Introduction
Janus kinase 2 (JAK2) belongs to the non-receptor
tyrosine kinase JAK family, which is composed of four kinases:
JAK1, JAK2, JAK3 and TYK2. JAK kinases transduce signals from
cytokine receptors to members of the signal transducer and
activator of transcription (STAT) protein family, including STAT3
and STAT5 (1). The JAK/STAT signaling
pathway regulates various physiological processes, including
proliferation and differentiation (1).
A previous study identified the JAK2
valine-617-phenylalanine (V617F) mutation in a significant
proportion of patients with myeloproliferative neoplasms (MPNs)
(2). The mutation resulted in a gain
of function for JAK2 and is located in the JH2 domain, a
pseudokinase domain involved in the autoinhibition of JAK2 activity
(3). The JAK2-V617F mutation and
other constitutively active forms of JAK2 were also identified in
hematological malignancies as an infrequent event (4,5), as
reviewed by Furqan et al (6).
Small molecule JAK2 inhibitors, including AZD1480, CYT387, SB1518
(pacritinib), INCB16562 and WP1066, have demonstrated preclinical
effects in various types of tumors, including leukemia, multiple
myeloma, prostate cancer, gastric cancer and neuroblastoma
(7–12). The aminopyridine derivative compound
KRC-180, in which the aminopyridine group is substituted with
benzoxazole, is a small molecule that inhibits c-Met and exhibits a
good structure-activity relationship (13). The current study examined the efficacy
of KRC-180 as a JAK2 inhibitor and an antiproliferative agent in
leukemic cells.
Materials and methods
Cell culture
The HEL92.1.7 human erythroleukemia cell line was
purchased from the American Type Culture Collection (Manassas, VA,
USA). The cells were cultured in RPMI 1640 medium (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) supplemented with 10% fetal bovine
serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) and 1%
penicillin/streptomycin The cultured cells were incubated at 37°C
in an atmosphere containing 5% CO2.
For the cytotoxicity assay, the cells were seeded
into a 96-well plate (10,000 cells/well) and incubated with KRC-180
(synthesized at Korea Research Institute of Chemical Technology,
Daejeon, Korea) for 72 h at 37°C. KRC-180 was three-fold serially
diluted in DMSO to create a 10-point curve at a starting
concentration of 100 µM. As a negative control, the cells were
treated with dimethyl sulfoxide. Cell viability was evaluated using
a tetrazolium-based assay with an EZ-Cytox Cell Viability Assay kit
(no. EZ-12000; Daeil Lab Service Co., Ltd., Seoul, Korea),
according to the manufacturer's protocol. The concentration that
inhibits growth by 50% (GI50) was calculated by
nonlinear regression using GraphPad Prism software (version 5.01;
GraphPad Software, Inc., La Jolla, CA, USA) (14).
For drug combination studies, the cells into a
96-well plate (10,000 cells/well) were treated with KRC-180,
cytarabine (also known as AraC; no. C6645; Sigma-Aldrich; Merck
KGaA) and a combination of KRC-180 and AraC. The cells were treated
with graded concentrations of AraC (0.0025, 0.05, 0.01, 0.02, 0.04,
0.08, 0.16, 0.32, 0.64 and 1.28 µM), KRC-180 (0.1875, 0.375, 0.75,
1.5, 3, 6, 12, 24, 48 and 96 µM), or a combination of AraC and
KRC-180 (ratio 1:75) for 72 h. Cell viability was measured and the
combination index (CI) was analyzed using CompuSyn software
(version 1.0; ComboSyn, Inc., Paramus, NJ, USA), as previously
described by Chou (15).
In vitro kinase assay
The inhibition of JAK2 recombinant kinase activity
was measured using homogeneous, time-resolved fluorescence (HTRF)
assays. Recombinant proteins containing the JAK2 kinase domain were
purchased from EMD Millipore (Billerica, MA, USA). Optimal enzyme,
ATP and substrate concentrations were established using an HTRF
KinEASE kit (no. 62TK0PEB; Cisbio Bioassays, Codolet, France),
according to the manufacturer's protocol. Test compounds are
three-fold serially diluted in DMSO to create a 10-point curve at a
starting concentration of 100 µM. The enzymes were mixed with
serially diluted compounds and 0.1 µM peptide substrates in a
kinase reaction buffer [50 mM HEPES (pH 7.0), 10 µM ATP, 0.1 mM
sodium orthovanadate, 5 mM MgCl2, 1 mM DTT, 0.01% bovine serum
albumin (BSA; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 0.02% NaN3]. Following the addition of reagents for
detection, the time-resolved fluorescence energy transfer (TR-FRET)
signal was measured using a VICTOR™ X5 2030 multilabel plate reader
(PerkinElmer, Inc., Waltham, MA, USA). The half maximal inhibitory
concentration (IC50) was calculated by nonlinear
regression using GraphPad Prism software. KRC-180 and its
derivatives were synthesized at the Korea Research Institute of
Chemical Technology (Daejeon, Korea) as previously described
(16).
Molecular docking
The molecular docking procedure was performed using
CDOCKER interfaced with BIOVIA Discovery Studio (version 3.5;
BIOVIA™ Corp; Dassault Systèmes, San Diego, CA, USA). The high
resolution (2.30 Å) crystal structure of human JAK2 in complex with
an inhibitor, N-[1-(3-chlorophenyl)-3-methyl-1H-pyrazol-5-yl]
pyrazolo [1,5-a] pyrimidine-3-carboxamide, was downloaded from the
RCSB Protein Data Bank (PDB code: 4HGE) (17). For protein preparation, the active
site for docking was identified from the binding site of the
inhibitor in the co-crystal structure. Following the removal of the
inhibitor, water molecules were removed, hydrogen atoms were added
and incomplete residues of the side chain were corrected.
Subsequently, the energy minimization of the protein structure was
achieved by the CHARMM force field of Discovery Studio (version 41;
Dassault Systems BIOVIA Corporation, San Diego, CA, USA). For
ligand preparation, energy minimization was performed for the
generation of 3-D conformers of KRC-180 with the Smart Minimizer
algorithm in the CHARMM force field. The original inhibitor of the
X-ray co-crystal structure was used as a reference compound to
verify the docking protocol, exhibiting a similar binding mode
compared with the original complex structure.
Flow cytometry analysis
The HEL92.1.7 cells were cultured in 6-well plates
(1×106 cells/well) and treated with 0, 1, 5 and 10 µM
KRC-180 at 37°C for 24 h. The cells were fixed with 4% formalin at
room temperature and treated with RNase A (4 mg/ml) at room
temperature. The cells were then stained with propidium iodide (PI;
Sigma-Aldrich; Merck KGaA) and subjected to flow cytometry using a
BD Accuri™ C6 flow cytometer (BD Biosciences, San Jose, CA, USA).
The data were analyzed using BD Accuri™ C6 software version
1.0.264.21 (BD Biosciences). For Annexin V staining (18), the cells were seeded in 6-well plates
(1×106 cells/well) and treated with 0, 1, 5 or 10 µM
KRC-180 for 48 h at 37°C. The cells were then stained using an
Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis kit (Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol and
subjected to flow cytometry.
Immunoblotting
HEL92.1.7 cells were lysed with SDS lysis buffer (12
mM Tris-Cl, pH 6.8, 5% glycerol, 0.4% SDS; USB Corporation,
Cleveland, OH, USA) and subjected to electrophoresis on 10%
SDS-PAGE. Polyvinylidene fluoride (PVDF) membranes (EMD Millipore)
were blocked in Tris-buffered saline (10 mM Tris-Cl, pH 7.4 and 140
mM NaCl; Bioneer Corporation, Daejeon, Korea) containing 0.1%
Tween-20 (TBST) and 5% nonfat dry milk or BSA. The membranes were
then incubated with blocking solution containing the indicated
antibodies for 1 h at room temperature. After washing three times
in TBST, the membranes were incubated with goat anti-rabbit IgG
(no. 111-035-003; dilution, 1:5,000) and anti-mouse IgG (no.
115-035-033; dilution, 1:5,000) secondary antibodies were purchased
from Jackson ImmunoResearch Laboratories, Inc., (West Grove, PA,
USA) and conjugated with horseradish peroxidase for 30 min at room
temperature. The blots were washed three times and developed using
Amersham™ ECL Select™ Western Blotting Detection Reagent (GE
Healthcare Life Sciences, Chalfont, UK), and the luminescent
signals were visualized using the ImageQuant™ LAS 4000 mini (GE
Healthcare Life Sciences). Antibodies against phospho-JAK2 [p-JAK2;
rabbit monoclonal immunoglobulin G (IgG); no. 3776; dilution,
1:1,000], p-STAT3 (rabbit polyclonal IgG; no. 9131; dilution,
1:1,000), p-STAT5 rabbit polyclonal IgG; no. 9351; dilution,
1:1,000) and poly (ADP ribose) polymerase (PARP; rabbit polyclonal
IgG; no. 9542; dilution, 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Antibodies against
JAK2 (rabbit polyclonal IgG; no. sc-278; dilution, 1:1,000), STAT3
(rabbit polyclonal IgG; no. sc-482; dilution, 1:1,000) and STAT5
(rabbit polyclonal IgG; no. sc-835; dilution, 1:1,000) were from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Goat anti-rabbit
IgG (no. 111-035-003; dilution, 1:5,000) and anti-mouse IgG (no.
115-035-033; dilution 1:5,000) secondary antibodies were purchased
from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA,
USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
The HEL92.1.7 cells were harvested and total RNA was
extracted using the ReliaPrep™ RNA Cell Miniprep System (Promega
Corporation, Madison, WI, USA). The cDNA was synthesized using a
Maxime RT PreMix kit (Intron Biotechnology, Inc., Seongnam, Korea).
The cDNA (1 µg) was amplified using 10 pmol primers (Integrated DNA
Technologies, Inc., Coralville, IA, USA) in the PCR reaction: 94°C
for 5 min followed by 30 cycles at 95°C for 30 sec, 60°C for 30 sec
and 74°C for 90 sec. The following primers were used in the study:
Elongation factor 1α (forward, 5′-AGGTGATTATCCTGAACCATCC-3′ and
reverse, 5′-AAAGGTGGATAGTCTGAGAAGC-3′; size 234 bp); Cyclin B1
(forward, 5′-AAGAGCTTTAAACTTTGGTCTGGG-3′ and reverse,
5′-CTTTGTAAGTCCTTGATTTACCATG-3′; size 319 bp). The PCR products
were analyzed by electrophoresis and stained with RedSafe™ nucleic
acid staining solution (iNtRON Biotechnology, Seongnam, Korea) on a
1.5% agarose gel.
Statistical analysis
The data from the experiments are presented as the
mean ± SEM.
Results
Structure-activity associations of
KRC-180 derivatives
In order to examine the structure-activity
associations of KRC-180 derivatives, the in vitro JAK2
kinase inhibitory activities toward a substituent effect on
R1, R2 and R3 positions of the
phenyl ring of benzoxazole were examined (Table I). In the initial assessment, a µM
range of potency was observed with no substitution on the phenyl
ring (entry 1, R1=R2=R3=H).
Introduction of simple CN, OMe CF3 at R1
position was observed to be detrimental to the activity (compound
6, 8, 9, 12). In particular, the lowest inhibitory activity was
observed in the case of CONMe2 at the R1
position, presumably due to steric congestion in the binding pocket
of the enzyme (compound 12). Introduction of piperidine, chlorine
and NMe2 at the R2 position of phenyl
slightly improved the inhibitory activity (compound 2, 3, 5). Of
the substituents, introduction of OH at the R2 position
significantly improved the inhibitory activity. In addition,
introduction of sulfate (compound 7) to the R2 position
exhibited a similar degree of activity to that of OH (compound 3).
Bulky substituents larger than methylsulfate (compound 7),
including phenoxy (compound 13) and pyrazinyloxy (compound 18), did
not significantly improve the activity, compared with that of OH.
To assess the substituent effect on the R3 position, OH
(compound 10), indazolyl (Table I;
compound 14) and pyrazinyloxy (compound 19) were evaluated, and the
results were disclosed as detrimental to increasing the inhibitory
activity, as the OH substituent at R2 (compound 1)
displayed potent activity, the OH substituent at R3
(compound 10) and pyrazine-2-oxy (compound 19) exhibited only
modest activity and the indazole substituent (compound 14) did not
contribute to potentency activity. To improve the inhibitory
activity, methyl and ethyl were introduced to the R1 and
R3 positions to induce hydrophobic interactions with
adjacent hydrophobic groups, but dual substitutions on phenyl did
not influence the inhibitory activity (compound 16, 17, 20). When
modifying the R3 position with a bulky substituent, such
as indazoly (compound 14), the compound exhibited moderate activity
compared with that of compound 3; however, the introduction of
pyrazinyloxy (compound 19) decreased the inhibitory activity.
 | Table I.Structure-activity relationship of
KRC-180 derivatives. |
Inhibition of JAK2 kinase activity in
vitro
The effect of KRC-180 on the kinase activity of the
purified recombinant JAK2 protein was measured using the TR-FRET
method. KRC-180 markedly inhibited the kinase activity of JAK2
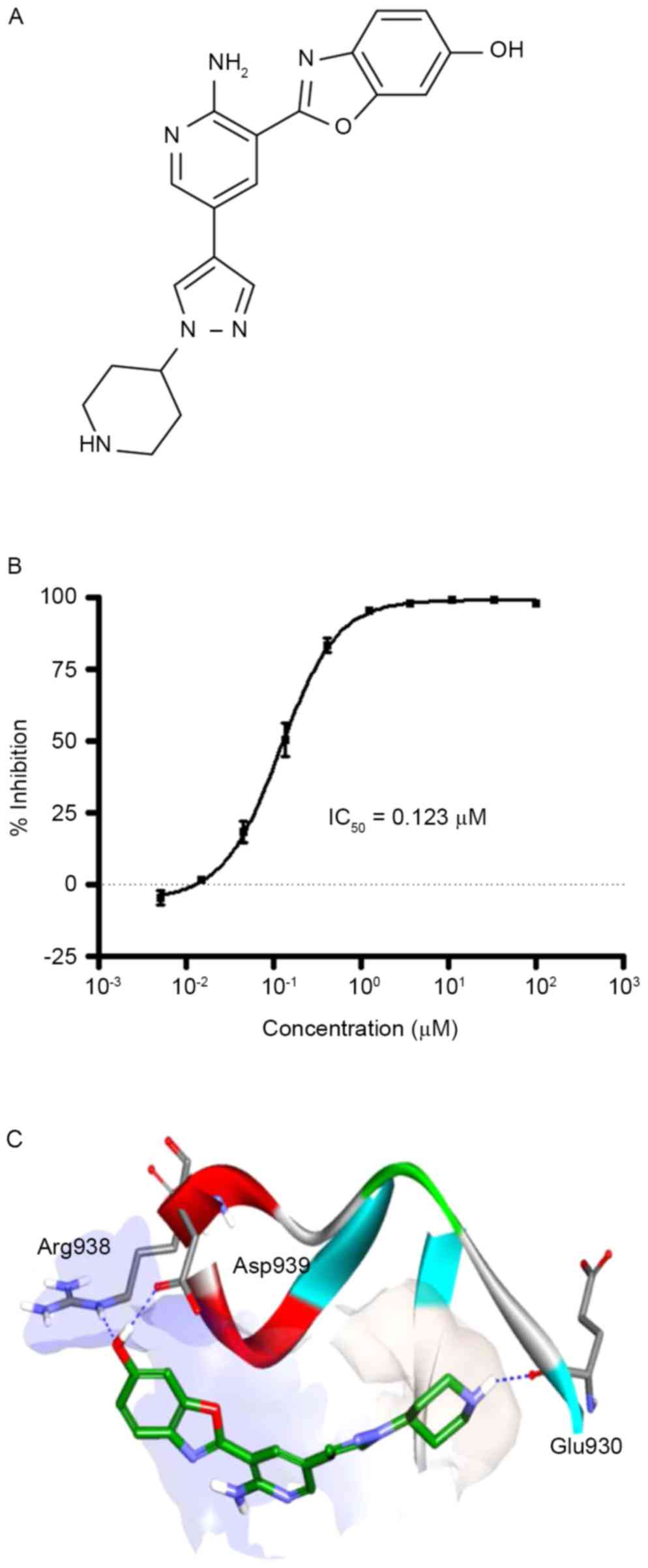
in vitro with an IC50 of 0.123 µM (Fig. 1A and B). The direct interaction
between JAK2 and KRC-180 in the docking analysis is presented in
Fig. 1C. A co-crystal protein
structure of JAK2 and a pyrazolopyrimidine-based inhibitor was
obtained from the protein data bank (PDB code: 4HGE). KRC-180 was
fitted into the active site of 4HGE. For example, the hydroxyl
benzoxazole moiety is positioned in a hydrophilic site composed of
aspartic acid and arginine, as indicated by the region of light
blue, and piperidine is located in a hydrophobic pocket, designated
in light gray. CDOCKER interaction energy values for ruxolitinib
(JAK1/2 inhibitor used for patients with myelofibrosis) (19) and KRC-180 were −50.10 kcal/mol and
−41.83 kcal/mol, respectively.
The current study has demonstrated that KRC-180 has
three important H-bonds. First, the oxygen of the OH group of
benzoxazole interacts as an H-bond acceptor to the NH of the Arg938
residue. Second, the hydrogen of the OH group acts as a hydrogen
bond donor to the COO− carboxylic acid side chain of the
Asp939 residue. Finally, the NH group of piperidine forms a
hydrogen bond with the backbone carbonyl group of Glu930. Although
piperidine is at a hydrophobic site, the hydrogen bond has a
bonding angle of optimum length.
KRC-180 inhibits JAK2 phosphorylation
in HEL92.1.7 cells
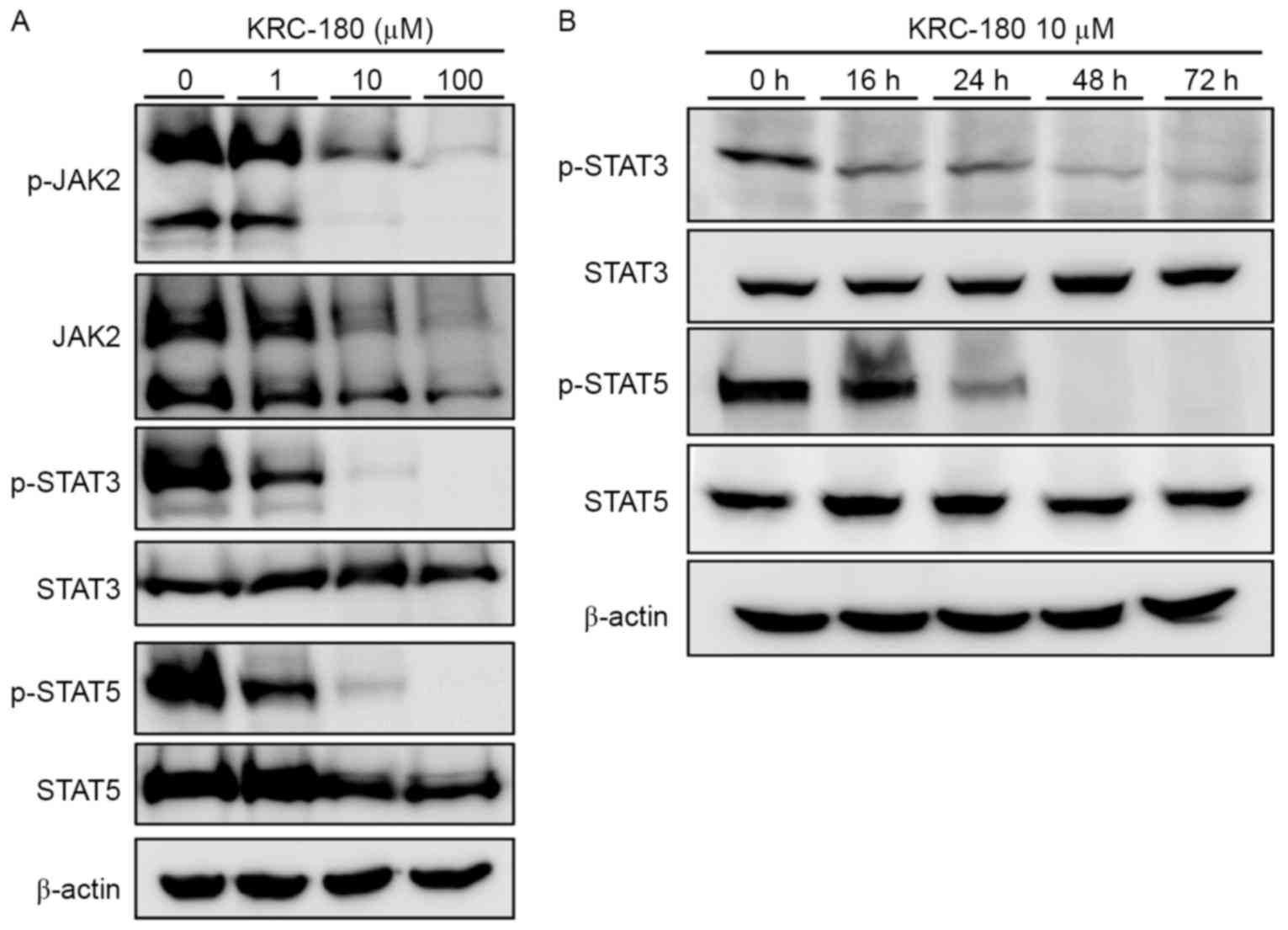
To examine the effect of KRC-180 JAK2
phosphorylation, HEL92.1.7 cells expressing the JAK2 mutant
(JAK2-V617F) were used. The JAK2 mutation in HEL92.1.7 cells
resulted in the constitutive activation and phosphorylation of JAK2
without stimulation. KRC-180 treatment decreased JAK2
phosphorylation and expression, as demonstrated via western
blotting analysis of p-JAK2 and JAK2 (Fig. 2A). Downstream of the JAK2 signaling
pathway, STAT5 and STAT3 phosphorylation was also dose-dependently
decreased by KRC-180 treatment. Phosphorylation of STAT3 and STAT5
was almost completely diminished following 10 µM KRC-180 treatment.
The levels of phosphorylated STAT3 and STAT 5 in HEL92.1.7 cells
treated with 10 µM of KRC-180 for 16 h, 24 h, 48 h and 72 h were
analyzed (Fig. 2B). STAT3 and STAT5
phosphorylation was not detected after 48 h. These results indicate
that direct kinase inhibition by KRC-180 results in the inhibition
of JAK2 activity and its downstream signaling in leukemic
cells.
KRC-180 inhibits HEL92.1.7 cell
proliferation and induces apoptosis
The effects of KRC-180 on the growth of HEL92.1.7
cells were examined. HEL92.1.7 cell proliferation was suppressed
dose-dependently by KRC-180, with a GI50 of 2.69 µM
(Fig. 3A). The effect of KRC-180 in
combination with AraC was investigated. The GI50 ratio
of AraC single treatment and KRC-180 single treatment
(GI50 ratio AraC:KRC-180) against HEL92.1.7 cells was
1:75. The CI calculated by CompuSyn software demonstrated the
additive effect of the 1:75 AraC and KRC-180 treatment on cell
viability (CI at the 50% effective dose, 1.05; CI at the 75%
effective dose, 1.01; CI at the 90% effective dose, 0.98; Fig. 3B).
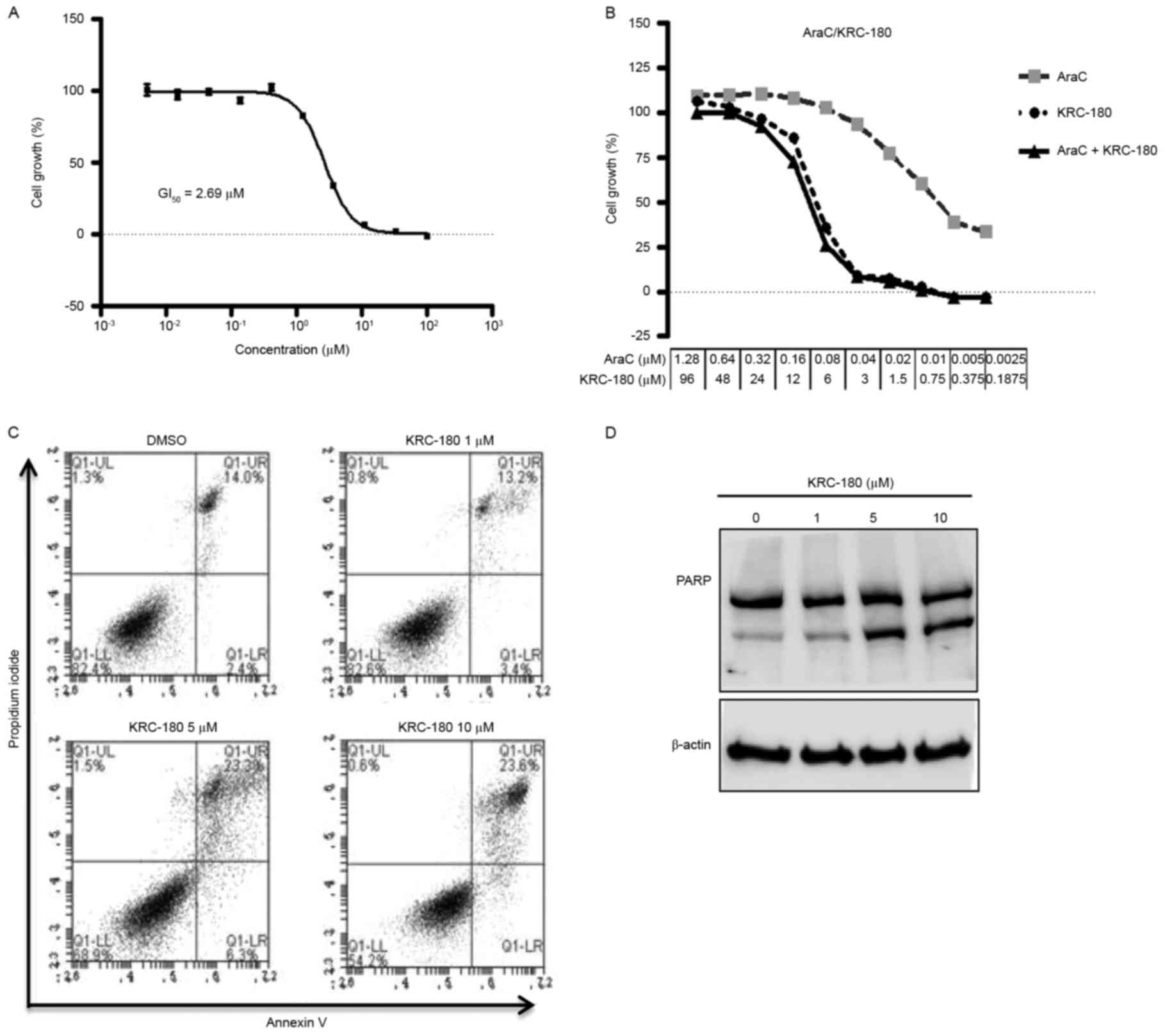 | Figure 3.Effect of KRC-180 on HEL92.1.7 cell
growth and cell death. (A) The indicated concentrations of KRC-180
were applied for 72 h, and the growth inhibition of HEL92.1.7 cells
was measured using a tetrazolium-based cell viability assay. The
cell growth (%) was calculated using 0.5% dimethyl sulfoxide
treatment as a negative control. The GI50 was calculated
by nonlinear regression. The data are presented as the mean ±
standard error of the mean of three independent experiments. (B)
The cells were treated with graded concentrations of AraC (0.0025,
0.05, 0.01, 0.02, 0.04, 0.08, 0.16, 0.32, 0.64, 1.28 µM), KRC-180
(0.1875, 0.375, 0.75, 1.5, 3, 6, 12, 24, 48, 96 µM), or a
combination of AraC and KRC-180 (ratio 1:75) for 72 h, and the cell
growth was subsequently determined using the cytotoxicity assay.
The combination index was calculated using CompuSyn software. (CI
value >1.0, antagonism; CI value=1.0, additivity; CI value
<1.0, synergism). (C) The cells were treated with the indicated
concentrations of KRC-180 for 48 h. The cells were then stained
with propidium iodide/Annexin V and subjected to flow cytometry.
(D) The cells were treated with KRC-180 for 24 h at the indicated
concentrations, and cleaved and uncleaved poly (ADP ribose)
polymerase was detected using western blot analysis. AraC,
cytarabine; GI50, concentration that inhibits growth by
50%. |
Apoptosis induction by KRC-180 was also
investigated. A dose-dependent increase in the apoptotic cell
population was observed with Annexin V/PI staining following
KRC-180 treatment (Fig. 3C). In
particular, 10 µM KRC-180 increased the early apoptotic population
from 14–23.6% (Fig. 3C, upper right
quadrant). The dose-dependent increase in PARP cleavage by KRC-180
confirmed that apoptotic cell death is induced by KRC-180 treatment
(Fig. 3D).
KRC-180 induces cell cycle arrest at
the G2/M phase
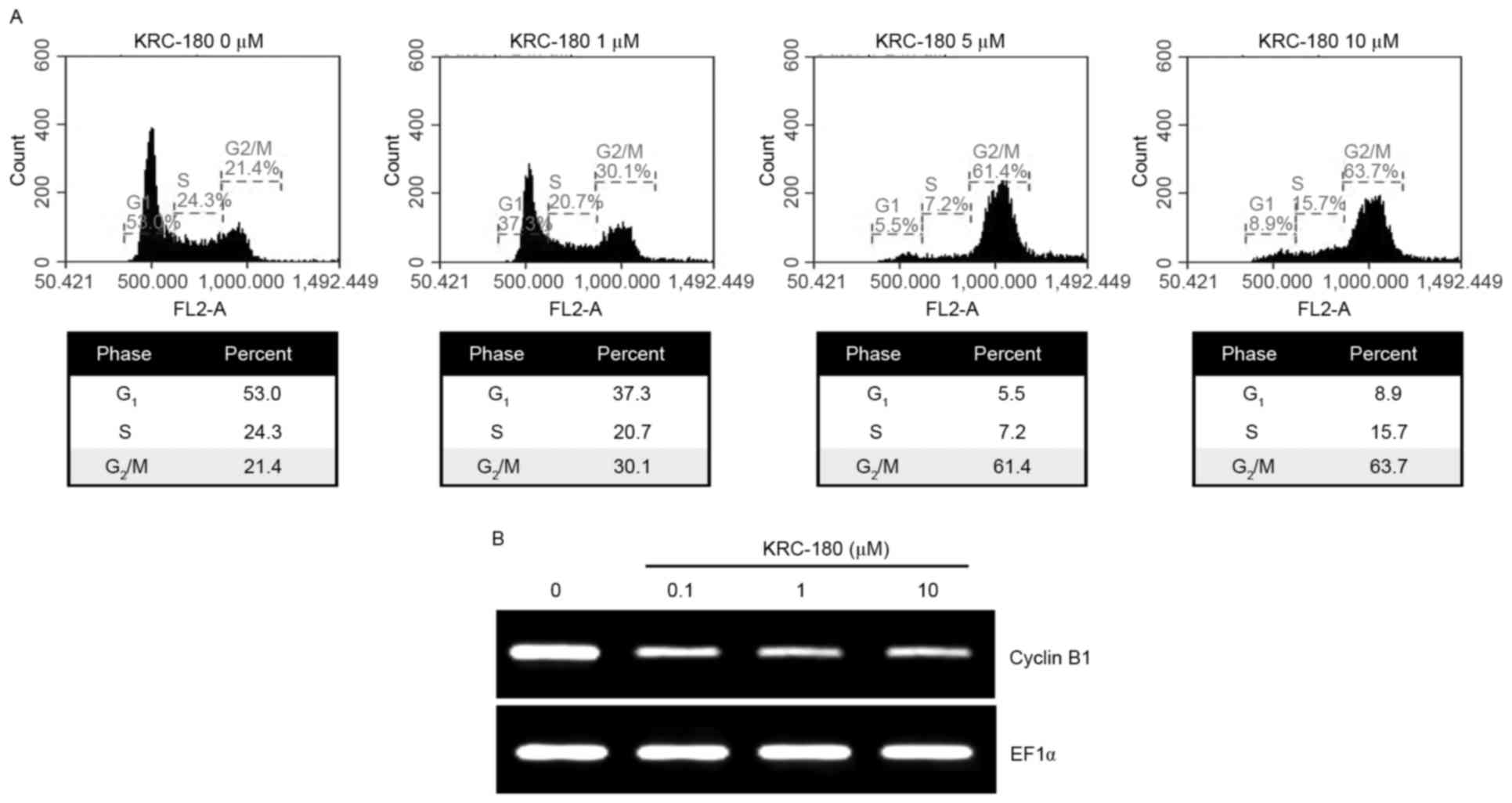
The HEL92.1.7 cells were subjected to cell cycle
analysis following treatment with KRC-180. KRC-180 treatment from
0.1–10 µM for 24 h resulted in cell cycle arrest at the
G2/M phase (Fig. 4A). The
cell population in the G2/M phase increased with rising
concentrations of KRC-180, from 23.3% (control) to 62.1% (10 µM
KRC-180). Cyclin B1 is one of the components required for the cell
cycle progression in the G2/M phase (20). KRC-180 treatment decreased the
expression levels of cyclin B1 mRNA, as demonstrated using RT-PCR
(Fig. 4B). The dose-dependent
reduction of cyclin B1 expression levels was consistent with cell
cycle arrest at the G2/M phase following KRC-180
treatment.
Discussion
In the present study, KRC-180 was characterized as a
direct inhibitor of JAK2 with antiproliferative activity in
erythroleukemia cells. KRC-180 directly inhibited the activity of
JAK2. A previous report demonstrated that KRC-180 also exhibits
inhibitory activity against c-Met, Flt3, Ron and Aurora A,
indicating that KRC-180 possesses multikinase inhibitory
characteristics (13). To evaluate
the JAK2-mediated effect of KRC-180 on cell survival, HEL92.1.7
erythroleukemia cells were used. HEL92.1.7 cells harbor the
JAK2-V617F mutation resulting in the constitutive activation of
JAK2 and associated downstream signaling. In cells carrying the
V617F mutation, cell survival depends on JAK2 signaling (2). Cell cytotoxicity induced by KRC-180 in
HEL92.1.7 cells confirms the JAK2-mediated effect of KRC-180.
To investigate the potential of KRC-180 in
combination treatment with current leukemic therapies, the CI of
KRC-180 and AraC was measured. A CI value of ~1 indicates an
additive effect of KRC-180 and AraC. Thus, this result provides a
basis for the combination therapy of KRC-180 and AraC.
KRC-108 (compound 4 in Table I), a compound structurally similar to
KRC-180, has been reported as a multikinase inhibitor with an
antitumor effect (21). The presence
of the OH residue at the benzoxazole ring in KRC-180 is the only
difference between KRC-180 and KRC-108, and the IC50 for
JAK2 inhibition of KRC-108 is 15-fold higher than that of KRC-180
(Table I). This suggests that the OH
residue on the benzo(d)oxazole ring is important for the inhibition
of JAK2 kinase activity. The importance of the hydroxyl residue was
confirmed by the docking analysis, which demonstrated an
interaction of the OH residue with the Arg938 and Asp939 residues
of the JAK2 protein.
At present, two JAK inhibitors have been approved
and clinically used: Ruxolitinib and tofacitinib. Ruxolitinib
(formerly known as INCB018424) is indicated for the treatment of
myelofibrosis and polycythemia vera, which are types of MPNs
(19). Ruxolitinib inhibits JAK1 and
JAK2 and clinically improves the symptoms associated with MPNs
(19). Tofacitinib is used for
rheumatoid arthritis with inhibitory activity against JAK3, in
addition to JAK1 and JAK2 (22). The
clinical development of various small molecules with
JAK2-inhibitory activity, which are indicated for MPNs, rheumatoid
arthritis and cancer, has been carried out (23). Although certain clinical trials are on
hold due to adverse effects (24),
the results acquired from these clinical studies will aid with the
future development of agents that inhibit JAK2. In the present
study, the JAK2-inhibitory activity and antiproliferative effect of
KRC-180 was investigated. JAK2 kinase inhibition and downstream
signaling pathway suppression resulted in apoptosis and cell cycle
arrest. Pharmacological and chemical data from these results may
aid the development of novel JAK inhibitors that could then be used
to treat cancer.
Acknowledgements
The present study was supported by grants from the
National Research Foundation of Korea (grant no.
2015R1C1A2A01053928) funded by the government of Korea (Ministry of
Education, Science and Technology).
References
|
1
|
Schindler C and Darnell JE Jr:
Transcriptional responses to polypeptide ligands: The JAK-STAT
pathway. Annu Rev Biochem. 64:621–651. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quintás-Cardama A, Kantarjian H, Cortes J
and Verstovsek S: Janus kinase inhibitors for the treatment of
myeloproliferative neoplasias and beyond. Nat Rev Drug Discov.
10:127–140. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saharinen P, Takaluoma K and Silvennoinen
O: Regulation of the Jak2 tyrosine kinase by its pseudokinase
domain. Mol Cell Biol. 20:3387–3395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Steensma DP, Dewald GW, Lasho TL, Powell
HL, McClure RF, Levine RL, Gilliland DG and Tefferi A: The JAK2
V617F activating tyrosine kinase mutation is an infrequent event in
both ‘atypical’ myeloproliferative disorders and myelodysplastic
syndromes. Blood. 106:1207–1209. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lacronique V, Boureux A, Valle VD, Poirel
H, Quang CT, Mauchauffé M, Berthou C, Lessard M, Berger R, Ghysdael
J and Bernard OA: A TEL-JAK2 fusion protein with constitutive
kinase activity in human leukemia. Science. 278:1309–1312. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Furqan M, Mukhi N, Lee B and Liu D:
Dysregulation of JAK-STAT pathway in hematological malignancies and
JAK inhibitors for clinical application. Biomark Res. 1:52013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gu L, Liao Z, Hoang DT, Dagvadorj A, Gupta
S, Blackmon S, Ellsworth E, Talati P, Leiby B, Zinda M, et al:
Pharmacologic inhibition of Jak2-Stat5 signaling By Jak2 inhibitor
AZD1480 potently suppresses growth of both primary and
castrate-resistant prostate cancer. Clin Cancer Res. 19:5658–5674.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hedvat M, Huszar D, Herrmann A, Gozgit JM,
Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, et
al: The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and
oncogenesis in solid tumors. Cancer cell. 16:487–497. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hart S, Goh KC, Novotny-Diermayr V, Hu CY,
Hentze H, Tan YC, Madan B, Amalini C, Loh YK, Ong LC, et al:
SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the
treatment of myeloid and lymphoid malignancies. Leukemia.
25:1751–1759. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scuto A, Krejci P, Popplewell L, Wu J,
Wang Y, Kujawski M, Kowolik C, Xin H, Chen L, Wang Y, et al: The
novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling,
resulting in suppression of human myeloma cell growth and survival.
Leukemia. 25:538–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan S, Li Z and Thiele CJ: Inhibition of
STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor
growth in Neuroblastoma and Pediatric Sarcomas In vitro and In
vivo. Oncotarget. 4:433–445. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Judd LM, Menheniott TR, Ling H, Jackson
CB, Howlett M, Kalantzis A, Priebe W and Giraud AS: Inhibition of
the JAK2/STAT3 pathway reduces gastric cancer growth in vitro and
in vivo. PLoS One. 9:e959932014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee J, Han SY, Jung H, Yang J, Choi JW,
Chae CH, Park CH, Choi SU, Lee K, Ha JD, et al: Synthesis and
structure-activity relationship of aminopyridines with substituted
benzoxazoles as c-Met kinase inhibitors. Bioorg Med Chem Lett.
22:4044–4048. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chung HJ, Kamli MR, Lee HJ, Ha JD, Cho SY,
Lee J, Kong JY and Han SY: Discovery of quinolinone derivatives as
potent FLT3 inhibitors. Biochem Biophys Res Commun. 445:561–565.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cho SY, Han SY, Ha JD, Ryu JW, Lee CO,
Jung H, Kang NS, Kim HR, Koh JS and Lee J: Discovery of
aminopyridines substituted with benzoxazole as orally active c-Met
kinase inhibitors. Bioorg Med Chem Lett. 20:4223–4227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hanan EJ, van Abbema A, Barrett K, Blair
WS, Blaney J, Chang C, Eigenbrot C, Flynn S, Gibbons P, Hurley CA,
et al: Discovery of potent and selective pyrazolopyrimidine janus
kinase 2 inhibitors. J Med Chem. 55:10090–11107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han MH, Park C, Kwon TK, Kim GY, Kim WJ,
Hong SH, Yoo YH and Choi YH: The histone deacetylase inhibitor
trichostatin a sensitizes human renal carcinoma cells to
TRAIL-induced apoptosis through down-regulation of c-FLIPL. Biomol
Ther (Seoul). 23:31–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vaddi K, Sarlis NJ and Gupta V:
Ruxolitinib, an oral JAK1 and JAK2 inhibitor, in myelofibrosis.
Expert Opin Pharmacother. 13:2397–2407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weinberg R: The Biology of Cancer. Second.
Taylor & Francis Group; 2013, View Article : Google Scholar
|
|
21
|
Han SY, Lee CO, Ahn SH, Lee MO, Kang SY,
Cha HJ, Cho SY, Ha JD, Ryu JW, Jung H, et al: Evaluation of a
multi-kinase inhibitor KRC-108 as an anti-tumor agent in vitro and
in vivo. Invest New Drugs. 30:518–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vyas D, O'Dell KM, Bandy JL and Boyce EG:
Tofacitinib: The first janus kinase (JAK) inhibitor for the
treatment of rheumatoid arthritis. Ann Pharmacother. 47:1524–1531.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dolgin E: Companies hope for kinase
inhibitor JAKpot. Nat Rev Drug Discov. 10:717–718. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ratner M: Setback for JAK2 inhibitors. Nat
Biotechnol. 32:1192014. View Article : Google Scholar : PubMed/NCBI
|