is involved in resistance to radiotherapy in pancreatic
cancer (29). Nuclear cyclin
B1-positive types of breast carcinoma are resistant to adjuvant
therapy (26). A previous clinical
study indicated that chemotherapy resistance in non-small cell lung
cancer may also be enhanced with the increased expression of cyclin
B1, and cyclin B1 inhibitors may increase the efficacy of
chemotherapy (30). In addition,
previous studies suggest that any blockade of the exit from
mitosis, such as degradation-resistant cyclin B1 expression, exerts
a lethal effect on cancer cells (31,32).
Consistent with the aforementioned findings, the knockdown of
cyclin B1 expression in A2780T cells increased the level of cancer
cell apoptosis and reversed the resistance to taxane in the present
study (Fig. 4).
It is also noteworthy that the combination of TSA
and PS-341 induced a marked increase in the expression level of
cyclin B1 in A2780 and A2780T cells, regardless of the basal
expression level of cyclin B1 (Fig.
2). Additionally, the combination of TSA and PS341 generated a
synergistic effect in terms of inducing G2/M arrest and
inhibiting cell proliferation in A2780 cells subsequent to the
overexpression of cyclin B1 (Fig. 3),
which indicates that the molecular mechanism underlining the
synergistic effect of TSA and PS-341 differs from the mechanism of
taxane.
In summary, the present study revealed that TSA and
PS-341 synergistically enhanced the level of G2/M arrest
and the cytotoxic potential in ovarian cancer cells. An additional
benefit of the combination of TSA and PS-341 is the potential
inhibition of the growth of taxane-resistant ovarian cancer, which
would increase the overall therapeutic effect. The present results
regarding the synergistic efficacy of TSA and PS-341 exposes a
novel perspective in terms of the therapeutic strategy for the
treatment of ovarian cancer, and potentially other solid
tumors.
Acknowledgements
The authors would like to thank the Biological
Sciences Collegiate Division of Huazhong University of Science and
Technology, Wuhan, China, for their support. The present study was
supported by grants from the National Science Foundation of China
(grant nos. 81302266, 81272859 and 81101962) and the ‘973’ Program
of China (grant no. 2015CB553903).
References
|
1
|
Fang F, Balch C, Schilder J, Breen T,
Zhang S, Shen C, Li L, Kulesavage C, Snyder AJ, Nephew KP and Matei
DE: A phase 1 and pharmacodynamic study of decitabine in
combination with carboplatin in patients with recurrent,
platinum-resistant, epithelial ovarian cancer. Cancer.
116:4043–4053. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bast RC Jr, Hennessy B and Mills GB: The
biology of ovarian cancer: New opportunities for translation. Nat
Rev Cancer. 9:415–428. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pchejetski D, Alfraidi A, Sacco K,
Alshaker H, Muhammad A and Monzon L: Histone deacetylases as new
therapy targets for platinum-resistant epithelial ovarian cancer. J
Cancer Res Clin Oncol. 142:1659–1671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ma X, Ezzeldin HH and Diasio RB: Histone
deacetylase inhibitors: Current status and overview of recent
clinical trials. Drugs. 69:1911–1934. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wagner JM, Hackanson B, Lubbert M and Jung
M: Histone deacetylase (HDAC) inhibitors in recent clinical trials
for cancer therapy. Clin Epigenetics. 1:117–136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miller CP, Singh MM, Rivera-Del Valle N,
Manton CA and Chandra J: Therapeutic strategies to enhance the
anticancer efficacy of histone deacetylase inhibitors. J Biomed
Biotechnol. 2011:5142612011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu K, Qu D, Sakamoto T, Fukasawa I,
Hayashi M and Inaba N: Telomerase expression and cell proliferation
in ovarian cancer cells induced by histone deacetylase inhibitors.
Arch Gynecol Obstet. 277:15–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shahshahan MA, Beckley MN and Jazirehi AR:
Potential usage of proteasome inhibitor bortezomib (Velcade,
PS-341) in the treatment of metastatic melanoma: Basic and clinical
aspects. Am J Cancer Res. 1:913–924. 2011.PubMed/NCBI
|
|
9
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: Pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Russo A, Fratto ME, Bazan V, Schiro V,
Agnese V, Cicero G, Vincenzi B, Tonini G and Santini D: Targeting
apoptosis in solid tumors: The role of bortezomib from preclinical
to clinical evidence. Expert Opin Ther Targets. 11:1571–1586. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kao C, Chao A, Tsai CL, Lin CY, Chuang WC,
Chen HW, Yen TC, Wang TH, Lai CH and Wang HS: Phosphorylation of
signal transducer and activator of transcription 1 reduces
bortezomib-mediated apoptosis in cancer cells. Cell Death Dis.
4:e5122013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Friday BB, Anderson SK, Buckner J, Yu C,
Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon
E, et al: Phase II trial of vorinostat in combination with
bortezomib in recurrent glioblastoma: A north central cancer
treatment group study. Neuro Oncol. 14:215–221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato A and Asano T, Ito K, Sumitomo M and
Asano T: Suberoylanilide hydroxamic acid (SAHA) combined with
bortezomib inhibits renal cancer growth by enhancing histone
acetylation and protein ubiquitination synergistically. BJU Int.
109:1258–1268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Spratlin JL, Pitts TM, Kulikowski GN,
Morelli MP, Tentler JJ, Serkova NJ and Eckhardt SG: Synergistic
activity of histone deacetylase and proteasome inhibition against
pancreatic and hepatocellular cancer cell lines. Anticancer Res.
31:1093–1103. 2011.PubMed/NCBI
|
|
16
|
Kim J, Guan J, Chang I, Chen X, Han D and
Wang CY: PS-341 and histone deacetylase inhibitor synergistically
induce apoptosis in head and neck squamous cell carcinoma cells.
Mol Cancer Ther. 9:1977–1984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nature
Protoc. 1:2315–2319. 2006. View Article : Google Scholar
|
|
18
|
Schägger H: Tricine-SDS-PAGE. Nat Protoc.
1:16–22. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bhatt S, Ashlock BM, Toomey NL, Diaz LA,
Mesri EA, Lossos IS and Ramos JC: Efficacious proteasome/HDAC
inhibitor combination therapy for primary effusion lymphoma. J Clin
Invest. 123:2616–2628. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hui KF, Lam BH, Ho DN, Tsao SW and Chiang
AK: Bortezomib and SAHA synergistically induce ROS-driven
caspase-dependent apoptosis of nasopharyngeal carcinoma and block
replication of Epstein-Barr virus. Mol Cancer Ther. 12:747–758.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karthik S, Sankar R, Varunkumar K and
Ravikumar V: Romidepsin induces cell cycle arrest, apoptosis,
histone hyperacetylation and reduces matrix metalloproteinases 2
and 9 expression in bortezomib sensitized non-small cell lung
cancer cells. Biomed Pharmacother. 68:327–334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Steg AD, Burke MR, Amm HM, Katre AA,
Dobbin ZC, Jeong DH and Landen CN: Proteasome inhibition reverses
hedgehog inhibitor and taxane resistance in ovarian cancer.
Oncotarget. 5:7065–7080. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Russell P, Hennessy BT, Li J, Carey MS,
Bast RC, Freeman T and Venkitaraman AR: Cyclin G1 regulates the
outcome of taxane-induced mitotic checkpoint arrest. Oncogene.
31:2450–2460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Androic I, Kramer A, Yan R, Rödel F, Gätje
R, Kaufmann M, Strebhardt K and Yuan J: Targeting cyclin B1
inhibits proliferation and sensitizes breast cancer cells to taxol.
BMC Cancer. 8:3912008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Soria JC, Jang SJ, Khuri FR, Hassan K, Liu
D, Hong WK and Mao L: Overexpression of cyclin B1 in early-stage
non-small cell lung cancer and its clinical implication. Cancer
Res. 60:4000–4004. 2000.PubMed/NCBI
|
|
26
|
Suzuki T, Urano T, Miki Y, Moriya T,
Akahira J, Ishida T, Horie K, Inoue S and Sasano H: Nuclear cyclin
B1 in human breast carcinoma as a potent prognostic factor. Cancer
Sci. 98:644–651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weng L, Du J, Zhou Q, Cheng B, Li J, Zhang
D and Ling C: Identification of cyclin B1 and Sec62 as biomarkers
for recurrence in patients with HBV-related hepatocellular
carcinoma after surgical resection. Mol Cancer. 11:392012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He Y, Zhou Z, Hofstetter WL, Zhou Y, Hu W,
Guo C, Wang L, Guo W, Pataer A, Correa AM, et al: Aberrant
expression of proteins involved in signal transduction and DNA
repair pathways in lung cancer and their association with clinical
parameters. PLoS One. 7:e310872012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cloos CR, Daniels DH, Kalen A, Matthews K,
Du J, Goswami PC and Cullen JJ: Mitochondrial DNA depletion induces
radioresistance by suppressing G2 checkpoint activation in human
pancreatic cancer cells. Radiat Res. 171:581–587. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stewart DJ: Tumor and host factors that
may limit efficacy of chemotherapy in non-small cell and small cell
lung cancer. Crit Rev Oncol Hematol. 75:173–234. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang HC, Shi J, Orth JD and Mitchison TJ:
Evidence that mitotic exit is a better cancer therapeutic target
than spindle assembly. Cancer Cell. 16:347–358. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kavallaris M: Microtubules and resistance
to tubulin-binding agents. Nat Rev Cancer. 10:194–204. 2010.
View Article : Google Scholar : PubMed/NCBI
|