Introduction
Lung cancer is the most frequently diagnosed cancer
with a poor prognosis; non-small cell lung cancer (NSCLC) remains
the predominant diagnosis among all cases of lung cancer. NSCLC
consists of ~85% of all lung cancer diagnoses, and these primarily
consist of the following morphological subtypes: Adenocarcinoma
(AC); squamous cell carcinoma (SCC); large cell carcinoma (LCC)
(1,2).
Although molecular-targeted cancer therapy has advanced
considerably, chemotherapy is the primary treatment for lung cancer
cases that are unresectable. For patients with advanced NSCLC, the
platinum-based two-drug combination regimen is a standard
chemotherapy (3,4). Cisplatin (CDDP)-based chemotherapy has
improved the survival rate of patients with NSCLC. However, the
resistance of tumor cells to this drug still limits its
efficacy.
Tumor protein p53 (p53 or TP53), the ‘guardian of
the genome’, was identified in 1979 (5) as the first tumor suppressor gene. p53 is
the most extensively studied tumor suppressor gene and has an
important role in suppressing tumor development. The function of
the p53 gene is to restrict cell proliferation in response to DNA
damage. Through the regulation of target genes, p53 is responsible
for various cellular responses, including growth arrest, senescence
and apoptosis (6,7). The investigation of p53-induced
apoptosis in response to genotoxic stress is of clinical
importance, as numerous traditional chemotherapeutic strategies
rely on the induction of DNA damage and the subsequent activation
of apoptosis (8). For example, the
mechanism for CDDP is to induce apoptosis via damaging the DNA of
cancer cells and activating the p53-dependent apoptosis pathway.
However, investigation of p53 biology has established that
manipulation of p53 function may provide further insight to the
potential of a cancer-free life (9).
p53 regulates the toxic effects of DNA damaging
drugs on cancer cells. However, the mutant TP53 proteins lose their
‘guardian of the genome’ function when mutated, including the
functions of arresting cell proliferation and inducing apoptosis
(10). Mutations in the p53 gene
occur in the majority of malignant tumors. Lung cancer cases have
high rates of specific p53 mutations (i.e., mutations to p53 that
are present in lung cancer but not other malignant tumors), with
~46% in lung adenocarcinoma and ~81% in squamous cell carcinoma of
lung (11). At the genetic level,
carcinogenesis is a multistep process, in which the activation of
oncogenes and the inactivation of tumor suppressors participate
(12). p53 gene mutations are
typically caused by alterations of a single amino acid that results
in the expression of mutant proteins with ‘gain of function’
activity (13). These mutant proteins
have been demonstrated to have a dominant oncogenic role (14,15). In
certain previous studies, p53 mutations have been reported to be
markers of poor survival in NSCLC (16,17). As
genetic alterations in the p53-dependent apoptosis pathway
frequently occur in NSCLC, the understanding of p53 activity in
treatment may be valuable for the development of novel therapeutic
methods (18).
In the present study, the exogenous wild-type p53α
gene was transfected into p53-null H1299 human lung adenocarcinoma
cells, and the cells were screened for those that stably expressed
the TP53 protein. The focus of the current study was to investigate
whether the expression of wild-type p53α resulted in an increase of
the sensitivity of cells to CDDP chemotherapy, and to explore the
associated underlying mechanisms.
Materials and methods
Cell line and cell culture
The human A549 lung adenocarcinoma cell line was
purchased from Sun Yat-sen University (Guangdong, China). The human
H1299 lung adenocarcinoma cell line was purchased from the Cell
Bank of Shanghai (Institute of Cell Biology, Shanghai, China). All
cells were cultured in RPMI-1640 (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA), supplemented with 10% fetal bovine serum
(Zhejiang Tianhang Biotechnology Co., Ltd., Hangzhou, China), 100
U/ml penicillin and 100 µg/ml streptomycin (Beyotime Institute of
Biotechnology, Shanghai, China) in a humidified atmosphere
containing 5% CO2 in an incubator at 37°C. All
experiments were performed on cells in the logarithmic phase of
growth.
Construction and identification of
recombinant vector pEGFP-p53α
Total RNA of A549 cells was isolated using TRIzol®
(Takara Biotechnology Co., Ltd., Dalian, China), according to the
manufacturer's protocol. Total RNA (1.0 µg) was reverse-transcribed
using AMV Reverse Transcriptase (Takara Biotechnology Co., Ltd.),
from which the cDNA was obtained for polymerase chain reaction
(PCR) amplification (DNase was not used). The primer sequences were
as follows: p53α forward, 5′-ACTAGAATTCATGGAGGAGCCGCAGTC-3′ and
reverse, 5′-CGCGGGATCCTCAGTCTGAGTCAGGCCCTT-3′. The PCR
amplifications were performed under the following thermocycling
conditions: The initial denaturation was at 50°C for 30 min and
94°C for 2 min, followed by 25–30 cycles of denaturation at 94°C
for 30 sec, annealing at 55–65°C for 30 sec and extension at 72°C
for 1 min. The PCR products were separated by electrophoresis on a
1% agarose gel and were stained by ethidium bromide and visualized
using ultraviolet light.
The PCR products were purified from agarose gel
using a MiniBest Agarose Gel DNA Extraction kit. (Takara
Biotechnology Co., Ltd.). The purified products were ligated into
the cloning vector pMD18-T and transformed into JM109
Escherichia coli (all from Takara Biotechnology Co., Ltd.).
The shuttle vector pEGFP-N1 and anti-sense fragments of
the p53α gene were digested using the restriction enzymes
EcoRI and BamHI, respectively (Takara Biotechnology
Co., Ltd.). The digested products were purified using the Takara
MiniBest Agarose Gel DNA Extraction kit (Takara Biotechnology Co.,
Ltd.) and ligated with T4 DNA ligase, and then co-transformed into
DH-5α E. coli (both from Takara Biotechnology Co., Ltd.).
The pEGFP-p53α was isolated and the successful construction was
validated via sequencing analysis.
Gene transfection and G418
selection
H1299 cells (3×105) were seeded in each
well of 6-well plates and cultured for 24 h at 37°C. The cells were
then co-incubated with 10 µl Lipofectamine® 2000 (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 4 µg plasmid pEGFP-p53α or
pEGFP-N1 at 37°C for 6 h. Subsequently, the cells were
treated with fresh medium followed by an additional 48 h of
incubation at 37°C. H1299 cells (5×103) were seeded in
24-well plates. On day 2, G418 (Amresco, LLC, Solon, OH, USA) was
added at the following various concentrations: 100, 200, 300, 400,
500, 600, 700, 800, 900, 1,000 and 1,100 µg/ml. The cells were
cultured with G418 for 2 weeks at 37°C. The medium was replaced
every 3 days. The cells that were resistant to 1,000 µg/ml G418
were selected using an Olympus BX-51 inverted fluorescent
microscope (Olympus Corporation, Tokyo, Japan). The Image Analysis
Software GM-SW2000 and Image Measurement Software GM-2000 M were
used to visualize the cells (Shanghai Guangmi Instrument Co., Ltd.,
Shanghai, China). The transfection efficiency was quantified by
assessing GFP fluorescence intensity at a wavelength of 590 nm.
Reverse transcription (RT)-PCR
analysis
Total RNA was extracted from the control group and
the stable-transfection H1299 cells. Total RNA was
reverse-transcribed using AMV reverse transcriptase, from which the
cDNA was used for PCR amplification with Takara Ex Taq polymerase.
The PCR reaction was performed using the following primers: p53α
forward, 5′-ACTAGAATTCATGGAGGAGCCGCAGTC-3′ and reverse,
5′-CGCGGGATCCTCAGTCTGAGTCAGGCCCTT-3. RT-PCR was performed for 1
cycle at 50°C for 30 min and 94°C for 2 min followed by 28 cycles
of 94°C for 30 sec, 55°C for 30 sec and 72°C for 1 min. In total, 5
µl PCR products stained with ethidium bromide were separated by
electrophoresis on a 1% agarose gel and were visualized under
ultraviolet light. All reagents were supplied by Takara
Biotechnology Co., Ltd.
Western blot analysis
Total protein was extracted from the control group
and stable-transfection H1299 cells on ice. Equal amounts of
protein from the samples (60 µg) were quantified using a
Bicinchoninic Protein Assay kit (Beyotime Institute of
Biotechnology, Haimen, China), and then separated on a 12% SDS-PAGE
gel prior to being transferred to polyvinylidene fluoride membranes
(Merck KGaA, Darmstadt, Germany). The immune complexes were formed
through the incubation of the proteins with 1:1,000 rabbit
monoclonal anti-p53 (cat. no. 9282; Cell Signaling Technology,
Inc., Danvers, MA, USA) and 1:1,000 rabbit anti-β-actin (cat. no.
4967; Bioworld Technology, Inc., St. Louis Park, MN, USA) primary
antibodies at 4°C overnight. The membranes were blocked in 5%
skimmed milk for at room temperature 1 h and incubated with
horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibodies
(cat. no. 7074; Cell Signaling Technology, Inc.) at a dilution of
1:3,000 at room temperature for 1 h. Next, the membrane was washed
three times with Tris-buffered saline-Tween-20 and the protein
bands were visualized using an enhanced chemiluminescence kit (cat.
no. 12630; Cell Signaling Technology, Inc.) and the signals on the
membranes were exposed to X-ray films.
Immunocytochemistry
Transfected and non-transfected H1299 cells were
seeded at density of 1.5×105 cells/well in 6-well plates
and cultured overnight at 37°C. The cells on slide inside 6-well
plates were washed with PBS, fixed with 4% paraformaldehyde for 30
min at room temperature and permeabilized with 0.1% Triton X-100
for 15 min at room temperature. The immune complexes were formed by
incubating the proteins with a 1:160 rabbit anti-p53 monoclonal
antibody at 4°C overnight. A horseradish peroxidase
(HRP)-conjugated anti-rabbit IgG antibody (cat. no. 7074; Cell
Signaling Technology, Inc.) was then added and the slides were
incubated at room temperature for 30 min and then washed with PBS
three times. Development of the slides was performed using
3,3′-diaminobenzidine solution for 8–10 min at room temperature.
The slides were washed in PBS for 5 min, counterstained with
hematoxylin for 2 min and dehydrated with ethanol for 5 min at room
temperature prior to the addition of the transparent neutral balsam
mounting medium and coverslips. Unless stated, all reagents were
supplied by Fuzhou Maixin Biotech Co., Ltd., Fuzhou, China.
MTT assay and estimation of
half-maximal inhibitory concentration (IC50) values
Transfected and non-transfected H1299 cells were
seeded at a density of 3.5×103 cells/well in 96-well
plates and incubated overnight at 37°C. CDDP was added at numerous
concentrations: 0.01, 0.05, 0.25, 1.25, 10, 20 and 30 µmol/l. CDDP
was administered for 48 h at 37°C. The MTT (Sigma-Aldrich; Merck
KGaA) reagent (20 µl of 5 mg/ml) was added and incubated for 4 h at
37°C. The reaction was stopped by the addition of 150 µl dimethyl
sulfoxide. The optical density at a wavelength of 570 nm was
determined on a microplate reader and the untreated cells were used
as the control for 100% viability. The IC50 values were
calculated from the dose-response data and was the concentration
required to inhibit cell growth by 50% compared with the untreated
control. Values were estimated using dose-concentration curves
showing the percent of cell count.
Clone formation assay
Transfected and non-transfected H1299 cells were
seeded in triplicate in 6-well plates at density of 200 cells/well.
After 24 h, cells in the growth phase were treated with 0.25 µmol/l
CDDP for 48 h at 37°C. They were then cultured for 14 days in an
atmosphere containing 5% CO2 at 37°C to facilitate
colony formation. Following washing with PBS twice, the colonies
were fixed with 4% paraformaldehyde for 30 min at room temperature.
Subsequently, the fixed solution was discarded, 1 ml diluted Giemsa
dye (1:9 dilution in PBS; Huamei Biotechnology Co., Ltd., Wuhan,
China) was added and the colonies were stained for 30 min at room
temperature. Finally, the positive colonies (>50 cells/colony)
were counted. The survival fraction was calculated using the
following formula: Survival fraction=number of colonies/200
cells.
DAPI staining of apoptotic nuclei
DAPI stain (Roche Applied Science, Rotkreuz,
Switzerland) was used to evaluate drug-associated apoptosis,
according to the manufacturer's protocol. Transfected and
non-transfected H1299 cells (80% confluent) were seeded onto the
slides in 6-well plates overnight and exposed to 20 µmol/l CDDP for
48 h at 37°C. Next, the cells were fixed with 4% paraformaldehyde
at room temperature for 30 min and stained with 0.5% DAPI for 15–20
min. The stained cells were visualized at ×100 magnification and
images were captured using an inverted fluorescence microscope
(Olympus BX-51; Olympus Corporation).
Statistical analysis
All data analysis was performed using SPSS v13.0
(SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ±
standard deviation. Differences between two groups were determined
using Student's or Welch's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Construction and identification of
recombinant vector pEGFP-p53α
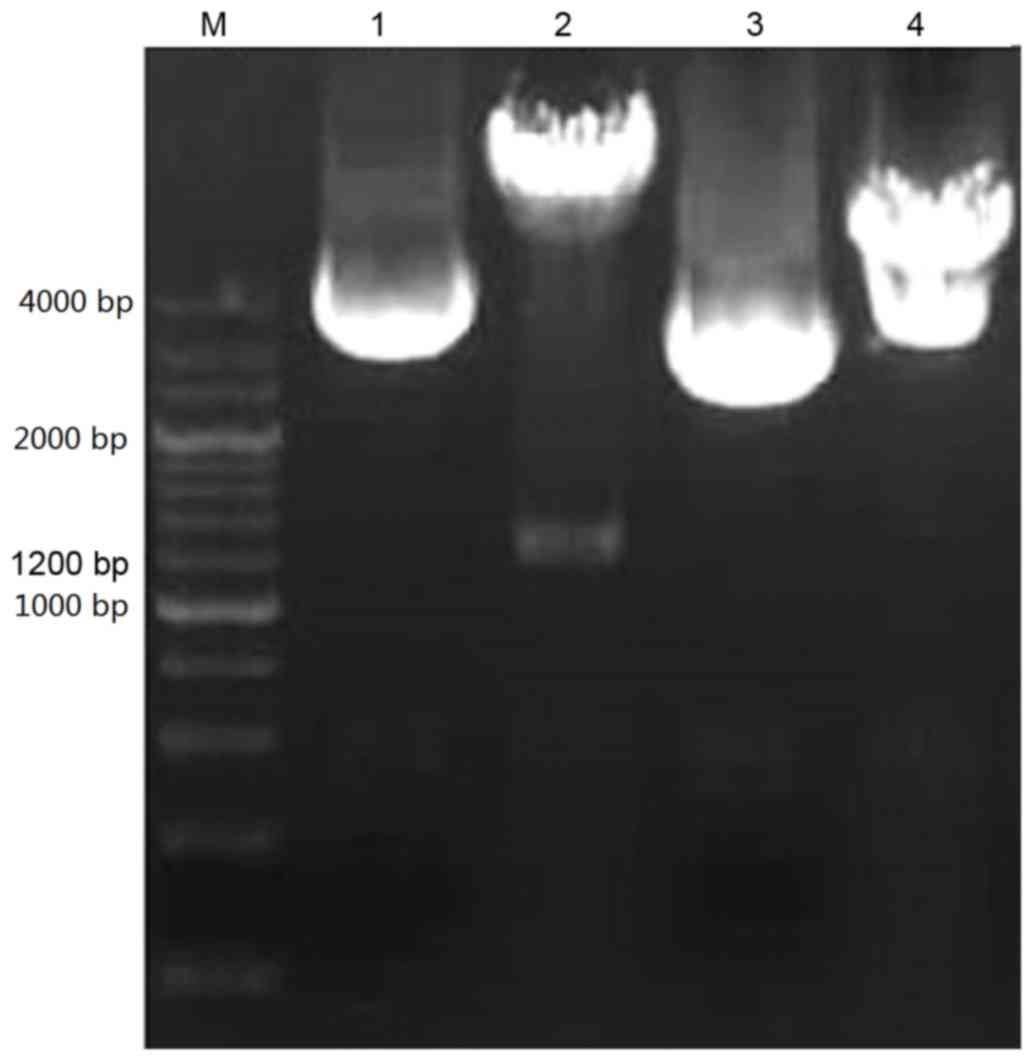
The PCR product of the plasmid was ~1,200 base pairs
(bp), and was separated using 1% agarose gel electrophoresis. As
presented in Fig. 1, the p53α gene
was isolated from the recombinant shuttle vector
pEGFP-N1 using restriction digestion with EcoRI and
BamHI. The 4.7 kb and 1.2 kb strips were detected using 1% agarose
gel electrophoresis, representing the shuttle vector
pEGFP-N1 and the fragment of the p53α gene,
respectively. The expression of the p53α gene was verified using
gene sequence examination (data not presented). The recombinant
vector pEGFP-p53α was established successfully.
G418 selection and gene
transfection
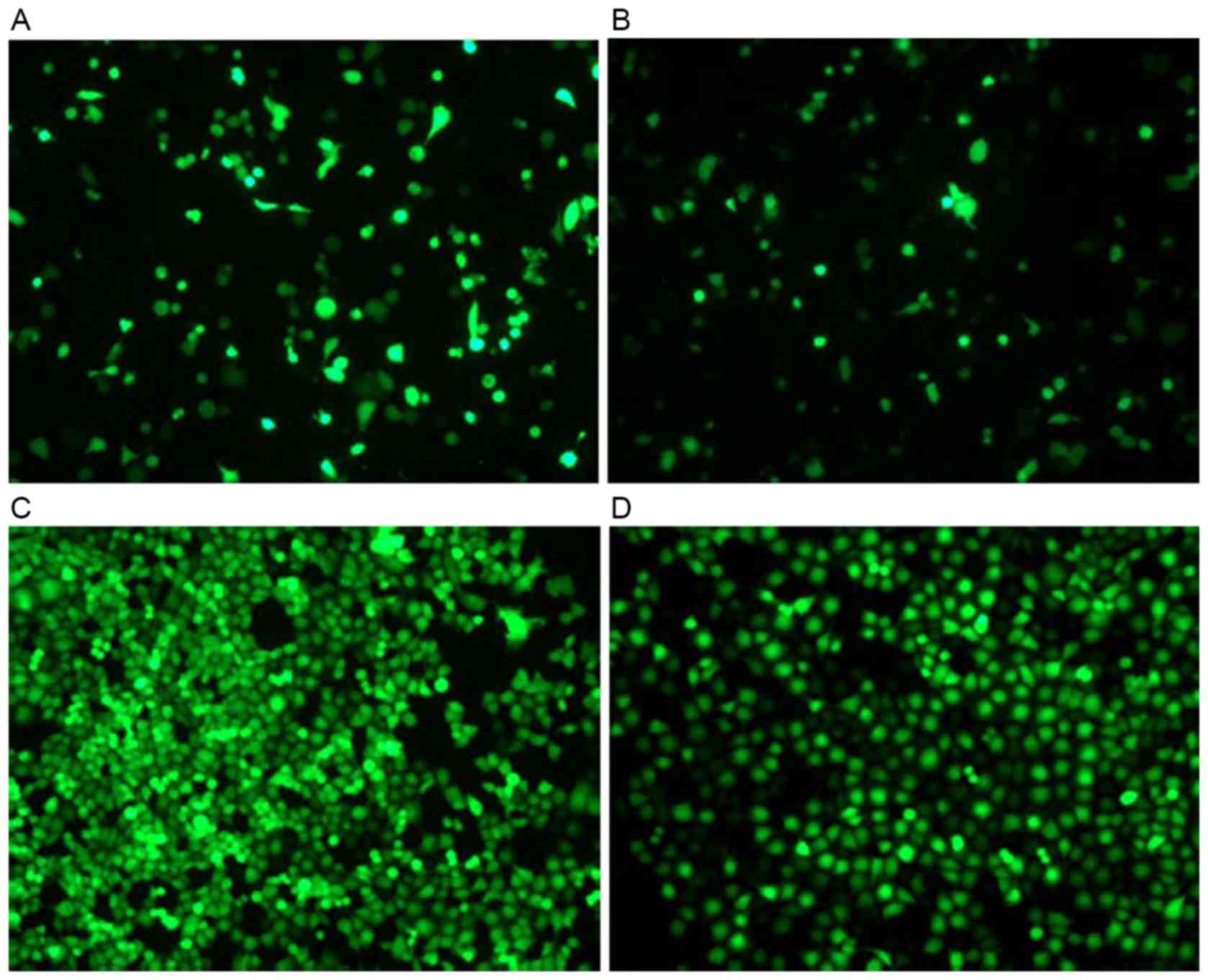
The H1299 cells were seeded in 24-well plates with
various concentrations of G418 and the pEGF-p53α screening
concentration of G418 was 1,000 µg/ml. Recombinant pEGFP-p53α was
transfected into H1299 cells with Lipofectamine 2000 for 2 days.
The green fluorescence was monitored using fluorescence microscopy.
The cells that were resistant to G418 were selected. As presented
in Fig. 2, the efficiency of the
pEGFP-N1 vector transfection was high, compared with
that of the pEGFP-p53α recombinant vector transfection.
Identification of the stable
expression cell line H1299/pEGFP-p53α
The expression of the p53α gene in the cells was
evaluated using RT-PCR. TP53 protein was evaluated using western
blot analysis and immunocytochemistry. As depicted in Fig. 3, the fragment of 1,200 bp was obtained
using RT-PCR amplification. The TP53 protein band was observed in
H1299/pEGFP-p53α cells using western blot analysis (Fig. 4). The expression of TP53 was also
investigated using immunocytochemistry in the control group and
stable-transfection H1299 cells. The H1299 and
H1299/pEGFP-N1 transfected groups demonstrated no
positive TP53 staining. However the H1299/pEGFP-p53α transfected
group exhibited the brown color of the TP53 stain (Fig. 5). Therefore, exogenous TP53 was
successfully expressed in H1299/pEGFP-p53α cells.
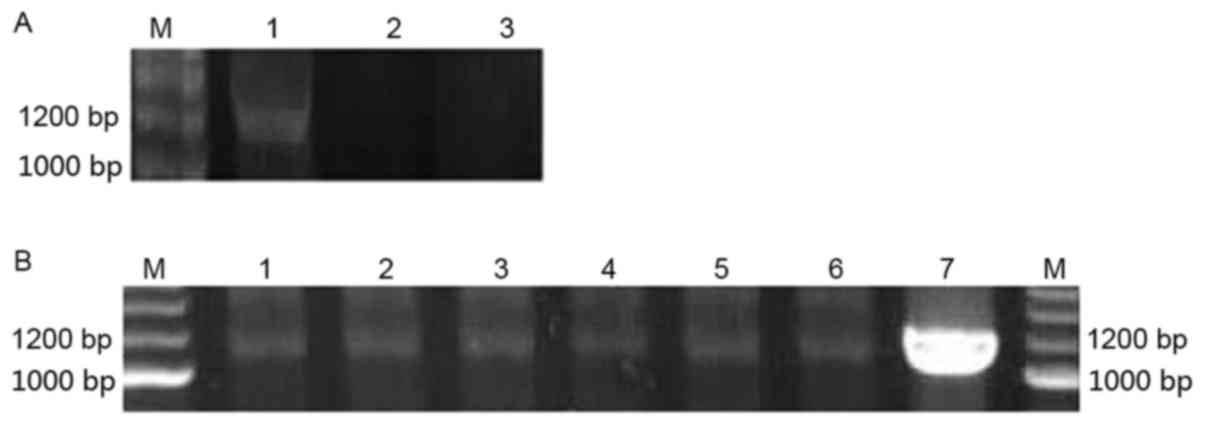 | Figure 3.p53α RNA expression levels evaluated
using RT-PCR. (A) p53α transient expression in H1299 cells at the
RNA level as determined by RT-PCR. M, marker; Lane 1,
H1299/pEGFP-p53α; Lane 2, H1299/pEGFP-N1; Lane 3, H1299.
(B) p53α stable expression in cells at RNA level by RT-PCR. M,
marker; Lanes, 1–6, H1299/pEGFP-p53α; Lane 7, A549. bp, base pairs;
RT-PCR, reverse transcription-polymerase chain reaction; p53, tumor
protein p53. |
Effects of p53α gene on
chemosensitivity
MTT assay results
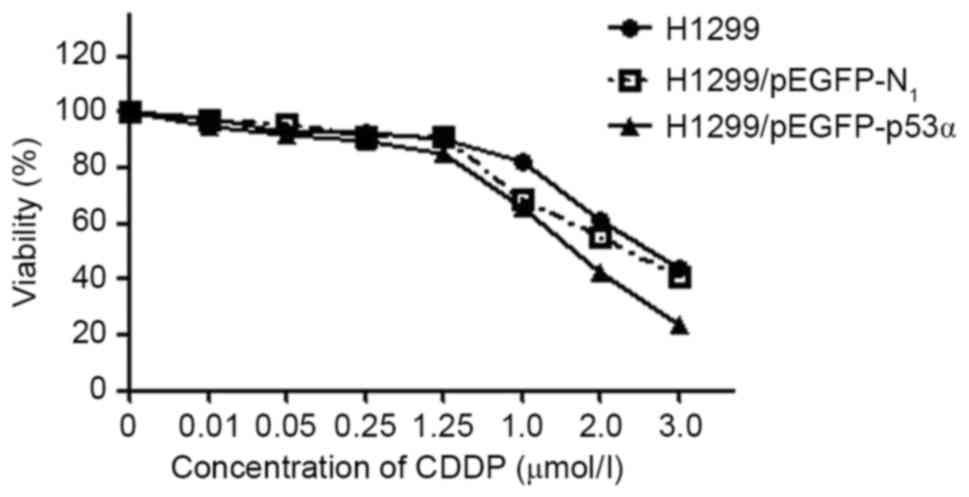
IC50 values in comparison with the
untreated control were estimated for the H1299 and
H1299/pEGFP-N1 control groups and the H1299/pEGFP-p53α
test group cells by deriving dose-response curves from the
percentage of cells that survived treatment, as obtained from the
cell counts (Fig. 6). The values for
H1299, H1299/pEGFP-N1 and H1299/pEGFP-p53α cells were
28, 24 and 18 µmol/l, respectively. Although the IC50
values did not differ significantly between the H1299 and
H1299/pEGFP-N1 cells (P>0.05), the value for the
H1299/pEGFP-p53α cells was significantly reduced compared with
those of H1299 and H1299/pEGFP-N1 cells (P<0.05).
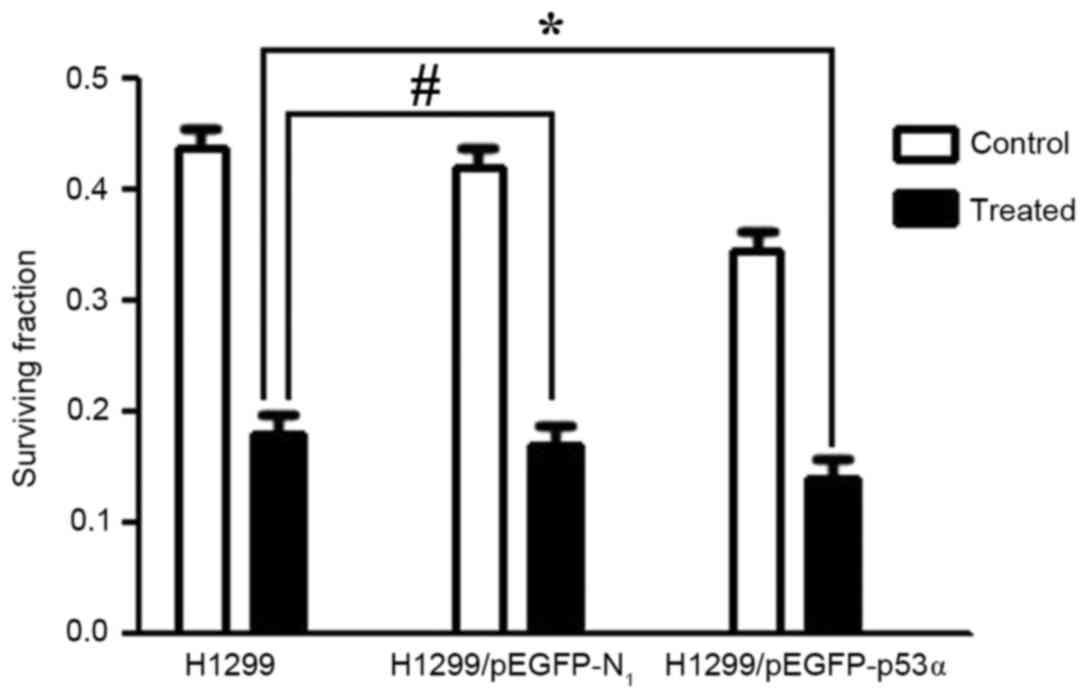
Clone formation assay results
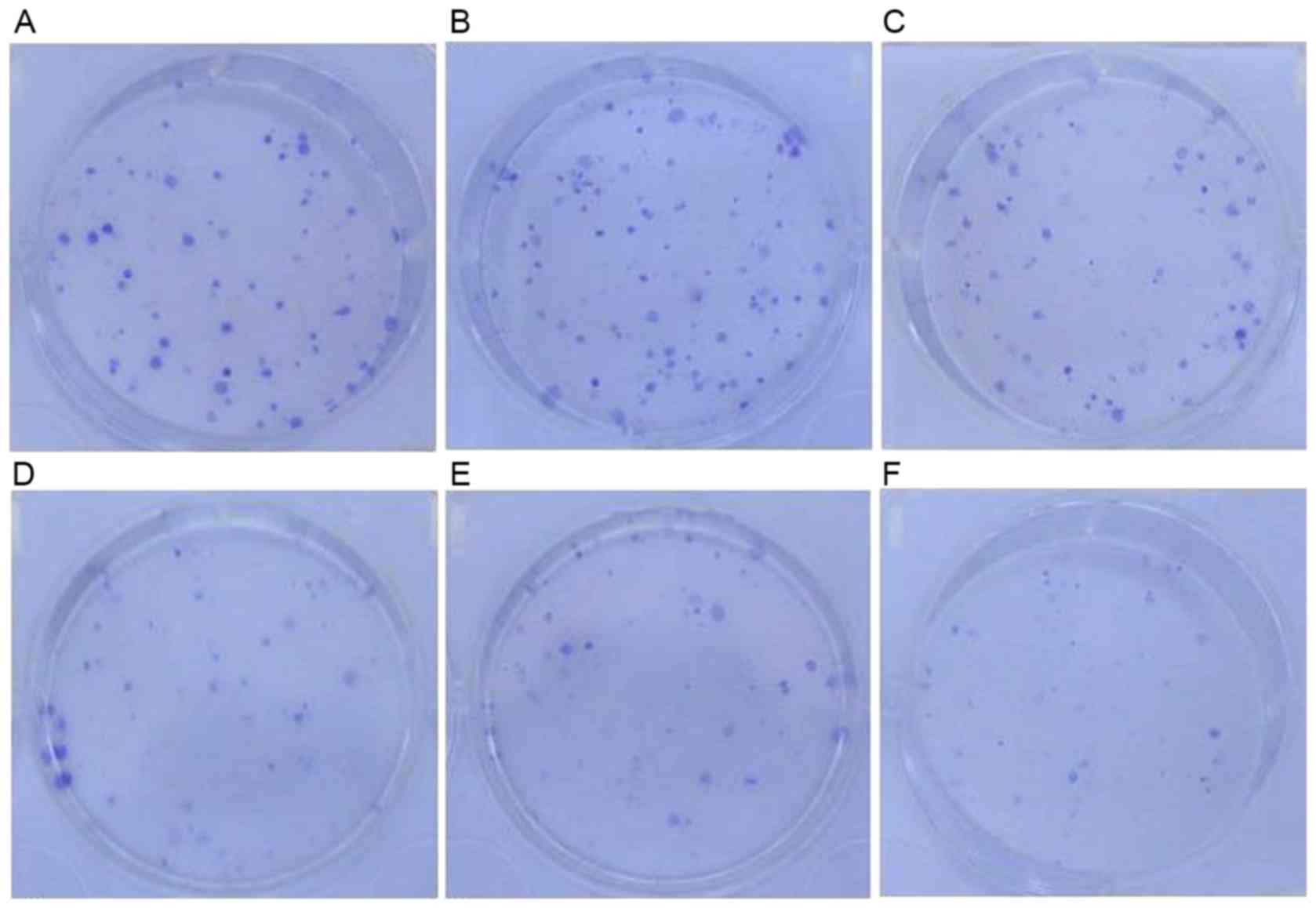
The survival fractions [calculated as follows:
Survival fraction (%) = number of colonies/200 cells] of control
groups prior to CDDP treatment were as follows: H1299, 43.6±0.23%;
H1299/pEGFP-N1, 41.8±0.13%. There was no significant
difference between these two groups. Following treatment with 0.25
µmol/l CDDP, the survival fractions in the H1299 and
H1299/pEGFP-N1 groups were 18.4±0.10 and 17.8±0.01%,
respectively, and were not significantly different (P>0.05).
However, the survival fraction in the H1299/pEGFP-p53α group
following CDDP treatment was 13.8±0.01%. When compared with the
control group H1299 (18.4±0.10%), there was a significant
difference in colony-formation rate (P<0.05; Figs. 7 and 8).
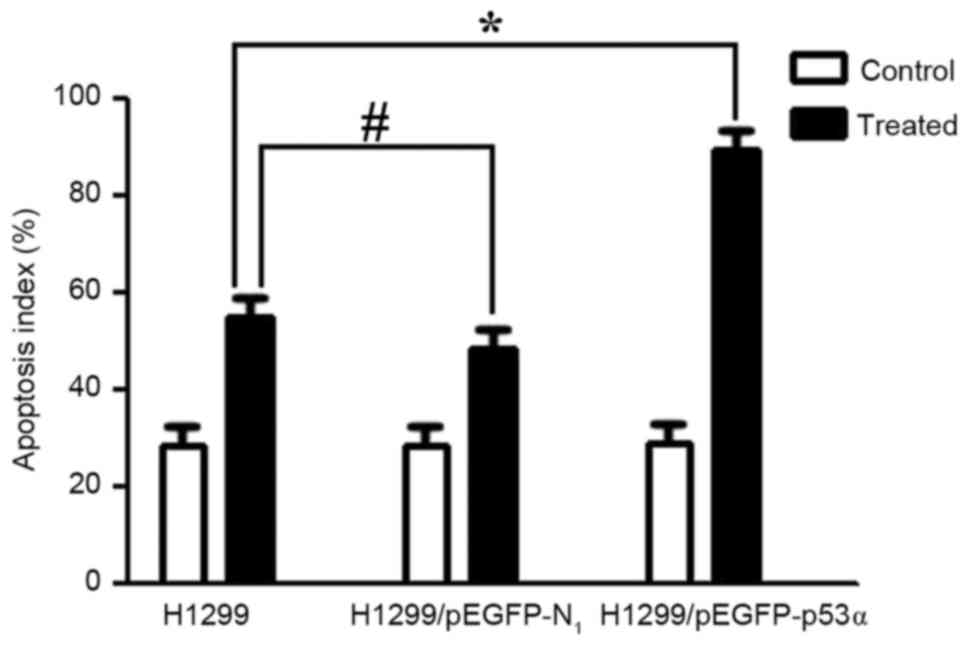
Cell apoptosis results
The apoptotic cells indicated by DAPI staining are
illustrated in Fig. 9. Following
treatment with CDDP for 48 h, the cells in the control group and
the H1299/pEGFP-p53α group appeared to reduce in size, and the
shapes of the cellular nuclei were irregular. There was no
significant difference in the mean number of apoptotic cells
between the H1299 (55.0±0.02) and the H1299/pEGFP-N1
groups (48.5±0.08). The highest mean number of apoptotic cells was
observed following CDDP therapy in the H1299/pEGFP-p53α group
(89.4±0.08), in which P<0.05 compared with the H1299 control
group (Fig. 10).
Discussion
p53 is associated with lung carcinogenesis and
treatment. In total ~40% of patients with NSCLC are candidates for
systemic chemotherapy, despite the majority of patients with NSCLC
being typically resistant to chemotherapy at the time of diagnosis
(19). Although chemical agents,
including CDDP, doxorubicin and gemcitabine, damage the DNA of
tumor cells and consequently activate p53, which leads to cell
death, a variety of p53 gene mutations may present an obstacle to
conventional chemotherapy. p53 gene mutations are typically caused
by alterations of a single amino acid, which results in the
expression of mutant proteins with gain-of-function activity. p53
protein loses its ‘guardian of the genome’ function when mutated,
preventing it from arresting cell proliferation or inducing
apoptosis (10,13). Therefore, maintaining the normal
function of p53 gene is essential for effective lung cancer
treatment.
In the present study, the full-length p53 gene was
successfully transfected into the H1299 cell line
(p53−/−; Figs. 1 and
2). The expression of transfected
p53α by H1299 cells was corroborated at the mRNA and protein levels
(Figs. 3–5). These results demonstrated that p53α gene
transfection inhibits the growth of H1299 cells. The
IC50 of CDDP decreased following p53α gene transfection
suggesting that p53 and the concentration of CDDP had synergistic
effects on inhibiting cell growth (Fig.
6). The p53α gene transfection also inhibited the colony
formation of H1299 cells (Fig. 7 and
8). The results of the current study
suggest that p53α cooperates with DNA-damaging drugs in inducing
apoptosis in H1299 cells (Figs. 9 and
10). Therefore, p53 gene
transfection may allow a reduction in the dose of chemotherapeutic
agents and minimize harmful side effects.
Various current strategies have been employed to
identify compounds that activate wild-type p53 or restore wild-type
function to mutated p53 (20).
However, there are certain obstacles for clinical translation
(21). For example,
adenovirus-mediated p53 (Ad-p53) is now commercially available as
Gendicine from Shenzhen SiBiono GeneTech Company, Ltd. (Shenzhen,
China) and is currently approved for clinical use in China
(6,22). However, Ad-p53 has not been approved
for use in Europe and the US due to its toxicity (23). In order to reduce the toxicity of
adenovirus-mediated transfection and maintain the effects of p53,
the current study investigated liposome-mediated p53α gene
transfection. Non-viral mediated gene therapy has the advantages of
safety and convenience. Combined therapy of agents with differing
mechanisms of action is feasible and effective in minimizing side
effects and avoiding resistance to chemotherapeutic drugs, whilst
still promoting the antitumor benefits, which may lead to novel
treatment regimes for NSCLC (24).
In conclusion, the transfection of H1299 cells with
the p53α gene resulted in an increase in sensitivity to CDDP
chemotherapy. The present study used the combination of p53 gene
therapy and chemotherapy in lung cancer cells and has provided an
experimental basis for future clinical trials. Further
chemotherapeutic drugs and experiments in vivo are required
to confirm the results of the present study.
Acknowledgements
Data published in this article are part of the
medical thesis of Ms. Weisong Gao.
References
|
1
|
Shao C, Lu C, Chen L, Koty PP, Cobos E and
Gao W: p53-dependent anticancer effects of leptomycin B on lung
adenocarcinoma. Cancer Chemother Pharmacol. 67:1369–1380. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Monroy-Estrada HI, Chirino YI,
Soria-Mercado IE and Sánchez-Rodríguez J: Toxins from the Caribbean
sea anemone Bunodeopsis globulifera increase cisplatin-induced
cytotoxicity of lung adenocarcinoma cells. J Venom Anim Toxins Incl
Trop Dis. 19:122013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ning X, Sun Z, Wang Y, Zhou J, Chen S,
Chen D and Zhang H: Docetaxel plus trans-tracheal injection of
adenoviral-mediated p53 versus docetaxel alone in patients with
previously treated non-small-cell lung cancer. Cancer Gene Ther.
18:444–449. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Taweel N, Varghese E, Florea A and
Büsselberg D: Cisplatin (CDDP) triggers cell death of MCF-7 cells
following disruption of intracellular calcium ([Ca (2+)]i)
homeostasis. J Toxicol Sci. 39:765–774. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saha MN, Qiu L and Chang H: Targeting p53
by small molecules in hematological malignancies. J Hematol Oncol.
6:232013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guan YS, Liu Y, Zou Q, He Q, La Z, Yang L
and Hu Y: Adenovirus-mediated wild-type p53 gene transfer in
combination with bronchial arterial infusion for treatment of
advanced non-small-cell lung cancer, one year follow-up. J Zhejiang
Univ Sci B. 10:331–340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma JT, Han CB, Zhao JZ, Jing W, Zhou Y,
Huang LT and Zou HW: Synergistic cytotoxic effects of recombinant
human adenovirus p53 and radiation at various time points in A549
lung adenocarcinoma cells. Oncol Lett. 4:529–533. 2012.PubMed/NCBI
|
|
8
|
Pietsch EC, Sykes SM, McMahon SB and
Murphy ME: The p53 family and programmed cell death. Oncogene.
27:6507–6521. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Selivanova G: Therapeutic targeting of p53
by small molecules. Semin Cancer Biol. 20:46–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rousseau B, Jacquot C, Le Palabe J,
Malleter M, Tomasoni C, Boutard T, Sakanyan V and Roussakis C: TP53
transcription factor for the NEDD9/HEF1/Cas-L gene: Potential
targets in non-small cell lung cancer treatment. Sci Rep.
5:103562015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Lin XJ, Wang CP, Yan KK, Zhao LY,
An WX and Liu XD: Association between smoking and p53 mutation in
lung cancer: A meta-analysis. Clin Oncol (R Coll Radiol). 26:18–24.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao J, Lu Y and Shen HM: Targeting p53 as
a therapeutic strategy in sensitizing TRAIL-induced apoptosis in
cancer cells. Cancer Lett. 314:8–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vaughan CA, Frum R, Pearsall I, Singh S,
Windle B, Yeudall A, Deb SP and Deb S: Allele specific
gain-of-function activity of P53 mutants in lung cancer cells.
Biochem Biophys Res Commun. 428:6–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gibbons DL, Byers LA and Kurie JM:
Smoking, p53 mutation, and lung cancer. Mol Cancer Res. 12:3–13.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mattioni M, Soddu S, Prodosmo A, Visca P,
Conti S, Alessandrini G, Facciolo F and Strigari L: Prognostic role
of serum p53 antibodies in lung cancer. BMC cancer. 15:1482015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Machado-Silva A, Perrier S and Bourdon JC:
p53 family members in cancer diagnosis and treatment. Semin Cancer
Biol. 20:57–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mogi A and Kuwano H: TP53 mutations in
nonsmall cell lung cancer. J Biomed Biotechnol. 2011:5839292011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vassilev LT, Vu BT, Graves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et
al: In vivo activation of the p53 pathway by small-molecule
antagonists of MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu X, Narayanan S, Vazquez A and Carpizo
DR: Small molecule compounds targeting the p53 pathway: Are we
finally making progress? Apoptosis. 19:1055–1068. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu M, Chen W and Zhang J: p53 gene therapy
for pulmonary metastasis tumor from hepatocellular carcinoma.
Anticancer Drugs. 21:882–884. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duffy MJ, Synnott NC, McGowan PM, Crown J,
O'Connor D and Gallagher WM: p53 as a target for the treatment of
cancer. Cancer Treat Rev. 40:1153–1160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Zhang Q, Zeng SX, Hao Q and Lu H:
Inauhzin sensitizes p53-dependent cytotoxicity and tumor
suppression of chemotherapeutic agents. Neoplasia. 15:523–534.
2013. View Article : Google Scholar : PubMed/NCBI
|