Introduction
Prostate cancer is the most prevalent type of male
cancer in Western countries and represents the fourth most common
type of cancer worldwide (1).
Prostate cancer growth is initially androgen-dependent; hence the
gold standard of treatment is hormone-ablation therapy with
anti-androgens and/or androgen-deprivation therapies (2). However, the efficacy of these treatments
in the majority of patients is short-lived and the cancer typically
recurs in a more aggressive form, termed castrate-resistant
prostate cancer, which remains challenging to treat, and is
fatal.
Phorbol esters, which are activators of protein
kinase C isozymes, can trigger distinct cellular processes,
including skin carcinogenesis in mice and migration of human glioma
cells (3). Despite their well-known
tumor-promoting effects, phorbol esters can also induce apoptosis
in several types of cancer cells, including prostate cancer cells
(4). Studies have demonstrated that
phorbol 12-myristate 13-acetate (PMA), one of the most active
phorbol esters, can induce apoptosis in LNCaP prostate cancer cells
(5). The prostate cancer LNCaP cell
line is considered to be a suitable model to investigate the
mechanism of apoptosis induced by phorbol esters in prostate cancer
cells (4,6,7), in which
the release of death factors, including tumor necrosis factor α
(TNFα), are necessary (7,8).
The Krüppel-like transcription factor 5
(KLF5/IKLF/BTEB2), a type of zinc-finger transcription factor, has
important roles in various cellular processes, including in cell
proliferation, cell cycle progression and apoptosis (9–11). Several
previous studies have demonstrated that KLF5 acts as an oncogene in
different types of cancer, including in bladder and breast cancer
(10,12). Patients with increased KLF5 expression
in breast cancer exhibit shorter disease-free survival times, and
increased KLF5 expression aids in cancer cell proliferation and
tumorigenesis (10,12). However, KLF5 has also been
demonstrated to act as a tumor suppressor under certain conditions,
including JNK activation-induced apoptosis in esophageal squamous
cell cancer (13). The loss of KLF5
expression has frequently been reported in prostate cancer;
furthermore, several studies have demonstrated that the deletion
and inactivation of KLF5 promotes tumor growth in prostate cancer
(14,15). However, the precise function of KLF5
in prostate cancer remains unclear.
TNFα is a type of inflammatory cytokine that
regulates normal cell and tissue functions, including immune
responses, hematopoiesis and morphogenesis; however, it has also
been implicated in deleterious processes, such as tumorigenesis,
transplant rejection, septic shock, viral replication and bone
resorption (16). The expression of
TNFα leads to the activation of mitogen-activated protein kinase
(MAPK) cascades, including the ERK1/2, p38, c-Jun N-terminal kinase
(JNK) signaling pathways (16). TNFα
has been reported to induce apoptosis in the LNCaP cell line
derived from prostate cancer cells (17,18), and
is an essential molecule involved in the autocrine loop through
PMA-induced apoptosis in LNCaP cells (7,8). Previous
studies have demonstrated that TNFα can upregulate the KLF5 mRNA
and protein levels (19), which
results in cell apoptosis via activation of the JNK signaling
pathway in esophageal cancer (13).
Therefore, it has been suggested that KLF5 participates in the
PMA-induced TNFα autocrine loop, which may be important in
apoptosis of LNCaP cells.
In the present study, the role of PMA in the
activation of KLF5 in LNCaP cells was investigated. Furthermore,
the role of KLF5 in the regulation of the JNK signaling pathway was
determined.
Materials and methods
Cell culture and reagents
Human prostate cancer LNCaP cells and human
embryonic kidney 293T cells were obtained from the American Type
Culture Collection (Manassas, VA, USA). The LNCaP cells were
cultured in RPMI-1640 (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS; Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA) and 293T cells were
cultured in Dulbecco's Modified Eagle's Medium (DMEM; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS at 37°C with 5%
CO2 in a humidified incubator. TNFα and anti-human TNF-α
antibody were purchased from PeproTech, Inc. (Rocky Hill, NJ, USA).
PMA was purchased from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). These reagents were dissolved in 0.1% bovine serum
albumin (Sigma-Aldrich; Merck KGaA) and stored at −20°C. The
antibody directed against human KLF5 has been previously described
(15).
Lentivirus preparation
PLKO.1 lentiviral vectors were used to package
encoding short hairpin RNAs (shRNAs) with the sequence,
5′-GGTTACCTTACAGTATCAACA-3′. To generate the lentivirus, PAX2,
VSV-G and the aforementioned plasmids were co-transfected into 90%
confluent 293T cells using Lipofectamine 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. Lentivirus expressing KLF5 was produced
using full-length KLF5 from the pcDNA3-KLF5 plasmid (20), which was sub-cloned into LV5 vectors
according to the manufacturer's protocol (Shanghai GenePharma Co.,
Ltd., Shanghai, China).
Western blot analysis
Western blot analysis was performed using a
previously described method (11). A
total of 2×106 LNCaP cells were washed once with cold
PBS and lysed in radioimmunoprecipitation assay buffer (50 mM Tris
pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40, and 0.5% sodium
deoxycholate) containing 1% protease inhibitor cocktail and 1 mM
phenylmethanesulfonyl fluoride (Sigma-Aldrich; Merck KGaA). The
concentration of protein was detected using a Bradford Assay
Protein Quantification kit (Abcam, Cambridge, UK). A total of 30 µg
protein was separated by 12% SDS-PAGE and transferred onto
nitrocellulose membranes. The membranes were blocked with 5%
skimmed milk reconstituted in Tris-buffered saline with 0.1%
Tween-20 (pH 7.6) at room temperature for 1 h, and washed with PBS
three times, followed by incubation at 4°C overnight with primary
antibody. The primary antibodies used were as follows: GAPDH
(1:10,000; cat. no. KC-5G4; Shanghai KangChen Bio-tech, Shanghai,
China); KLF5 [1:500, a gift from Dr Jin-Tang Dong, Emory
University, Atlanta, GA, USA (15)];
poly (ADP-ribose) polymerase (PARP, dilution 1:1,000; cat. no.
9532), caspase-8 (dilution, 1:1,000; cat. no. 9746), caspase-3
(dilution, 1:1,000; cat. no. 9662), JNK (dilution, 1:1,000; cat.
no. 9258) and phosphorylated-JNK (dilution, 1:1,000; cat. no. 4668;
all from Cell Signaling Technology, Inc. Danvers, MA, USA).
Following incubation with the primary antibodies, membranes were
incubated with secondary antibody [horseradish
peroxidase-conjugated affnipure goat anti-Rabbit IgG (cat. no.
ZB-2301; dilution, 1:2,000; OriGene Technologies, Beijing, China)
and horseradish peroxidase-conjugated affinipure goat anti-Mouse
IgG (cat. no. ZB-2305; dilution, 1:2,000; OriGene Technologies)]
for 1 h at room temperature. Immunoreactive signals were detected
using a WesternBright Quantum HRP substrate kit (Advansta, Inc.,
Menlo Park, CA, USA), visualized by a Molecular Imager ChemiDoc XRS
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Immunoblotting for GAPDH was performed as an internal control.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was performed using a previously described
method (11). Briefly, total RNA from
2×105 LNCaP cells was isolated using TRIzol reagent
(Thermo Fisher Scientific, Inc.) and reverse transcribed into cDNA
using the PrimeScript™ RT reagent kit (Takara Biotechnology Co.,
Ltd., Dalian, China). A total of 50 ng of cDNA was analyzed using a
CFX96 real-time PCR system (Bio-Rad Laboratories, Inc.) with
SYBR-Green PCR Master mix (Takara Biotechnology Co., Ltd.) to
determine the transcriptional expression of specific genes. The
thermocycling conditions were as follows: 95°C for 30 sec, followed
by 40 cycles at 95°C for 5 sec, and 60°C for 30 sec. GAPDH was used
for normalization. Relative gene expression was calculated by the
2−ΔΔCq method (21). The
primer sequences used were as follows: KLF5 forward,
5′-CAGAGGACCTGGTCCAGACAAG-3′ and reverse,
5′-GAGGCCAGTTCTCAGGTGAGTG-3′; TNFα forward,
5′-AGCCCATGTTGTAGCAAACC-3′ and reverse, 5′-GGAAGACCCCTCCCAGATAG-3′;
GAPDH forward, 5′-ATGGGGAAGGTGAAGGTCGG-3′ and reverse,
5′-GACGGTGCCATGGAATT-TGC-3′.
Apoptosis assays
Cells were trypsinized and collected by
centrifugation (1,000 × g for 5 min at 4°C), and then washed with
cold PBS twice. Next cells were stained with Annexin V following
the protocol of Annexin-V-FLUOS staining kit (Roche Diagnostics
GmbH, Mannheim, Germany). The stained cells were analyzed by
fluorescence activated cell sorting using a FACSCalibur™ flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and BD
CellQuest Pro software (version 6.0; BD Biosciences).
Collection of conditional medium
(CM)
Cells were treated with PMA (100 nM) or vehicle
(DMSO) for 1 h at room temperature, and then washed twice with
RPMI-1640 medium to remove the PMA or DMSO. Following incubation
for 24 h, the cell debris was removed from the CM using a 0.22-µm
Miliex filter (Merck KGaA).
Statistical analysis
GraphPad Prism software (version 5.0; GraphPad,
Inc., La Jolla, CA, USA) was used for analyzing differences between
two groups using a one-tailed Student's t-test. Data are presented
as the mean ± standard deviation. P<0.05 was considered to
indicate a statistically significant difference.
Results
PMA upregulates KLF5 expression in
LNCaP prostate cancer cells
LNCaP cells were treated with 100 nM PMA for various
periods of time (0–24 h) and the mRNA and protein expression levels
of KLF5 were detected through RT-qPCR and western blot analyses,
respectively. As presented in Fig.
1A, KLF5 protein and mRNA expression levels gradually increased
throughout the first 0–8 h of PMA treatment, suggesting that PMA
activates KLF5 expression in LNCaP cells. The increase in KLF5 was
transient, possibly as the induction of KLF5 is dependent on the
phosphorylation of PKC-δ, whose increase is transient.
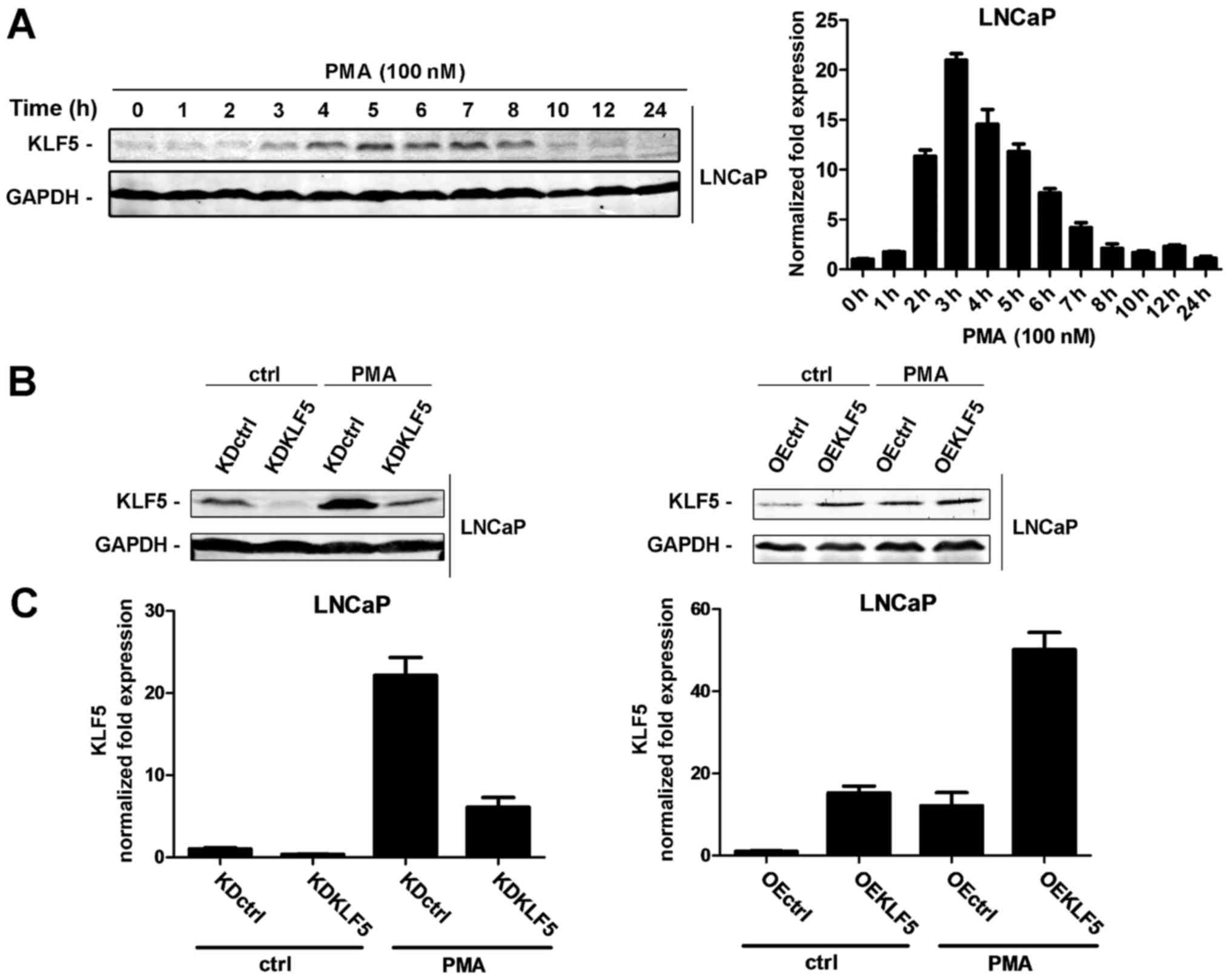 | Figure 1.Effect of PMA treatment on the
activation of KLF5 in LNCaP cells. (A) LNCaP cells were treated
with 100 nM PMA for various amounts of time (0–24 h). KLF5
expression levels were detected at the protein (left) and mRNA
(right) levels through western blot analysis and RT-qPCR,
respectively. (B) The following LNCaP cell clones were established:
Left, KDctrl and KDKLF5; Right, OEctrl and OEKLF5. KLF5 protein
expression was determined in the presence or absence of treatment
with 100 nM PMA for 4 h using western blot analysis. (C) Expression
of KLF5 mRNA normalized to GAPDH in the following LNCaP clones as
detected using RT-qPCR array: Left, KDctrl and KDKLF5; Right,
OEctrl and OEKLF5. Similar results were observed in two additional
experiments. Data are presented as mean ± standard deviation
following three replicates. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; KDctrl, KLF5
knockdown control; KDKLF5, KLF5 knockdown; OEctrl, overexpression
control; OEKLF5, KLF5 overexpression; PMA, phorbol 12-myristate
13-acetate; KLF5, Krüppel-like transcription factor 5; ctrl,
control. |
Establishing the KLF5 knockdown and
KLF5 overexpressing LNCaP cell lines
To investigate whether KLF5 is involved in
PMA-induced apoptosis of LNCaP cells, stable KLF5 knockdown, KLF5
overexpression cell lines and the corresponding controls were
established using lentiviral techniques. The expression of KLF5 was
verified using western blot and RT-qPCR analyses. The following
clones were designed (Fig. 1B and C):
KLF5 knockdown control (LNCaP/KDctrl), KLF5 knockdown
(LNCaP/KDKLF5), overexpression control (LNCaP/OEctrl) and KLF5
overexpression (LNCaP/OEKLF5).
KLF5 is required for PMA-induced
apoptosis in LNCaP cells
Different LNCaP cell clones were treated with 100 nM
of PMA for 24 h. Subsequently, cells were harvested and cell
apoptosis was determined using flow cytometry. Following
stimulation with PMA, the percentage of apoptotic cells was
significantly decreased in LNCaP/KDKLF5 compared with LNCaP/KDctrl
cells (Fig. 2A), while the cell
apoptosis rate was significantly increased in the LNCaP/OEKLF5
group compared with that of the LNCaP/OEctrl group (Fig. 2B). These results suggest that KLF5
modulates the apoptotic response of LNCaP cells to PMA.
Furthermore, the expression levels of proteins involved in the
apoptotic cascade following PMA stimulation were measured using
western blot analysis. Cleaved PARP and cleaved caspase-3 protein
expression levels were decreased following KLF5 knockdown (Fig. 2C), and increased following KLF5
overexpression (Fig. 2D) compared
with the corresponding control groups. However, no differences in
the expression of cleaved caspase-8 were observed in the KLF5
knockdown and overexpression groups, indicating that KLF5 knockdown
has no significant effect upstream of the TNFα extrinsic apoptosis
signaling pathway.
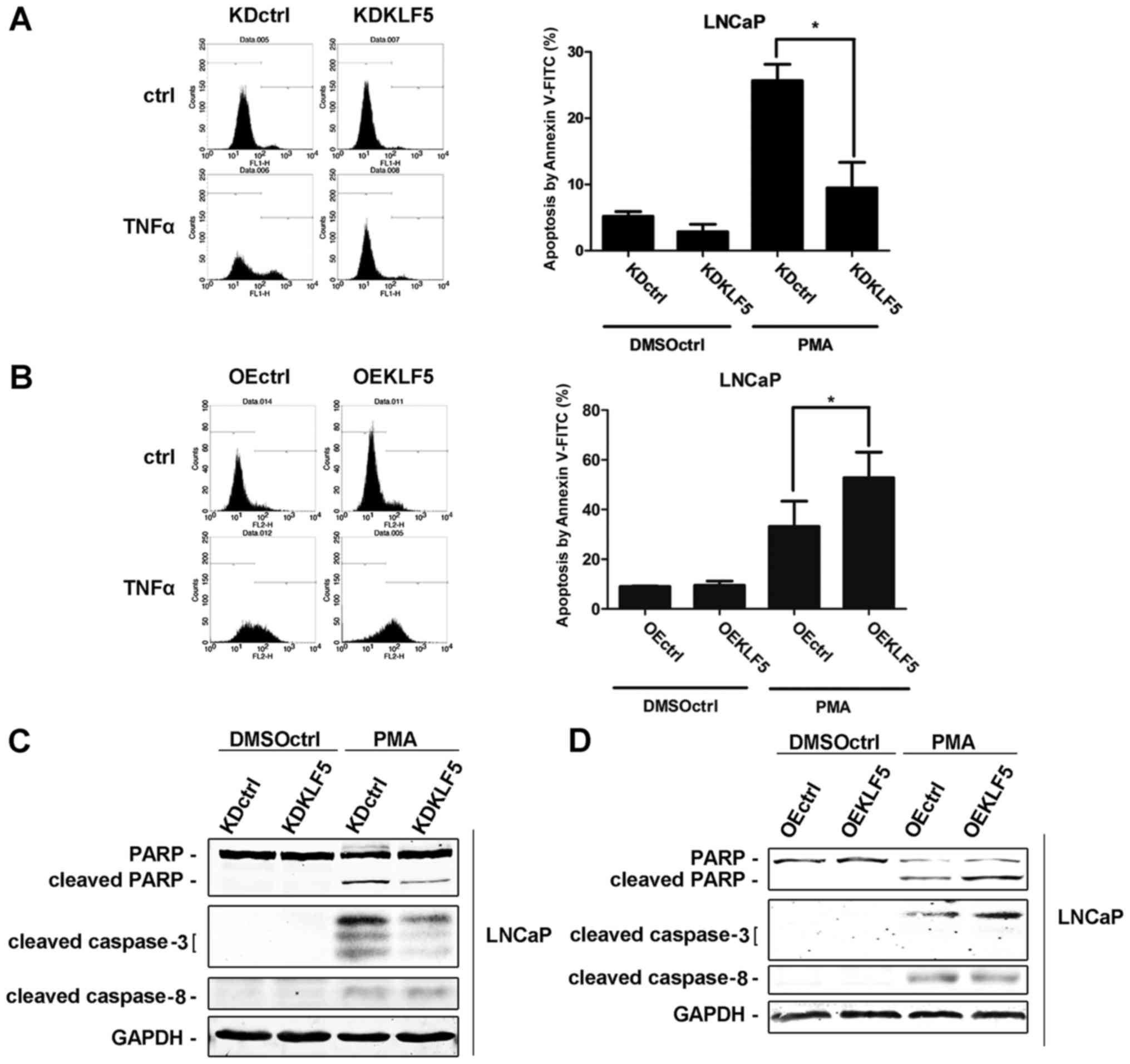 | Figure 2.Effect of KLF5 on apoptosis in LNCaP
cells induced by PMA. The following LNCaP clones were treated with
100 nM PMA or vehicle control (DMSO) for 24 h: (A) KDctrl and
KDKLF5; (B) OEctrl, and OEKLF5. Then the apoptotic cells were
detected using flow cytometry analysis. (C) KDctrl and KDKLF5, and
(D) OEctrl and OEKLF5 cells were treated with 100 nM PMA or vehicle
control for 24 h. Then the differences in specific
apoptotic-associated protein expression levels were determined
using western blot analysis. Data are presented as mean ± standard
deviation following three replicates. *P<0.05. KDctrl, KLF5
knockdown control; KDKLF5, KLF5 knockdown; OEctrl, overexpression
control; OEKLF5, KLF5 overexpression; DMSOctrl, DMSO control; PMA,
phorbol 12-myristate 13-acetate; KLF5, Krüppel-like transcription
factor 5; PARP, poly(ADP-ribose) polymerase. |
KLF5 is required for CM-PMA-induced
apoptosis in LNCaP cells
PMA-induced apoptosis is triggered by the secretion
of death factors. Therefore, LNCaP cells were treated with PMA (100
nM) or vehicle (DMSO) for 24 h, and the CM-PMA and CM-control (Con)
was collected. CM-Con and different doses of CM-PMA (dose ratios of
CM-Con/CM-PMA were 4:0, 3:1, 2:2, 0:4) were added to a new culture
of LNCaP cells. KLF5 protein (Fig.
3A) and mRNA (Fig. 3B) expression
levels were increased in a dose-dependent manner following PMA
treatment, indicating that KLF5 responds to death factors secreted
by PMA-stimulated cells. Furthermore, different LNCaP cell clones
were treated with CM-Con and CM-PMA for 24 h and the apoptotic
response were determined using flow cytometry and western blot
analysis. The percentage of apoptotic cells was significantly
decreased in LNCaP/KDKLF5 compared with LNCaP/KDctrl cells
(Fig. 3C), while the cell apoptosis
was significantly increased in LNCaP/OEKLF5 (Fig. 3D) compared with LNCaP/OEctrl cells.
The cleaved PARP and cleaved caspase-3 protein expression levels
were decreased in the KLF5 knockdown group (Fig. 3E), and increased following KLF5
overexpression (Fig. 3F) compared
with the corresponding control groups. These results suggest that
KLF5 is an important molecule for regulating the apoptotic response
to death factors secreted by PMA stimulating cells. However, no
differences in the expression of cleaved caspase-8 were observed in
the KLF5 knockdown and overexpression groups, indicating that KLF5
is downstream of death factor activation.
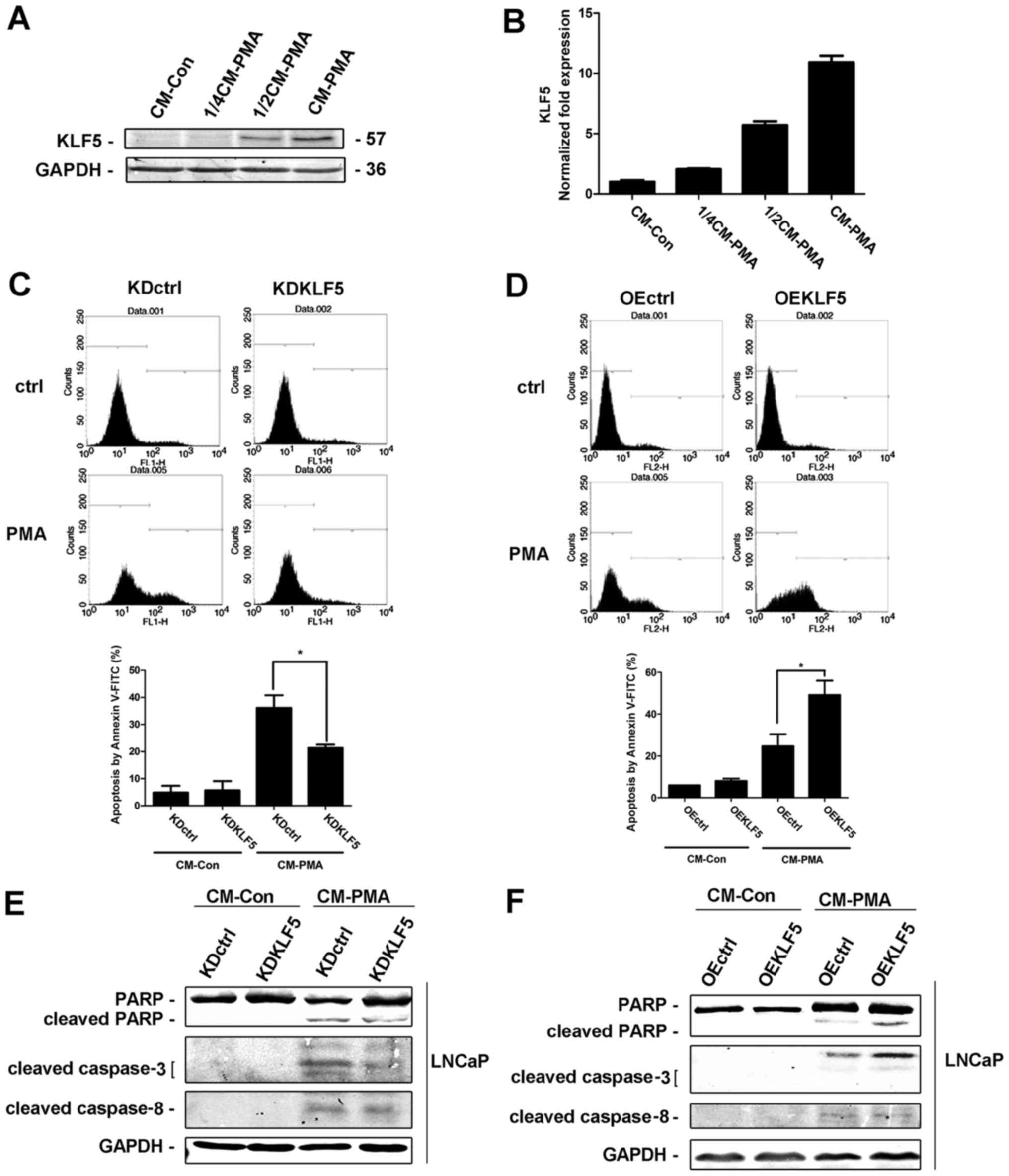 | Figure 3.KLF5 is required for CM to induce
apoptosis in LNCaP cells. CM from PMA-treated cells enhanced
expression of KLF5 (A) protein and (B) mRNA in a dose-dependent
manner, as detected using western blot and reverse
transcription-quantitative polymerase chain reaction analyses,
respectively. (C) Knockdown of KLF5 significantly decreased, but
(D) overexpression of KLF5 significantly increased LNCaP cell
apoptosis induced by CM-PMA, as detected using flow cytometry
assays. Expression of apoptotic proteins in (E) KLF5 knockdown and
(F) KLF5-expressing cells treated with CM, as detected through
western blot analysis. Similar results were observed in two
additional experiments. Data are presented as mean ± standard
deviation following three replicates. *P<0.05. KLF5,
Krüppel-like transcription factor 5; CM, control medium; KDctrl,
KLF5 knockdown control; KDKLF5, KLF5 knockdown; OEctrl,
overexpression control; OEKLF5, KLF5 overexpression; PMA, phorbol
12-myristate 13-acetate; PARP, poly(ADP-ribose) polymerase; CM-Con,
control medium from control cells; CM-PMA, control medium from PMA
cells. |
KLF5 modulates apoptosis induced by
PMA and CM through regulation of the JNK signaling pathway in LNCaP
cells
KLF5 has been determined as an important mediator of
death factor-induced apoptosis under stimulation of PMA. PMA
activates various signaling cascades, in which the JNK signaling
pathway is one of the most important apoptotic pathways (7,22).
Furthermore, a previous study demonstrated that JNKs are activated
by KLF5 (13). Thus, in the present
study, the importance of KLF5 to the JNK signaling pathway was
investigated. Phosphorylation of JNKs was detected using western
blot analysis in different LNCaP cell clones. Following treatment
with PMA (0–2 h), the levels of phosphorylation of the JNKs were
decreased in LNCaP/KDKLF5 cells (Fig.
4A) and increased in LNCaP/OEKLF5 cells (Fig. 4B) compared with the control groups.
Similar results were obtained for LNCaP/KDKLF5 (Fig. 4C) and LNCaP/OEKLF5 (Fig. 4D) cells treated with CM-PMA. These
results suggest that KLF5 modulates the apoptotic response to PMA
and CM via regulation of the JNK signaling pathway.
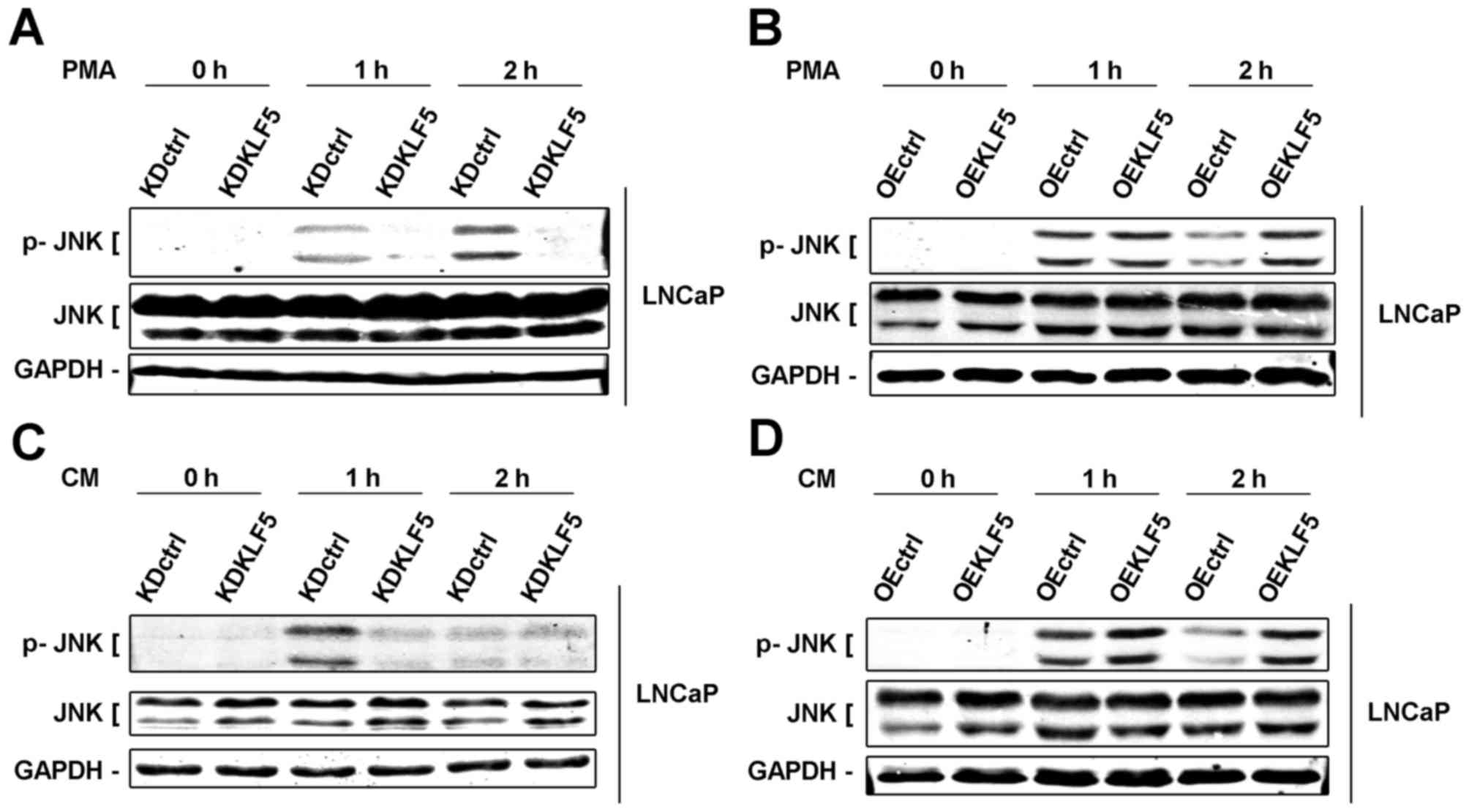 | Figure 4.KLF5 modulates apoptosis induced by
PMA and CM through regulating the activity of JNK signaling pathway
in LNCaP cells. (A) Knockdown of KLF5 decreased, but (B)
overexpression of KLF5 increased phosphorylation of JNKs induced by
PMA in LNCaP cells. (C) Knockdown of KLF5 decreased, but (D)
overexpression of KLF5 increased phosphorylation of JNKs induced by
CM in LNCaP cells. Similar results were observed in two additional
experiments. Data are presented as mean ± standard deviation
following three replicates. KDctrl, KLF5 knockdown control; KDKLF5,
KLF5 knockdown; OEctrl, overexpression control; OEKLF5, KLF5
overexpression; PMA, phorbol 12-myristate 13-acetate; KLF5,
Krüppel-like transcription factor 5; CM, control medium; p,
phosphorylated; JNK, c-Jun N-terminal kinase. |
Autocrine factor TNFα mediates the
apoptosis induced by PMA, independent of KLF5 expression
It has been reported that TNFα is the one of most
important autocrine death factors stimulated by PMA in LNCaP cells
(7). The stimulation of death
receptors activates the extrinsic apoptotic pathway. To confirm the
role of TNFα, an antibody directed against TNFα was used to inhibit
TNFα expression following PMA or CM-PMA treatment. TNFα inhibition
results in decreased KLF5 mRNA expression (Fig. 5A), suggesting that KLF5 is activated
by TNFα. Furthermore, inhibition of TNFα decreased the PMA- and
CM-induced apoptosis in LNCaP cells (Fig.
5B), indicating that autocrine factor TNFα mediates PMA-induced
apoptosis, which is consistent with a previous study (7). Furthermore, the involvement of KLF5 in
the release of TNFα was investigated. All cell groups were treated
with PMA for different times (0–2 h). The release of TNFα protein
was determined using western blot analysis (Fig. 5C and D) and the mRNA expression was
measured using RT-qPCR analysis (Fig. 5E
and F). However, with the presence or absence of PMA, no
differences were identified in LNCaP/KDKLF5 or LNCaP/OEKLF5 cells
compared with the control groups, suggesting that KLF5 does not
regulate the secretion of TNFα, and thus does not promote the
PMA-induced apoptosis through increasing the expression of
TNFα.
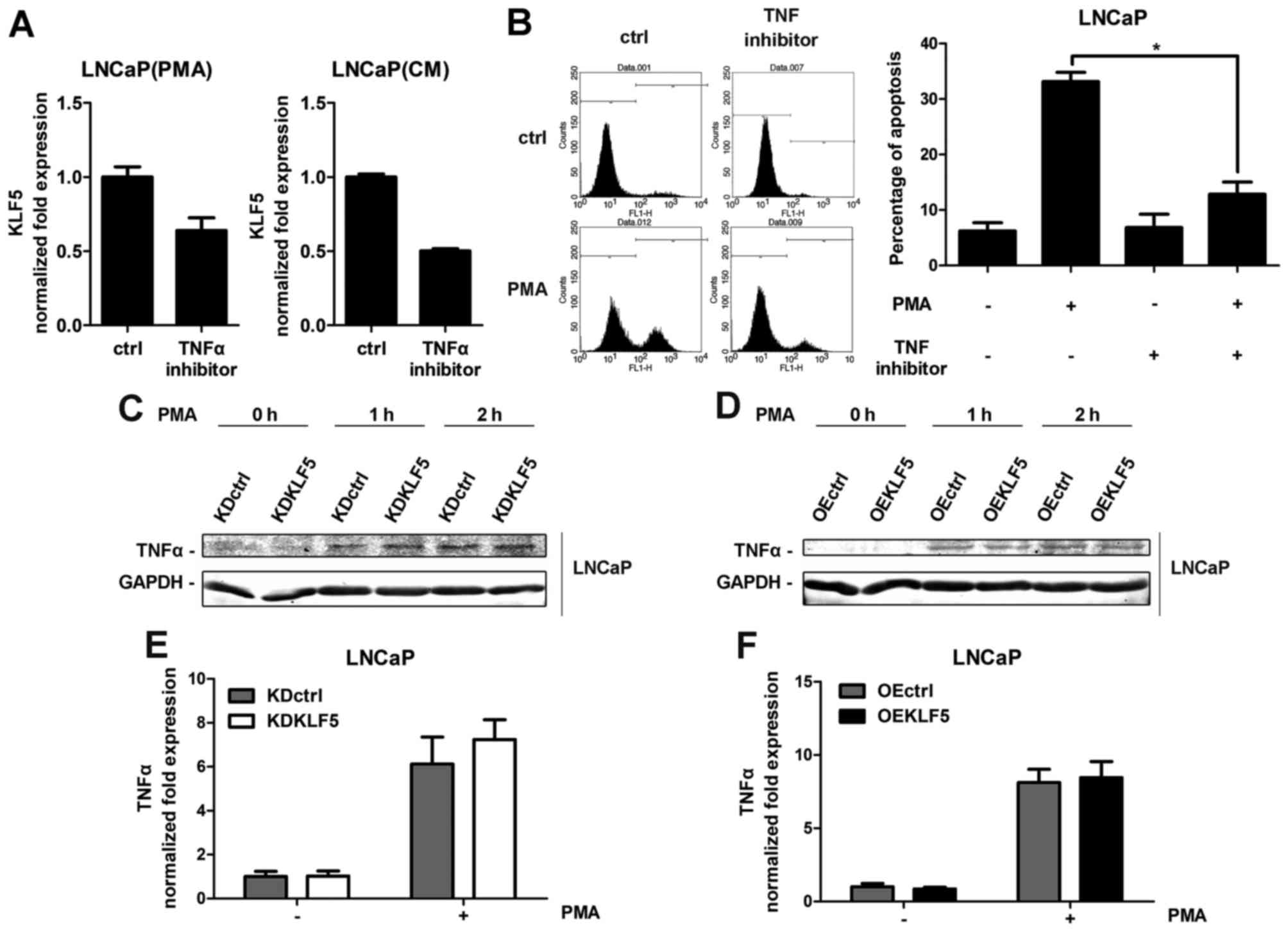 | Figure 5.PMA- and CM-induced apoptosis through
the autocrine factor TNFα. (A) Inhibition of TNFα using an antibody
suppressed KLF5 expression induced by PMA and CM, as detected
through RT-qPCR arrays. LNCaP cells were treated with PMA or CM for
4 h. (B) TNFα inhibitor suppressed apoptosis induced by PMA and CM,
as detected through flow cytometry assays. LNCaP cells were treated
with PMA or CM for 24 h. (C) Knockdown of KLF5 and (D)
overexpression of KLF5 did not change the expression of TNFα
protein induced by PMA treatment in LNCaP cells as detected through
western blot analysis. (E) Knockdown of KLF5 and (F) overexpression
of KLF5 did not change the expression of TNFα mRNA induced by PMA
treatment in LNCaP cells as detected using RT-qPCR arrays and
normalized to GAPDH. Cells were treated with 100 nM PMA for 2 h.
Similar results were observed in two additional experiments. Data
are presented as mean ± standard deviation following three
replicates. *P<0.05. KDctrl, KLF5 knockdown control; KDKLF5,
KLF5 knockdown; OEctrl, overexpression control; OEKLF5, KLF5
overexpression; PMA, phorbol 12-myristate 13-acetate; KLF5,
Krüppel-like transcription factor 5; CM, control medium; TNFα,
tumor necrosis factor α; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction. |
Discussion
Several previous studies have reported that phorbol
esters induce an apoptotic response in prostate cancer LNCaP cells
(4–7).
Treatment of LNCaP cells with PMA leads to the autocrine release of
death factors, including cytokine TNFα (7,8).
Furthermore, the CM collected from PMA-treated LNCaP cells has been
demonstrated to promote the activation of the extrinsic apoptotic
cascade, which activates death effectors, including p38 MAPK, JNK
and nuclear factor-κB (7,8). Inhibition of JNKs has been revealed to
decrease the apoptotic effect of PMA (6,23).
However, the mechanisms involved remain unclear. In the present
study, the results suggest that KLF5 is an essential modulator of
PMA-induced apoptosis in LNCaP prostate cancer cells. KLF5 was also
identified to be required as an effector of the death factor TNFα
and was able to activate the JNK signaling pathway to promote the
apoptotic response. Thus, suggesting that KLF5 could be a
therapeutic target for prostate cancer. However, the results of the
present study revealed that KLF5 exhibited no regulation on the
secretion of TNFα.
It has been reported that KLF5 has a role in
different cell processes, including in cell proliferation, cell
cycle progression and apoptosis (9–11).
Previous studies have demonstrated that KLF5 acts as an oncogene in
different cancer cells, and is a potential tumor suppressor in
prostate cancer cells (24). In the
current study, it was revealed that PMA could activate KLF5, and
the knockdown or overexpression of KLF5 modulated the apoptotic
response to PMA in LNCaP cells. This is similar to the results
demonstrated in the CM-treated LNCaP cells, indicating that KLF5 is
required for PMA-induced apoptosis.
Death receptors are necessary for the extrinsic
apoptotic cascade triggered by death factors, which contain
cytoplasmic death domains that enable the receptors to engage the
cell apoptotic machinery. Stimulation of these receptors results in
the activation of the initiator caspase-8, whose cleavage
propagates the apoptotic signal (16). However, in the present study, no
differences in cleaved caspase-8 expression were identified between
KLF5 knockdown or overexpression cells that were treated with PMA
or CM and their corresponding control groups. Thus, it may be
suggested that stimulation of the death receptor is not influenced
by KLF5 or PMA. Furthermore, as TNFα is regarded as the most
important autocrine death factor (7),
the level of TNFα following PMA stimulation was detected. The
results revealed that the protein release or mRNA expression of
TNFα was not regulated by KLF5; thus, KLF5 may only be an effector
of the cytokine TNFα.
The involvement of JNK in various forms of cell
death has been reported; however, whether JNK is required for
apoptotic cell death induced by TNFα in prostate cancer cells has
not been previously elucidated (25).
Previous studies have determined that JNK can be activated
following phorbol ester treatment in LNCaP cells (7,26). In the
current study, KLF5 was detected to regulate phosphorylation of
JNKs. Either under stimulation of PMA or treatment with CM, loss of
KLF5 was able to significantly decrease the phosphorylation of
JNKs, whereas levels were significantly increased in the
overexpression group, suggesting that KLF5 is a modulator of JNKs
in response to TNFα in LNCaP cells.
In conclusion, the results of the present study
suggest that KLF5 is essential for the PMA-induced apoptosis in
LNCaP prostate cancer cells. Furthermore, KLF5 is indispensable for
the autocrine factor TNFα, which is secreted by cells treated with
PMA, to induce cell apoptosis through regulating the activity of
JNK signaling pathway. These observations provide novel insights
into the complexity of the signaling pathways and the mechanisms
regulating cell apoptosis in prostate cancer cells, which could aid
in developing novel treatments for patients with prostate
cancer.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81672557 and
81372279; received by Peng Guo).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mukherji D, Omlin A, Pezaro C, Shamseddine
A and de Bono J: Metastatic castration-resistant prostate cancer
(CRPC): Preclinical and clinical evidence for the sequential use of
novel therapeutics. Cancer Metastasis Rev. 33:555–566.
2014.PubMed/NCBI
|
|
3
|
Barry OP and Kazanietz MG: Protein kinase
C isozymes, novel phorbol ester receptors and cancer chemotherapy.
Curr Pharm Des. 7:1725–1744. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garcia-Bermejo ML, Leskow FC, Fujii T,
Wang Q, Blumberg PM, Ohba M, Kuroki T, Han KC, Lee J, Marquez VE
and Kazanietz MG: Diacylglycerol (DAG)-lactones, a new class of
protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate
cancer cells by selective activation of PKCalpha. J Biol Chem.
277:645–655. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tanaka Y, Gavrielides MV, Mitsuuchi Y,
Fujii T and Kazanietz MG: Protein kinase C promotes apoptosis in
LNCaP prostate cancer cells through activation of p38 MAPK and
inhibition of the Akt survival pathway. J Biol Chem.
278:33753–33762. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao L, Eto M and Kazanietz MG: ROCK
mediates phorbol ester-induced apoptosis in prostate cancer cells
via p21Cip1 up-regulation and JNK. J Biol Chem. 284:29365–29375.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gonzalez-Guerrico AM and Kazanietz MG:
Phorbol ester-induced apoptosis in prostate cancer cells via
autocrine activation of the extrinsic apoptotic cascade: A key role
for protein kinase C delta. J Biol Chem. 280:38982–38991. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiao L, Gonzalez-Guerrico A and Kazanietz
MG: PKC-mediated secretion of death factors in LNCaP prostate
cancer cells is regulated by androgens. Mol Carcinog. 48:187–195.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo P, Dong XY, Zhang X, Zhao KW, Sun X,
Li Q and Dong JT: Pro-proliferative factor KLF5 becomes
anti-proliferative in epithelial homeostasis upon
signaling-mediated modification. J Biol Chem. 284:6071–6078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao Y, Wu K, Chen Y, Zhou J, Du C, Shi Q,
Xu S, Jia J, Tang X, Li F, et al: Beyond proliferation: KLF5
promotes angiogenesis of bladder cancer through directly regulating
VEGFA transcription. Oncotarget. 6:43791–43805. 2015.PubMed/NCBI
|
|
11
|
Gao Y, Shi Q, Xu S, Du C, Liang L, Wu K,
Wang K, Wang X, Chang LS, He D and Guo P: Curcumin promotes KLF5
proteasome degradation through downregulating YAP/TAZ in bladder
cancer cells. Int J Mol Sci. 15:15173–15187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tong D, Czerwenka K, Heinze G, Ryffel M,
Schuster E, Witt A, Leodolter S and Zeillinger R: Expression of
KLF5 is a prognostic factor for disease-free survival and overall
survival in patients with breast cancer. Clin Cancer Res.
12:2442–2448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tarapore RS, Yang Y and Katz JP: Restoring
KLF5 in esophageal squamous cell cancer cells activates the JNK
pathway leading to apoptosis and reduced cell survival. Neoplasia.
15:472–480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen C, Bhalala HV, Vessella RL and Dong
JT: KLF5 is frequently deleted and down-regulated but rarely
mutated in prostate cancer. Prostate. 55:81–88. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen C, Sun X, Ran Q, Wilkinson KD, Murphy
TJ, Simons JW and Dong JT: Ubiquitin-proteasome degradation of KLF5
transcription factor in cancer and untransformed epithelial cells.
Oncogene. 24:3319–3327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aggarwal BB: Signalling pathways of the
TNF superfamily: A double-edged sword. Nat Rev Immunol. 3:745–756.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chopra DP, Menard RE, Januszewski J and
Mattingly RR: TNF-alpha-mediated apoptosis in normal human prostate
epithelial cells and tumor cell lines. Cancer Lett. 203:145–154.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Q, Gao Y, Xu S, Du C, Li F, Tang XS,
Jia J, Wang X, Chang L, He D and Guo P: Kruppel-like factor 5
promotes apoptosis triggered by tumor necrosis factor α in LNCaP
prostate cancer cells via up-regulation of mitogen-activated
protein kinase kinase 7. Urol Oncol. 34:58.e11–e18. 2016.
View Article : Google Scholar
|
|
19
|
Bafford R, Sui XX, Wang G and Conte M:
Angiotensin II and tumor necrosis factor-alpha upregulate survivin
and Kruppel-like factor 5 in smooth muscle cells: Potential
relevance to vein graft hyperplasia. Surgery. 140:289–296. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen C, Bhalala HV, Qiao H and Dong JT: A
possible tumor suppressor role of the KLF5 transcription factor in
human breast cancer. Oncogene. 21:6567–6572. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lorenzo PI and Saatcioglu F: Inhibition of
apoptosis in prostate cancer cells by androgens is mediated through
downregulation of c-Jun N-terminal kinase activation. Neoplasia.
10:418–428. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gavrielides MV, Gonzalez-Guerrico AM,
Riobo NA and Kazanietz MG: Androgens regulate protein kinase Cdelta
transcription and modulate its apoptotic function in prostate
cancer cells. Cancer Res. 66:11792–11801. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xing C, Ci X, Sun X, Fu X, Zhang Z, Dong
EN, Hao ZZ and Dong JT: Klf5 deletion promotes Pten
deletion-initiated luminal-type mouse prostate tumors through
multiple oncogenic signaling pathways. Neoplasia. 16:883–899. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Engedal N, Korkmaz CG and Saatcioglu F:
C-Jun N-terminal kinase is required for phorbol ester- and
thapsigargin-induced apoptosis in the androgen responsive prostate
cancer cell line LNCaP. Oncogene. 21:1017–1027. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ikezoe T, Yang Y, Taguchi H and Koeffler
HP: JNK interacting protein 1 (JIP-1) protects LNCaP prostate
cancer cells from growth arrest and apoptosis mediated by
12–0-tetradecanoylphorbol-13-acetate (TPA). Br J Cancer.
90:2017–2024. 2004. View Article : Google Scholar : PubMed/NCBI
|














