Introduction
Chronic myeloid leukemia (CML) is a clonal bone
marrow stem cell disorder in which the proliferation of mature
granulocytes and their precursor myeloid cells occurs. CML accounts
for 15% of all leukemia cases in adults (1). The global annual incidence rate of CML
is between 1.6 and 2 cases/100,000 people (2), while epidemiological surveys demonstrate
an annual incidence of 0.39–0.55 cases/100,000 people in several
areas of China (2). Patients with CML
in China are younger compared with those in western countries
(3).
CML is associated with the presence of the
Philadelphia (Ph) chromosome. This chromosomal translocation is
termed t(9, 22)(q34, q11). This translocation generates an
oncogenic BCR-ABL fusion gene, encoding the BCR-ABL fusion protein
(BCR-ABL) of 210 kDa (4). As the ABL
gene encodes a domain that can add phosphate groups to tyrosine
residues, BCR-ABL possesses tyrosine kinase activity (5,6). Other
molecules typically control the activity of tyrosine kinases, but
the mutant tyrosine kinase of the BCR-ABL transcript encodes for a
continuously activated protein, resulting in unregulated cell
division and consequent tumor development. Therefore, tyrosine
kinase inhibitors such as imatinib (IM), became the first-line
treatment against a variety of cancers, including CML. Although IM
demonstrated a successful effect in the majority of patients,
10–15% of patients developed drug resistance or exhibited disease
progression into the CML accelerated phase due to a late and thus
less effective treatment. Once the tumor progresses into the blast
phase, the median progression-free survival time of patients is 4
months with a median overall survival time of 7 months (7).
At present, allogenic stem cell transplantation,
bone marrow transplantation and tyrosine kinase inhibitors (TKIs)
represent the treatments available for patients with advanced CML.
In addition to these treatments, other targeted drugs have been
considered as potential candidates. The demethylating drug
decitabine (DAC) has been successfully used in the treatment of
myelodysplastic syndrome and acute myeloid leukemia in the elderly
(8). Several clinical studies
(9–13)
demonstrated that DAC monotherapy exhibits a substantial effect
during all the phases of CML. DAC is a DNA methylation inhibitor,
enabling the expression of silenced tumor suppressor genes
(8). Src homology region 2 (SH2)
domain-containing tyrosine phosphatase-1 (SHP-1) is a gene
primarily expressed in hematopoietic cells and is considered as a
hematopoietic cell malignancy tumor suppressor gene (14,15). It
serves an important role in the negative regulation of growth
factor receptor-mediated signal transduction processes. (14,15). The
results of a previous study (16)
demonstrated that there is a significant decrease in SHP-1
expression in patients with advanced CML compared with chronic
phase CML, suggesting that SHP-1 serves a role in the onset of the
blast crisis of CML. Previous studies have suggested that the
decreased SHP-1 expression in a variety of hematologic malignancies
is associated with the methylation of the SHP-1 gene promoter
(17–21). A previous study demonstrated that
SHP-1 forms a complex with BCR-ABL through immunoprecipitation in
immortalized myeloblast-like 32D and fibroblast 3T3 cell lines,
suggesting that SHP-1 regulates BCR-ABL (22). Imbalanced levels of tyrosine
phosphatase and tyrosine kinase serve an important role in cell
proliferation and apoptosis. As BCR-ABL is continuously activated,
patients with CML-blastic phase (CML-BP) exhibit enhanced tyrosine
kinase activity (23). As DAC enables
the expression of silenced tumor suppressor genes due to its DNA
methylation inhibitor function, it may possess a role in SHP-1
expression and function. Therefore, the present study aimed to
investigate the mechanism of action of DAC on restoring the
expression of SHP-1 in CML, compared with the effect of tyrosine
kinase inhibitors, to provide a novel strategy to prevent the
progression of CML and treat patients with CML.
Materials and methods
Drugs and reagents
IM and nilotinib were provided by Novartis
International AG (Basel, Switzerland). DAC was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The Cell Counting
kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies,
Inc. (Kumamoto, Japan).
Cell culture
The human immortalized myelogenous leukemia K562
cell line (Chinese Academy of Medical Sciences and Peking Union
Medical College, Tianjin, China) was preserved in the Province Key
Laboratory of Experimental Hematology (The Second Affiliated
Hospital of Hebei Medical University, Shijiazhuang, China). The
K562 cells were suspended in RPMI-1640 medium containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and incubated at 37°C with 5% CO2. Subsequently,
cells were seeded into 96-well plates at a density of
1×105 cells/well and incubated for 2–3 days prior to
treatment with 0.1 µM IM, 5 nM nilotinib, 5 µM DAC, 0.1 µM IM + 5
µM DAC, or 5 nM nilotinib + 5 µM DAC at 37°C for 48 h.
Subsequently, the cells were incubated with 10 µl CCK-8 reagent for
2 h at 37°C and the optical density (UV-visible NanoDrop 8000
spectrophotometer; Thermo Fisher Scientific, Inc.) was measured at
450 nm. The % of cell proliferation was calculated as follows:
(Untreated negative control group-experimental group)/(untreated
negative control group-blank group)x100%.
Primary cell culture of mononuclear
cells from patients with CML
A total of 10 patients (4 male, 6 female; median
age, 40.5 years; age range, 21–60 years) with CML-chronic phase
(CML-CP) and 6 patients (5 male, 1 female; median age, 42.5 years;
range, 35–50 years) with CML-BP were selected between October 2013
and October 2014 at The Second Affiliated Hospital of Hebei Medical
University (Shijiazhuang. China). This study was approved by the
ethics committee of The Second Hospital of Hebei Medical
University. The definitions of chronic and blast phase CML were
obtained from European Leukemia Net 2013 (24). A total of 4 ml fresh heparin marrow
was taken from patients with chronic, accelerated or blast phase
CML using an aseptic technique. An equal volume of RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) was added to each sample,
then mixed and dried. Subsequently, the bone marrow was diluted
with 4 ml of lymphocyte separation medium (Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China) and centrifuged at
670.8 × g for 20 min at 4°C. The middle layer containing the
mononuclear cells was collected. Following joining the
high-pressure liquid PBS 167.7 (x g) rinsing 5 min at 4°C.
Subsequently, mononuclear cells were seeded into the flasks at a
density of 1×106 in RPMI-1640 medium containing 20%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.).
Measuring SHP-1 expression through
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis
Following harvesting of each group, total RNA was
extracted using the TRIzol method. Reverse transcription was
performed using the All-in-One™ First-Strand cDNA Synthesis kit
(GeneCopoeia Inc., Rockville, MD, USA), according to the
manufacturer's protocol. The protocol was as follows: RNA
degeneration at 65°C for 10 min and reverse transcription at 37°C
for 60 min. Complementary (c)DNA was synthesized in a total volume
of 25 µl of working solution (5 µl 5X RT Reaction Buffer, 1 µl 250
µM random primers, 1 µl 25 mM dNTP, 1 µl 25 U/µl RNase inhibitor, 1
µl 200 U/µl Moloney murine leukemia virus reverse transcriptase, 1
µg total RNA and deionized H2O to make up to 25 µl). The
qPCR reaction was performed in a total volume of 20 µl of working
solution [9 µl SYBR Green Real-Time MasterMix (Invitrogen; Thermo
Fisher Scientific, Inc.) solution, 0.5 µl SHP-1 forward primer (AAC
AGC CGT GTC ATC GTC AT), 0.5 µl SHP-1 reverse primer (ATC AGG TCT
CCA TTG TCC AGC), 2 µl cDNA, 8 µl deionized H2O] at 95°C
for 2 min, followed by 40 cycles of 95°C for 15 sec, 60°C for 30
sec and 68°C for 45 sec. The reaction was performed in a Rotor-Gene
Q 6000/3000 Quantitative PCR instrument (Qiagen GmbH, Hilden,
Germany). Relative SHP-1 expression in the treated and control
groups was calculated using the 2−ΔΔCq method (25) using β-actin (forward primer, GAG CTA
CGA GCT GCC TGA C; reverse primer, GGT AGT TTC GTG GAT GCC ACA G)
as a reference gene.
RT-qPCR determination of absolute
BCR-ABL gene expression
Following harvesting each group of cells, RNA was
extracted using the TRIzol method and the RNA concentration was
measured using the NanoDrop 8000 spectrophotometer. The RT-qPCR
reaction to amplify the BCR-ABL gene was performed in a total
volume of 25 µl working solution using a BCR-ABL210 Fusion Gene
Detection kit (Shanghai YuanQi Biomedical Technology Co., Ltd.,
Shanghai, China), according to the manufacturer's protocol. The
reaction was performed in a Rotor-Gene Q 6000/3000 Quantitative PCR
instrument. BCR-ABL expression was calculated as follows: BCR-ABL
(p210) mRNA levels (%)=(BCR-ABL copy number/ABL copy
number)x100.
Immunoprecipitation and western blot
analysis
A total of 200 µl RIPA lysis buffer (150 mM NaCl, 50
mM Tris-HCl, 1% NP40, 0.5% sodium deoxycholate) and 2 µl
phenylmethylsulfonyl fluoride were added to the CML and K562 cells,
and incubated on ice for 30 min to obtain the cell lysate. Next,
the cells were centrifuged at 10,732.8 × g for 10 min at 4°C, and
the supernatant containing the proteins was collected. The protein
was quantified using a Coomassie (Bradford) Protein Assay kit
Nanjing Jiancheng Bioengineering Institute, China). Subsequently,
10 µl c-ABL rabbit polyclonal IgG antibody (cat. no. sc-131;
dilution, 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
was added to 100 µg of each protein sample. Intracellular pH (IPH)
buffer [10 ml 1 M Tris-HCl (pH 8.0), 15 ml 2 M NaCl, 2 ml 0.5 M
EDTA, 1 ml NP-40, 172 ml triple-distilled water] was added to reach
a total volume of 300 µl in 1.5 ml eppendorf (EP) tubes. The EP
tubes were subsequently shaken for 1 h at 4°C. Then, 25 µl protein
A/G PLUS-Agarose (cat. no. sc-2003; Santa Cruz Biotechnology, Inc.)
was added and the EP tubes were shaken overnight at 4°C. EP tubes
were subsequently washed with IPH buffer 3–4 times.
The supernatant was discarded and 35 µl 2X SDS
buffer (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added,
centrifuged at 24148.8 × g for 1 min at 4°C, boiled at 100°C for
7–8 min, and separated on an 8% gel using SDS-PAGE (300 mA) for 2
h, prior to being transferred onto a polyvinylidene difluoride
(PVDF) membrane. The PVDF membrane was removed and blocked in 5%
bovine serum albumin (Roche Diagnostics, Basel, Switzerland) for 1
h at 4°C. The PVDF membrane was transferred to a hermetic bag,
rabbit polyclonal IgG primary antibody directed against SHP-1 (cat.
no. sc-287; dilution, 1:200; Santa Cruz Biotechnology, Inc.) was
added and the membrane was incubated in the dark for 1 h at 4°C.
The membrane was washed with 1X TBS-Tween 20 (TBS-T) 3 times and
incubated with goat anti-rabbit IgG HRP-conjugated secondary
antibody (cat. no. sc-2004; dilution, 1:5,000; Santa Cruz
Biotechnology, Inc.) for 1 h at 4°C. Finally, the membrane was
washed with 1X TBS-T 3 times, and the fluorescence was developed
using Potent ECL kit [Multi Sciences (Lianke) Biotech Co., Ltd.,
Hangzhou, China]. The software used to analyze the protein band
density was Image-ProPlus (version 6.0; Media Cybernetics, Inc.,
Rockville, MD, USA).
Methylation-specific PCR (MSP)
The following SHP-1 primers (26) were used: SHP-1 methylated MSP forward,
5′-GAACGTTATTATAGTATAGCGTTC-3′ and SHP-1 methylated MSP reverse,
5′-TCACGCATACGAACCCAAACG-3′; SHP-1 demethylation MSP forward,
5′-GTGAATGTTATTATAGTATAGTGTTTGG-3′ and SHP-1 demethylation MSP
reverse, 5′-TTCACACATACAAACCCAAACAAT-3′. MSP was performed
according to the EZ-DNA Methylation kit (Zymo Research Corp,
Irvine, CA, USA) protocol. The total PCR reaction volume was 25 µl
and consisted of the following: 5 µl bisulfite-converted DNA; 2 µl
forward primers; 2 µl reverse primers; 12.5 µl Green PCR system
(Thermo Fisher Scientific, Inc.); and 3.5 µl ddH2O. PCR
thermocycling conditions were as follows: Initial denaturation at
94°C for 5 min; 40 cycles of denaturation at 94°C for 45 sec;
annealing at 54°C for 45 sec; extension at 72°C for 45 sec; and a
final extension at 72°C for 10 min. A total of 10 µl of the PCR
amplification product was collected from each group and the
products were detected using GoldView™ nucleic acid dyes in a 2%
agarose gel electrophoresis running at 90 V for 60 min, then
visualized using a gel imaging system (Dinco Ltd., Tirat Carmel,
Israel).
Calculation of combination index
(CI)
The CI of two drugs was calculated to evaluate their
potential synergistic effect, using the calculation previously
described by Soriano et al (27): DA/ICX, A + DB/ICX, B. A and B
represent two drugs separately; ICX, A and ICX, B are the
concentrations of two drugs used alone to achieve the drug growth
inhibition rate X; and DA and DB are the combinations of two drugs
that achieve growth inhibition rate X. According to Soriano et
al (27) the synergistic effect
is classified as follows: 0.9≤CI≤1.1, additive effect;
0.8≤CI<0.9, slight synergism; 0.6≤CI<0.8, moderate synergism;
0.4≤CI<0.6, synergism; 0.2≤CI<0.4, strong synergism.
Statistical analysis
Each experiment was repeated ≥3 times. All data are
presented as the mean ± standard deviation, and the statistical
analysis was performed using GraphPad Prism software (version 5.0;
GraphPad Software, Inc., La Jolla, CA, USA). The one-way analysis
of variance with Tukey's post hoc test was used for multiple
comparisons. The Student's t-test was used to compare two groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
K562 cell proliferation inhibition
rate is significantly increased in the DAC+TKI groups (IM+DAC, and
nilotinib+DAC) compared with the TKI monotherapy group
OD values of K562 cells following 48 h drug
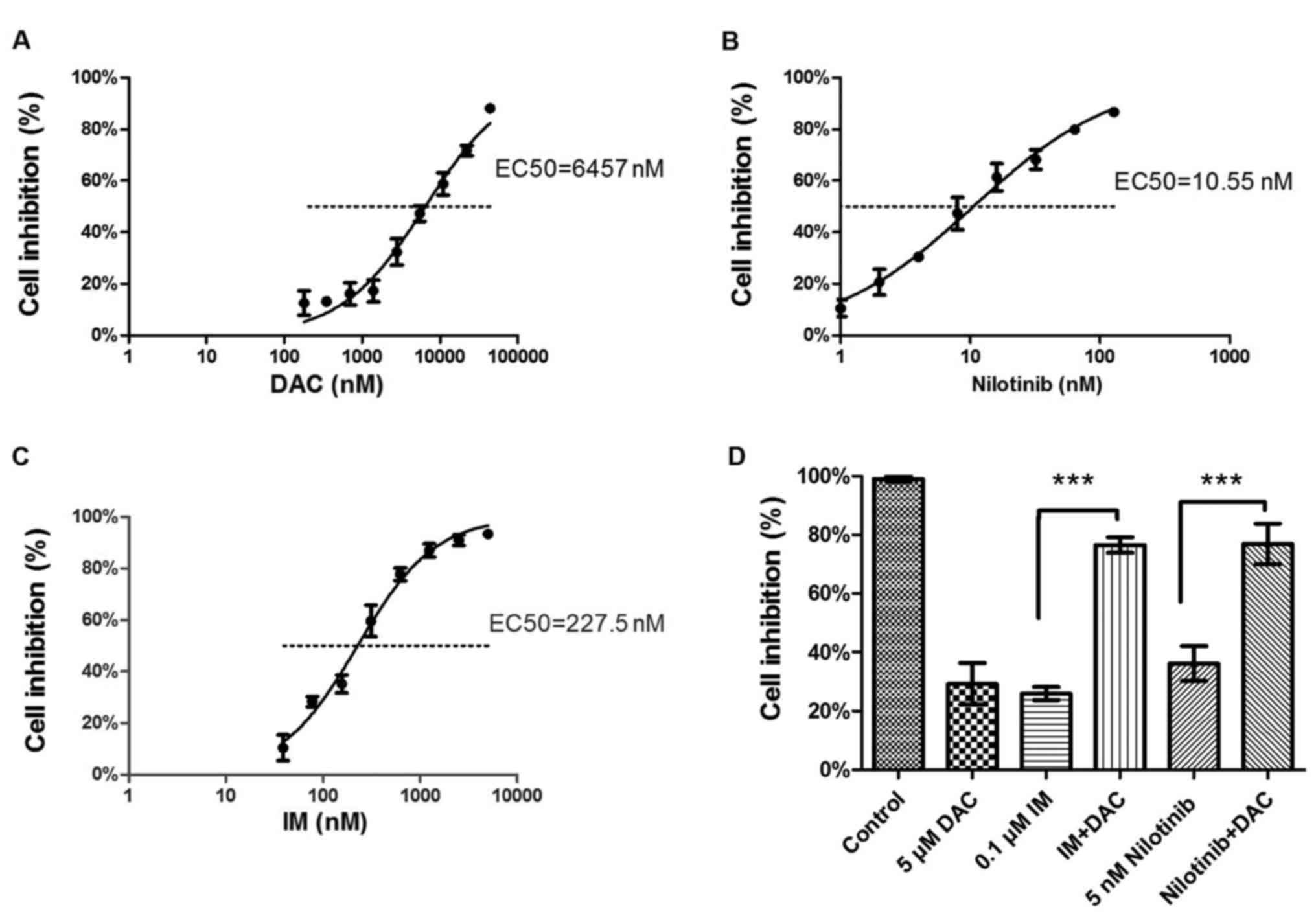
treatment were measured using a CCK-8, and the EC50
values were calculated (DAC, 6,457 nM, Fig. 1A; nilotinib EC50, 10.55 nM,
Fig. 1B; IM EC50, 227.5
nM, Fig. 1C). K562 cells treated with
5 µM DAC combined with 0.1 µM IM (P<0.0001; vs. IM monotherapy)
or 5 nM nilotinib (P=0.0015; vs. nilotinib monotherapy)
demonstrated a significant inhibition in proliferation compared
with the corresponding IM or nilotinib monotherapy treatment groups
(Fig. 1D). According to the Chou and
Talalay calculation method, the results of the present study
revealed that the drug combinations used exhibited a synergistic
effect as follows: IM+DAC CI=(0.1/0.625)+(5/20)=0.41, synergism;
nilotinib+DAC CI=(0.01/0.1)+(5/22)=0.33, strong synergism.
Determination of SHP-1 protein
expression in K562 cells, and CML-CP and CML-BP mononuclear cells
through immunoprecipitation
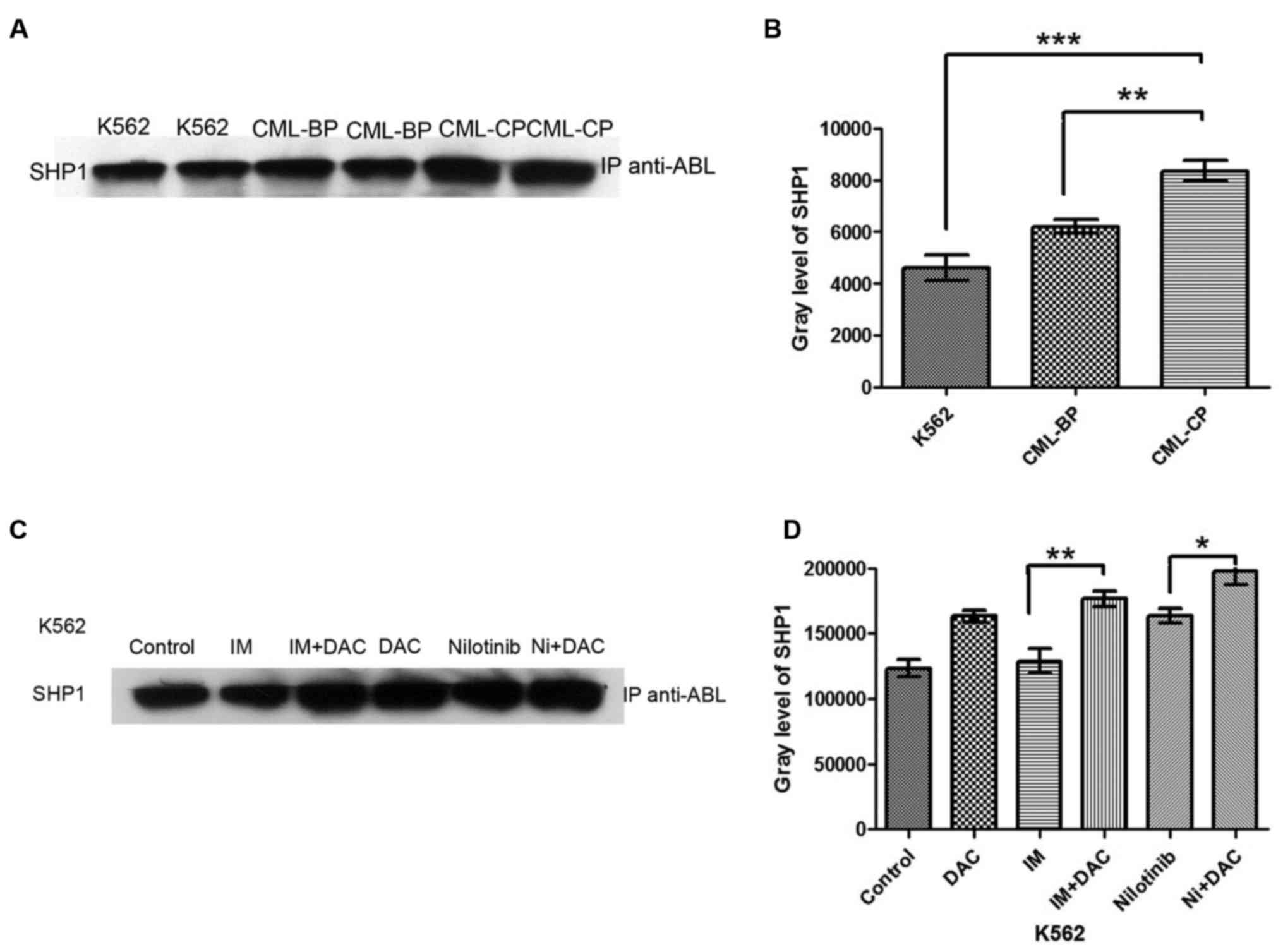
The immunoprecipitation analysis revealed that SHP-1
protein was expressed in K562 cells, and CML-CP and CML-BP
mononuclear cells (Fig. 2A). The
expression of SHP-1 was significantly lower in K562 cells compared
with CML-CP mononuclear cells (P<0.001), and significantly lower
in CML-BP compared with CML-CP cells (P<0.01; Fig. 2B). These results indicate that
decreased SHP-1 expression is associated with CML disease
progression.
SHP-1 protein expression is
significantly increased in the TKI+DAC group compared with the TKI
monotherapy group in K562 cells
SHP-1 protein expression was significantly increased
in the IM+DAC group compared with the IM monotherapy group
(P<0.01; Fig. 2C and D). SHP-1
protein expression was significantly increased in the nilotinib+DAC
group compared with the nilotinib monotherapy group (P<0.05). No
significant difference was identified in SHP-1 protein expression
between the control and IM monotherapy group (Fig. 2C and D).
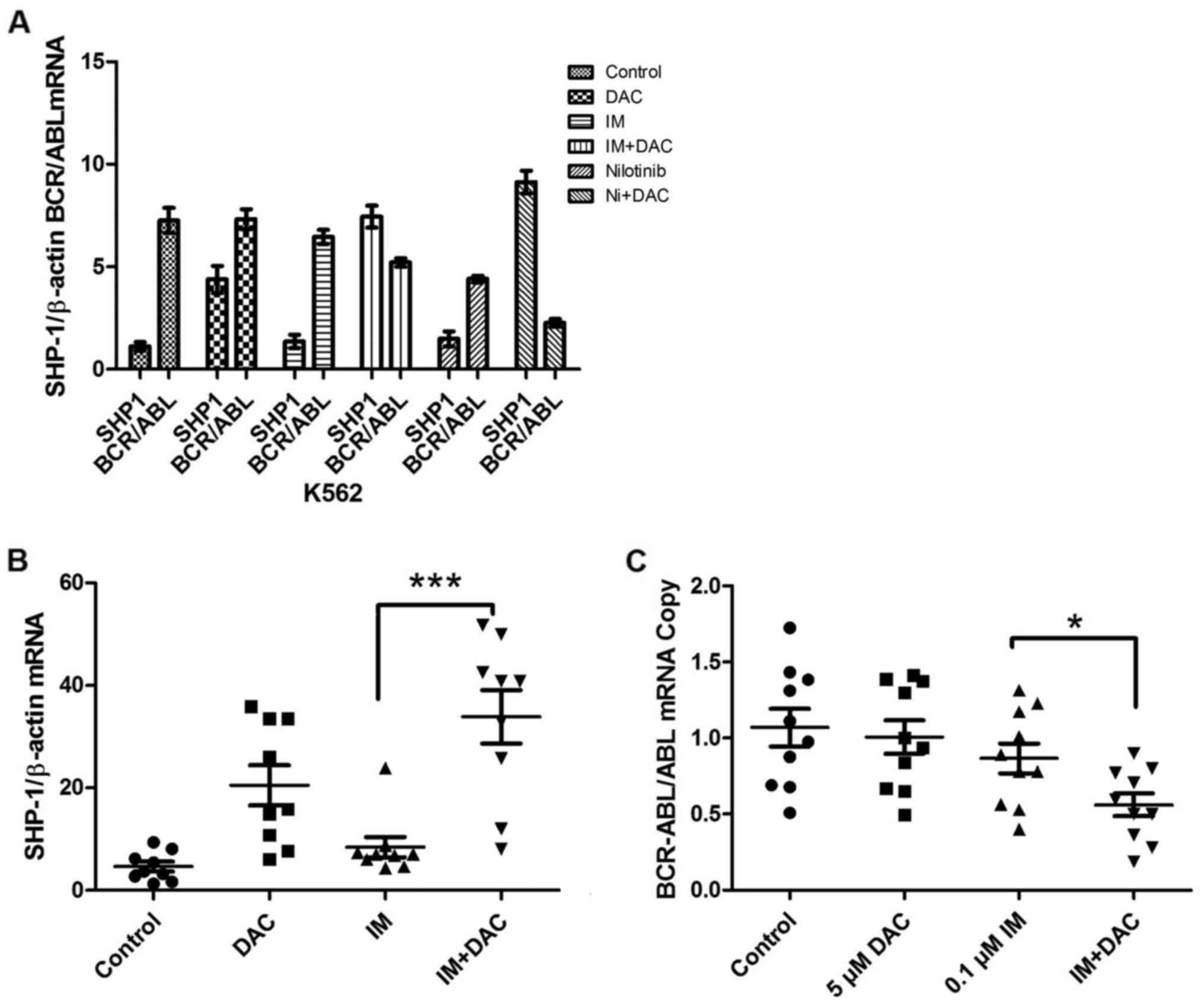
SHP-1 and BCR-ABL mRNA expression in
K562 cells
RT-qPCR results revealed no significant difference
in SHP-1 gene expression between IM and nilotinib monotherapy
treatment groups and the control group (Fig. 3A). The DAC group demonstrated a marked
increase in SHP-1 expression compared with the control group.
IM+DAC group demonstrated a significant increase in SHP-1
expression compared with IM monotherapy group, while BCR/ABL mRNA
expression decreased in a marked manner in the combined group
compared with the IM monotherapy group. The nilotinib+DAC group
revealed a significant increase in the expression of SHP-1 compared
with in the nilotinib monotherapy group, while BCR-ABL and ABL copy
number ratio levels decreased in a significant manner in the
combined group respect nilotinib monotherapy group. No significant
differences were identified in the nilotinib monotherapy BCR-ABL
and ABL copy number ratio compared with in the DAC+IM group
(Fig. 3A).
SHP-1 and BCR-ABL gene and protein
expression in patients with CML 48 h following drug treatment
SHP-1 gene expression was measured using
RT-qPCR analysis in patients with CML (control), IM, DAC and IM+DAC
groups. The results revealed that SHP-1 gene expression was
significantly increased in the IM+DAC group compared with the IM
monotherapy group (P<0.001; Fig.
3B), while BCR-ABL gene expression was significantly
decreased in IM+DAC group compared with the IM monotherapy group
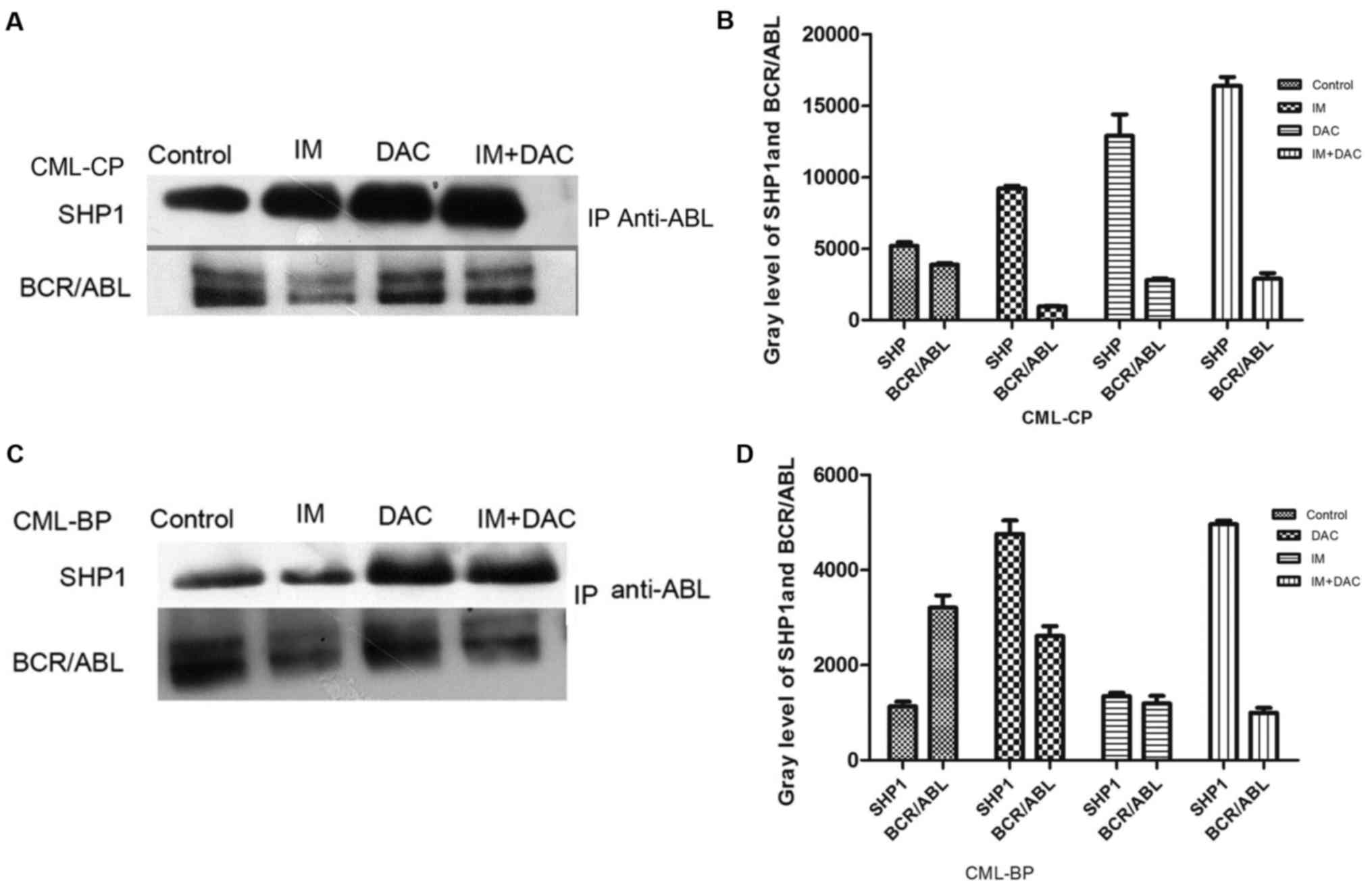
(P<0.05; Fig. 3C). SHP-1 protein
expression was measured through immunoprecipitation in patients
with CML (control group), IM, DAC and IM+DAC groups. SHP-1 protein
expression was markedly increased and BCR-ABL (p210) protein
expression was markedly decreased in the IM+DAC group compared with
the IM monotherapy group (Fig.
4).
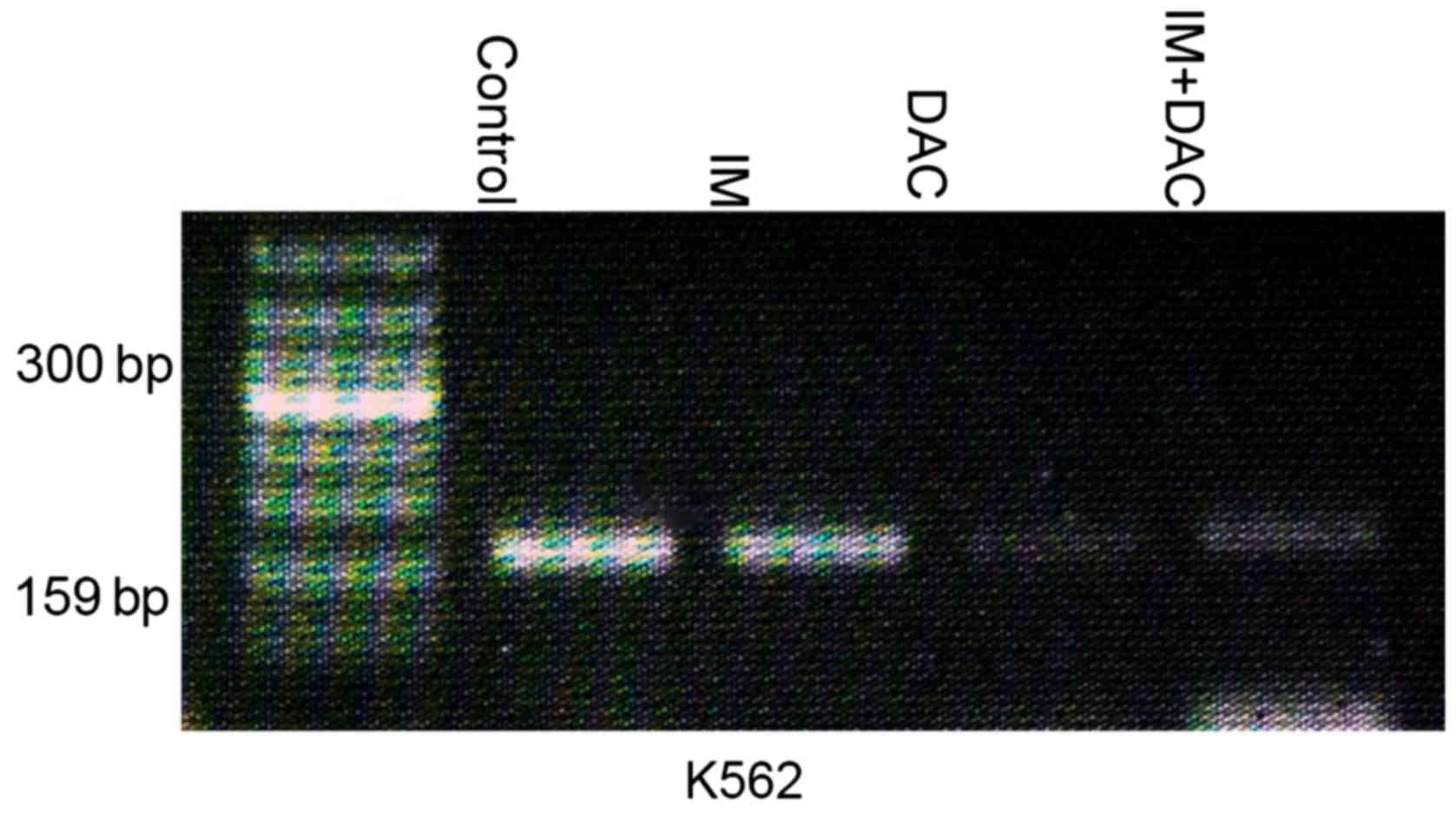
SHP-1 gene methylation levels in K562
cells 48 h following drug treatment
SHP-1 gene methylation levels of K562 cells
were measured using MSP analysis in the control, IM, DAC and IM+DAC
groups. The results demonstrated the presence of SHP-1 gene
methylation in the control and IM monotherapy group, while no
methylation was demonstrated in the IM+DAC and DAC monotherapy
group (Fig. 5).
Discussion
K562 cells are derived from Philadelphia
chromosome-positive chronic myelogenous leukemia blast
erythro-megakaryoblastic leukemia cell lines (28). A CCK-8 assay was used to investigate
the proliferative ability of the K562 control and the IM,
nilotinib, DAC, IM+DAC, and nilotinib+DAC treatment groups. DAC
demonstrated a proliferation inhibition, which was
dosage-dependent. A significant difference in cell proliferation
was identified between 0.1 µM IM and IM + 5 µM DAC, in addition to
between 10 nM nilotinib and nilotinib + 5 µM DAC. Therefore, the
combinations of these drugs possess high synergy. The concentration
of DAC used was according to the results obtained through CCK-8
analysis, and confirmed by a previous study by Momparler et
al (29). The concentration of IM
and nilotinib used was according to the result obtained through
CCK-8 analysis, and confirmed by a previous study by Savage and
Antman (30). The effects of TKIs in
combination with low dose DAC were more effective than monotherapy
in the K562 cell line.
DAC is a hypomethylating cytosine analogue used in
the treatment of myeloid malignancies due to its ability to
activate silent tumor suppressor genes through demethylation. SHP-1
is a non-transmembrane protein tyrosine phosphatase containing two
N-terminal SH2 domains that is primarily expressed in hematopoietic
cells (31). It has been demonstrated
to be associated with the negative regulation of signaling pathways
mediated by growth factor, cytokine and antigen receptors (31). Several studies have demonstrated that
the expression of SHP-1 is decreased in lymphoma (17), leukemia (18), multiple myeloma (19), cervical cancer (20) and colorectal cancer (21) due to the methylation of the promoter
region of the SHP-1 gene. In a previous study, SHP-1 was identified
in a complex with BCR-ABL in 3T3 cells and 32D/Bcr-Abl cells
through immunoprecipitation analysis, and an association between
SHP-1 and Bcr-Abl expression was demonstrated (22). The results of the present study
obtained through immunoprecipitation demonstrated that SHP-1 forms
a complex with BCR-ABL in K562 cells and primary cells derived from
patients with CML. SHP-1 may be a type of tyrosine phosphatase,
which control the activity of BCR/ABL (p210) tyrosine kinase since
in the present study, the expression of SHP-1 was demonstrated to
be significantly decreased in K562 cells compared with in patients
with CML-CP. A previous study revealed that there was no expression
of SHP-1 in K562 cells (32). The
expression of SHP-1 was restored by drug induced differentiation,
therefore inactivation of SHP-1 may serve a role in the progression
to blast crisis in CML. The results of the present study revealed
that the expression of SHP-1 is low as opposed to non-existent. The
conflict between the two results may be due to the sensitivity of
the test methods used. In addition, the expression of SHP-1 was
identified to be decreased in CML-BP compared with CML-CP primary
cells, thus it may be associated with CML disease progression and
is consistent with the studies of Amin et al (16). The results of the present study
revealed that SHP-1 mRNA expression increased while BCR-ABL
expression decreased in the IM+DAC treated group compared with the
IM group. This was confirmed through analysis of SHP-1 and BCR-ABL
protein expression. SHP-1 mRNA and protein expression increased,
while BCR-ABL expression decreased in the nilotinib+DAC compared
with the nilotinib group. No significant difference was identified
in SHP-1 and BCR-ABL expression between the nilotinib and IM+DAC
treatment groups. These results demonstrated that treatment with
DAC combined with TKIs possesses an improved ability to enhance
SHP-1 mRNA and protein expression, and decreasing BCR-ABL
expression. This suggests that DAC restores SHP-1 expression by
demethylation, which enhances TKIs anti-tumor effect. A previous
study revealed that SHP-1 expression is significantly lower in
TKI-resistant compared with TKI-sensitive cell lines (33). In KCL22-R cells, SHP-1 ectopic
expression restores responsiveness to imatinib (IM) (33). Data from the present study
demonstrated that the increase in SHP-1 and decrease in BCR-ABL
expression in response to IM+DAC, as compared with IM monotherapy,
in K562 cells was consistent with that in primary cells from
patients with CML in vitro.
In conclusion: i) DAC was demonstrated to inhibit
the proliferation of K562 cells in a dose-dependent manner. ii)
IM+DAC and nilotinib+DAC exhibited high synergy. iii)
Immunoprecipitation demonstrated that SHP-1 and BCR-ABL (p210)
proteins form a complex in K562 cells and in the primary cells
derived from patients with CML, suggesting there is an interaction
between SHP-1 and BCR-ABL (p210) protein. Therefore, SHP-1 may be
considered a tyrosine phosphatase that controls the activity of
BCR-ABL (p210) tyrosine kinase. SHP-1 expression was identified to
be decreased in K562 cells and CML-BP primary cells compared with
the expression in CML-CP primary cells, suggesting that it is also
associated with CML disease progression. iv) SHP-1 mRNA and protein
expression increased in the combined groups (IM+DAC/nilotinib+DAC)
compared with the monotherapy groups, while BCR-ABL expression
decreased. Methylation of SHP-1 was observed in K562 cells,
although it was not identified in the IM+DAC therapy group. Hence,
it is suggested that DAC recovers SHP-1 expression through
demethylation, thereby enhancing the therapeutic effect of TKIs.
The results of the present study suggest that DAC combined with
TKIs provide a novel strategy for managing TKI tolerance and the
progression of CML in patients.
Acknowledgements
The authors of the present study would like to thank
Dr Luo Jian Min (Department of Hematology, The Second Affiliated
Hospital of Hebei Medical University, Shijiazhuang, China) for
reading the manuscript.
References
|
1
|
National Comprehensive Cancer Network, .
NCCN Clinical Practice Guidelines in Oncology: Chronic Myelogenous
Leukemia, V.2.[DB/OL].(2007-10-25). 2008.
|
|
2
|
Druker BJ and Lee SJ: Chronic myelogenous
leukemia. Cancer: Principles and Practice of Oncology. 2007.
|
|
3
|
Incidence survey of leukemia in China, .
Chinese Epidemiologic Study Group of Leukemia and Aplastic Anemia.
Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 14:12–19. 1992.(In Chinese).
PubMed/NCBI
|
|
4
|
Goldman JM and Melo JV: Chronic myeloid
leukemia-advances in biology and new approaches to treatment. N
Engl J Med. 349:1451–1464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McWhirter JR and Wang JY: An actin-binding
function contributes to transformation by the Bcr-Abl oncoprotein
of Philadelphia chromosome-positive human leukemias. EMBO J.
12:1533–1546. 1993.PubMed/NCBI
|
|
6
|
van Etten RA, Jackson PK, Baltimore D,
Sanders MC, Matsudaira PT and Janmey PA: The COOH terminus of the
c-Abl tyrosine kinase contains distinct F- and G-actin binding
domains with bundling activity. J Cell Biol. 124:325–340. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Foroni L, Gerrard G, Nna E, Khorashad JS,
Stevens D, Swale B, Milojkovic D, Reid A, Goldman J and Marin D:
Technical aspects and clinical applications of measuring BCR-ABL1
transcripts number in chronic myeloid leukemia. Am J Hematol.
84:517–522. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hackanson B and Daskalakis M: Decitabine.
Recent Results Cancer Res. 201:269–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kantarjian HM, O'Brien SM, Keating M,
Beran M, Estey E, Giralt S, Kornblau S, Rios MB, de Vos D and
Talpaz M: Results of decitabine therapy in the accelerated and
blastic phases of chronic myelogenous leukemia. Leukemia.
11:1617–1620. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sacchi S, Kantarjian HM, O'Brien S, Cortes
J, Rios MB, Giles FJ, Beran M, Koller CA and Keating MJ: Chronic
myelogenous leukemia in nonlymphoidblastic phase: Analysis of the
results of first salvage therapy with three different treatment
approaches for 162 patients. Cancer. 86:2632–2641. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kantarjian HM, O'Brien S, Cortes J, Giles
FJ, Faderl S, Issa JP, Garcia-Manero G, Rios MB, Shan J, Andreeff
M, et al: Results of decitabine (5-aza-2-deoxycytidine) therapy in
130 patients with chronic myelogenousleukemia. Cancer. 98:522–528.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Issa JP, Garcia-Manero G, Giles FJ,
Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS,
Cortes J and Kantarjian HM: Phase 1 study of low-dose prolonged
exposure schedules of the hypomethylating agent
5-aza-2-deoxycytidine (decitabine) in hematopoietic malignancies.
Blood. 103:1635–1640. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Issa JP, Gharibyan V, Cortes J, Jelinek J,
Morris G, Verstovsek S, Talpaz M, Garcia-Manero G and Kantarjian
HM: Phase II study of low-dose decitabine in patients with chronic
myelogenous leukemia resistant to imatinib mesylate. J Clin Oncol.
23:3948–3956. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu C, Sun M, Liu L and Zhou GW: The
function of the protein tyrosine phosphatase SHP-1 in cancer. Gene.
306:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oka T, Yoshino T, Hayashi K, Ohara N,
Nakanishi T, Yamaa Y, Hiraki A, Sogawa CA, Kondo E, Teramoto N, et
al: Reduction of hematopoietic cell specific tyrosine phosphatase
SHP-1 gene expression in natural killer cell lymphoma and various
types of lymphom as/leukemia. Combination analysic with cDNA
expression array and tissue micrearray. Am J Patho. 159:1495–1505.
2001. View Article : Google Scholar
|
|
16
|
Amin HM, Hoshino K, Yang H, Lin Q, Lai R
and Garcia-Manero G: Decreased expression level of SH2
domain-containing protein tyrosine phosphatase-1 (Shp1) is
associated with progression of chronic myeloid leukaemia. J Pathol.
212:402–410. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Q, Wang HY, Marzec M, Raghunath PN,
Nagasawa T and Wasik MA: STAT3- and DNA methyltransferase
1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor
suppressor gene in malignant T lymphocyte. Proc Natl Acad Sci USA.
102:pp. 6948–6953. 2005; View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Uhm KO, Lee ES, Lee YM, Park JS, Kim SJ,
Kim BS, Kim HS and Park SH: Differential methylation pattern of
ID4, SFRP1, and SHP1 between acute myeloid leukemia and chronic
myeloid leukemia. J Korean Med Sci. 24:493–497. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chim CS, Fung TK, Cheung WC, Liang R and
Kwong YL: SOCS1 and SHP1 hypermethylation in multiple myeloma:
Implications for epigenetic activation of the Jak/STAT pathway.
Blood. 103:4630–4635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim JH, Choi YD, Lee JS, Lee JH, Nam JH
and Choi C: Assessment of DNA methylation for the detection of
cervical neoplasiain liquid-based cytology specimens. Gynecol
Oncol. 116:99–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu SB, Liu XH, Li BH, Zhang Y, Yuan J,
Yuan Q, Li PD, Yang XZ, Li F and Zhang WJ: DNA methylation
regulates constitutive expression of Stat6 regulatory genes SOCS-1
and SHP-1 in colon cancer cells. J Cancer Res Clin Oncol.
135:1791–1798. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liedtke M, Pandey P, Kumar S, Kharbanda S
and Kufe D: Regulation of Bcr-Abl-induced SAP kinase activity and
transformation by the SHPTP1 protein tyrosine phosphatase.
Oncogene. 17:1889–1892. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cramer K, Nieborowska-Skorska M, Koptyra
M, Slupianek A, Penserga ET, Eaves CJ, Aulitzky W and Skorski T:
BCR/ABL and other kinases from chronic myeloproliferative disorders
stimulate single-strand annealing, an unfaithful DNA double-strand
break repair. Cancer Res. 68:6884–6888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baccarani M, Deininger MW, Rosti G,
Hochhaus A, Soverini S, Apperley JF, Cervantes F, Clark RE, Cortes
JE, Guilhot F, et al: European LeukemiaNet recommendations for the
management of chronic myeloid leukemia: 2013. Blood. 122:872–884.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oka T, Ouchida M, Koyama M, Ogama Y,
Takada S, Nakatani Y, Tanaka T, Yoshino T, Hayashi K, Ohara N, et
al: Gene silencing of the tyrosine phosphatase SHP1 gene by
aberrant methylation in leukemias/lymphomas. Cancer Res.
62:6390–6394. 2002.PubMed/NCBI
|
|
27
|
Soriano AF, Helfrich B, Chan DC, Heasley
LE, Bunn PA Jr and Chou TC: Synergistic effects of new
chemopreventive agents and conventional cytotoxic agents against
human lung cancer cell lines. Cancer Res. 59:6178–6184.
1999.PubMed/NCBI
|
|
28
|
Lozzio CB and Lozzio BB: Human chronic
myelogenous leukemia cell-line with positive Philadelphia
chromosome. Blood. 45:321–334. 1975.PubMed/NCBI
|
|
29
|
Momparler RL, Côté S and Eliopoulos N:
Pharmacological approach for optimization of the dose schedule of
5-Aza-2-deoxycytidine (Decitabine) for the therapy of leukemia.
Leukemia. 11 Suppl 1:S1–S6. 1997.PubMed/NCBI
|
|
30
|
Savage DG and Antman KH: Imatinib
mesylate-a new oral targeted therapy. N Eng J Med. 346:683–693.
2002. View Article : Google Scholar
|
|
31
|
Tonks NK and Neel BG: From form to
function: Signaling by protein tyrosine phosphatases. Cell.
87:365–368. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bruecher-Encke B, Griffin JD, Neel BG and
Lorenz U: Role of the tyrosine phosphatase SHP-1 in K562 cell
differentiation. Leukemia. 15:1424–1432. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Esposito N, Colavita I, Quintarelli C,
Sica AR, Peluso AL, Luciano L, Picardi M, Del Vecchio L, Buonomo T,
Hughes TP, et al: SHP-1 expression accounts for resistance to
imatinib treatment in Philadelphia chromosome-positive cells
derived from patients with chronic myeloid leukemia. Blood.
118:3634–3644. 2011. View Article : Google Scholar : PubMed/NCBI
|