Introduction
Mammalian cells are exposed to numerous sources of
DNA damage, and have therefore evolved DNA damage response
signaling pathways to monitor genome integrity (1–3). In the
last 30 years, a number of studies investigated the molecular
mechanisms underlying the DNA damage response, and previous studies
have revealed complex and profound changes in genome integrity and
gene expression levels (4–6). More than 180 genes have been
demonstrated to be directly or indirectly involved in the
biological processes underlying DNA repair. Individuals with
inherited deficiencies in the three major components involved in
the DNA damage response, sensors, signal transducers and effectors
have been identified (7,8). These components function by detecting
several forms of DNA damage and triggering DNA damage response
cascades (9). Examples of core
sensors include the ataxia telangiectasia mutated (ATM) (10), ATM-Rad3-related (11) and DNA-dependent protein kinases, which
are crucial to the DNA damage response signaling pathway (12,13).
Signal transducers include the tumor protein p53, which has
numerous roles in various DNA repair signaling pathways (14). Effectors that respond to the cellular
stress of DNA damage include cyclin-dependent kinase inhibitor 1A
(15), growth arrest and
DNA-damage-inducible α, p53 dependent G2 arrest mediator
candidate and damage-specific DNA binding protein 2, all of which
are regulated by p53 (16,17). Among the 180 genes involved in several
repair signaling pathways, numerous genes are regulated by
epigenetic mechanisms and are frequently downregulated or silenced
in various types of cancer (18).
Defects in the DNA damage response frequently occur in types of
human cancer and further elucidation of this process may facilitate
the development of effective personalized cancer therapy (19–25).
In the present study, changes in the expression
levels of DNA damage repair genes were determined using gene
expression profile analysis through second-generation sequencing in
cluster of differentiation (CD) 133+/CD133−
colorectal cancer cells (26,27). The current study, therefore, aimed to
enhance the understanding of the complex gene regulation network in
cancer cells and to identify novel candidate genes for cancer
therapy.
Materials and methods
Cell lines, animal models and
reagents
The COLO 205 human colorectal cancer cell line was
obtained from the Cell Resource Center of the Shanghai Institutes
for Biological Sciences, Chinese Academy of Sciences (Shanghai,
China). BALB/c nude mice were obtained from the Laboratory Animal
Center of Daping Hospital of the Third Military Medical University
(Chongqing, China) and kept in a specific-pathogen-free animal
facility. A total of twelve 5-week-old, 18–22 g
specific-pathogen-free male Balb/c nude mice were purchased from
the Laboratory Animal Center of Daping Hospital of the Third
Military Medical University (Chongqing, China) and maintained in a
pathogen-free animal facility. The study protocol was approved by
Animal ethics committee of Zunyi Medical University (Zunyi, China).
The present study followed guidelines of the Zunyi Medical
University for Biomodel Organisms during this study.
A human CD133 MicroBead Kit (cat. no., 130-050-801)
was purchased from Miltenyi Biotec GmbH (Bergisch Gladbach,
Germany). B27 supplement, cell factors including recombinant human
alkaline fibroblast growth factor (rh-AFGF), recombinant human
epidermal cell growth factor (rh-ECGF), and recombinant human
leukemia inhibitory factor (rh-LIF) and kits for RNA isolation,
purification and quantification were all purchased from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Fetal bovine
serum, media (Dulbecco's modified Eagle's medium/nutrient mixture
F-12, DMEM/F-12; RPMI-1640) and trypsin were purchased from Gibco
(Thermo Fisher Scientific, Inc.). Dimethyl sulfoxide and all other
laboratory chemicals were purchased from Sigma-Aldrich (Merck
Millipore, Darmstadt, Germany). The kits for sequencing library
construction, Low Input Library Prep Kit HT (cat. no., 634900), DNA
Standards for Library Quantification (cat. no., 638325) and gene
expression profile analysis were obtained from Illumina, Inc. (San
Diego, CA, USA). Reagents for reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis were purchased from
Takara Biotechnology Co., Ltd. (Dalian, China). The primers used
for gene expression analysis (Table
I) were synthesized by Invitrogen (Thermo Fisher Scientific,
Inc.) and preserved in Zunyi Key Laboratory of Genetic Diagnosis
& Targeted Drug Therapy.
 | Table I.Primers for RT-qPCR. |
Table I.
Primers for RT-qPCR.
| Primer | Primer sequence,
5′-3′ | Gene name |
|---|
| ActB F |
CAGCAAGCAGGAGTATGACGAGT | β-actin |
| ActB R |
GCTGTCACCTTCACCGTTCC | β-actin |
| BCL-11A F |
GACAGGGTGCTGCGGTTGA | BCL11A |
| BCL-11A R |
GGCTTGCTACCTGGCTGGAA | BCL11A |
| Zf1 F |
GAGGAATGGGAACAACTGGACC | Rad 51 |
| Zf1 R |
ATTGCCTCGCCTGCTCGTC | Rad 51 |
| P53 F |
CCCTCCTCAGCATCTTATCCG | P53 |
| P53 R |
CAACCTCAGGCGGCTCATAG | P53 |
Cell culture and isolation
These processes were performed according to previous
protocol (28,29). The COLO 205 cells were maintained in
RPMI-1640 supplemented with 10% fetal bovine serum until the
logarithmic growth phase and were subsequently transferred to
serum-free medium (SFM; DMEM/F-12 supplemented with 2% B27
supplement, 2 ng/ml recombinant human alkaline fibroblast growth
factor, 2 ng/ml recombinant human epidermal cell growth factor, and
2 ng/ml recombinant human leukemia inhibitory factor) for 10 days
until cell spheres formed. Cell morphology was observed by inverted
fluorescence microscopy in order to detect cell adherence and
growth.
Single cells separated from spheres were collected
for CD133 cell isolation using a cell isolation kit (Miltenyi
Biotec Inc., Shanghai, China) according to the manufacturer's
protocol. Briefly, the single cell suspension was centrifuged at
800 × g for 10 min at room temperature, washed 3 times with PBS and
resuspended in 300 µl isolation buffer (Miltenyi Biotec Inc.,
Shanghai, China). Subsequently, 100 µl FcR blocking reagent and 100
µl CD133 immunomagnetic beads (Miltenyi Biotec Inc., Shanghai,
China) were added. The cell suspension was incubated at 4°C for 30
min. The cells were washed with 2 ml isolation buffer and
centrifuged at 800 × g for 10 min at room temperature followed by
resuspension in 500 µl isolation buffer. An MS column (Miltenyi
Biotec Inc., Shanghai, China) was washed with 500 µl isolation
buffer, to which the cells were added for isolation.
Cell differentiation and
tumorigenicity assay in nude mice
Upon isolation of the CD133+ and
CD133− cells, cell densities were adjusted to 5,000
cells/ml. For the differentiation assay, 1 ml isolated
CD133+ and CD133− cell suspensions were
inoculated in SFM and cultured at 37°C with 5% CO2 for
one week. Every two days, 2 ml SFM was added until differentiated
cells formed under fluorescence microscope. For the tumorigenicity
assay, the cells were divided into three groups including the
isolated CD133+ and CD133− cells and the
unisolated COLO 205 cells, each were diluted with normal saline to
a density of 1,000, 10,000, and 10,0000 cells/ml, respectively.
Subsequently, various numbers of cells (1,000, 10000, and 10,0000)
were injected into the axilla and the left groin of each nude mouse
by subcutaneous injection. For the control mice, 1 ml normal saline
was injected into the right groin. A total of 45 days subsequent to
injection with cells or normal saline, all the animals were
sacrificed via cervical dislocation.
RNA extraction, cDNA synthesis and
expression profile sequencing
In total, 20 µg RNA was extracted separately from
the isolated CD133+ and CD133− cells and the
unisolated COLO 205 cells using TRIzol® reagent (Thermo Fisher
Scientific, Inc.), and mRNA was purified from total RNA using the
Dynabeads® mRNA Purification kit, following the manufacturer's
protocol (Thermo Fisher Scientific, Inc.). For cDNA synthesis and
sequencing library construction, 5 µg RNA was processed using the
TruSeq™ RNA Sample Prep kit (Illumina, Inc., San Diego, CA, USA).
The library was constructed with an anchored oligo-dT primer, which
initiated cDNA synthesis and added a universal primer sequence
(TGCGAATT). The cDNA was polynucleotide tailed by the RT, producing
a 3′ overhanging tail. The cDNA was amplified by PCR with the
reverse complementary sequence of the 3′ overhanging tail and
quantified using a TBS-380 Picogreen kit (Illumina, Inc. San Diego,
CA, USA.). The library DNA was recovered using Certified Low Range
Ultra Agarose (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Briefly, DNA fragments were separated by agarose gel
electrophoresis and the band was cut out and placed in a dissolving
buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA and 200 mM KCl] with an
equal volume of phenol. Subsequently, the mixture was frozen and
thawed 3 times on dry ice. Aqueous supernatant was collected by
centrifugation at 10,000 × g for 5 min at room temperature. Then,
dilution buffer [100 mM Tris, 15% EtOH and 900 mM KCl (pH 6.3)] was
added to the final optimized concentration [33 mM Tris, 5% EtOH and
300 mM KCl (pH 6.3)] and the DNA was precipitated by adding of
isopropanol (final concentration: 40%, v/v), followed by
centrifugation at 10,000 × g for 15 min at 4°C. The pellet was
washed with 70% ethanol, air-dried and dissolved in an appropriate
buffer [10 mM Tris-HCl 1 mM EDTA (pH 7.6)] prior to further
experiments.
Bridge amplification was performed using a cBot
TruSeq PE Cluster kit v3-cBot-HS (Illumina, Inc., San Diego, CA,
USA) followed by 200 cycles of DNA sequencing with a HiSeq 2000
TruSeq SBS kit v3-HS (Illumina, Inc. San Diego, CA, USA).
Gene expression profile analysis. Subsequent to
sequencing, raw reads were trimmed by stripping the adaptor
sequences and ambiguous nucleotides using SeqPrep (v 1.2.1;
https://github.com/jstjohn/SeqPrep)
and Sickle (v 0.3; https://github.com/najoshi/sickle). Following
background subtraction and quantile normalization, the signal
intensity values from the three group cells were exported to TopHat
software (v 2.0.0-beta5; http://tophat.cbcb.umd.edu) for genome mapping,
respectively. Gene expression analysis was performed using the
Cuffdiff program (v 2.2.0) from the Cufflinks suite (http://cufflinks.cbcb.umd.edu) and differentially
expressed genes were identified using the NOISeq workflow (v
2.16.0) in R software. Gene ontology analysis was conducted using
goatools (v 0.6.10) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway analysis was performed using KEGG-Orthhology Based
Annotation System (KOBAS; v 2.1.1). For a gene to be classified as
differentially expressed, Statistical significance of the gene
expression model was evaluated with an ANOVA (analysis of variance)
test that comparatively evaluated the variance in isolated
CD133+ and CD133− cells as well as the
unisolated COLO 205 cells of gene expression. Differential
expression was assessed using moderated t-statistics and false
discovery rate <5%. q<0.05 was considered to indicate a
statistically significant difference, whilst fold-changes were
required to be >2 or <0.5.
RT-qPCR
To evaluate the differentially expressed genes
observed during expression profile sequencing analysis, 11 genes
were analyzed by real-time RT-qPCR using an MyiQ™ 2 Two-Color
Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The β-actin gene (accession number
NM_0011101.3; National Center for Biotechnology Information) was
used as the endogenous control. The primers (Table I) were designed using Primer Premier
Software version 5.0 (Premier Biosoft International, Palo Alto, CA,
USA). Total RNA (2.5 µg) was used as a template for cDNA synthesis
in 50 µl RT reactions using the SuperScript® IV Reverse
Transcriptase kit (Thermo Fisher Scientific, Inc.). In total, 1 µl
cDNA was added into SYBR Green Real-Time PCR Master Mixes (Thermo
Fisher Scientific, Inc.) for 25 µl PCR reactions. The following
thermal profile was used for RT-qPCR amplification: initial
denaturation for 3 min at 95°C, followed by 40 cycles of
denaturation for 10 sec at 95°C, annealing for 30 sec at 58°C and a
thermal denaturing step from 55 to 95°C (duration, 5 sec/0.5°C) to
generate melt curves for the determination of amplification
specificity. The data were statistically analyzed using the
2−ΔΔCq method (30).
Results
Identification of cancer stem
cells
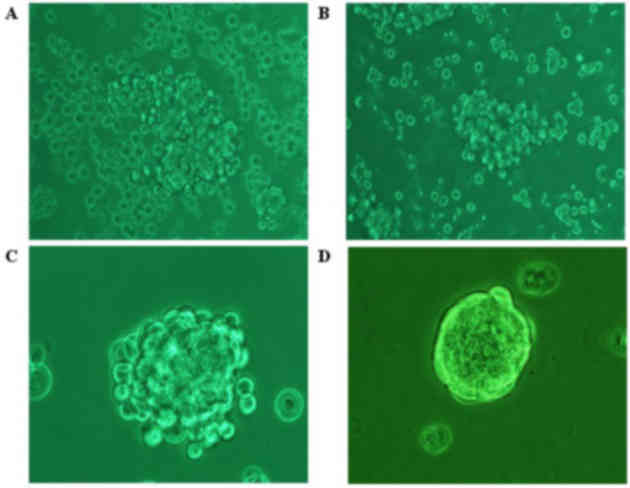
Following incubation with SFM for 1 day, the COLO
205 cells showed characteristics of low cell adherence and growth
(Fig. 1A). The cells in the
suspension culture demonstrated logarithmic growth and
significantly increased (P=0.03) numbers of floating spheres after
5 days (Fig. 1B). After 7 days, the
floating spheres began to collapse, with a small number of
regular-shaped cells remaining strongly attached to one another
(Fig. 1C). The surface of floating
spheres appeared smoother and denser as the cell generation number
increased (Fig. 1D).
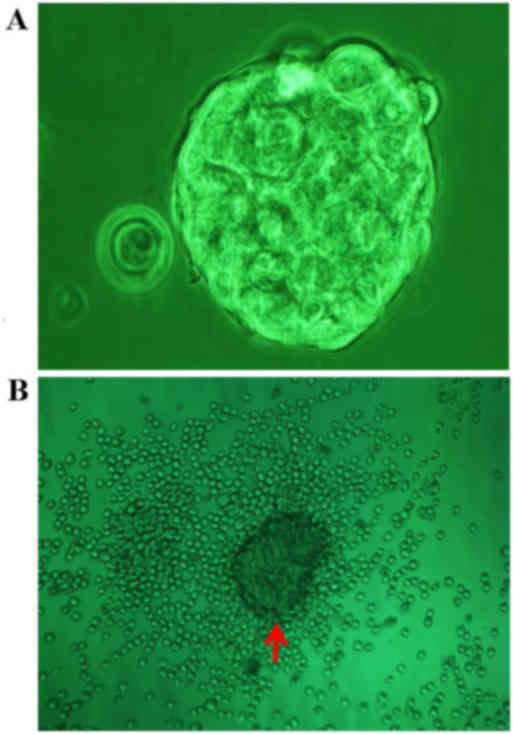
Following isolation with immunomagnetic beads and
culture in SFM for 1 day, the CD133+ cells exhibited
suspended growth, and grouped to form typical tumor spheres after 4
days, whereas spheres were absent from the CD133− cell
cultures (Fig. 2A). Once fetal calf
serum was added to the SFM, all the spheres derived from the
CD133+ cells adhered within 1 day and numerous
differentiated cells with the same shape as the COLO 205 cells
emerged in the region surrounding the spheres after three days
(Fig. 2B).
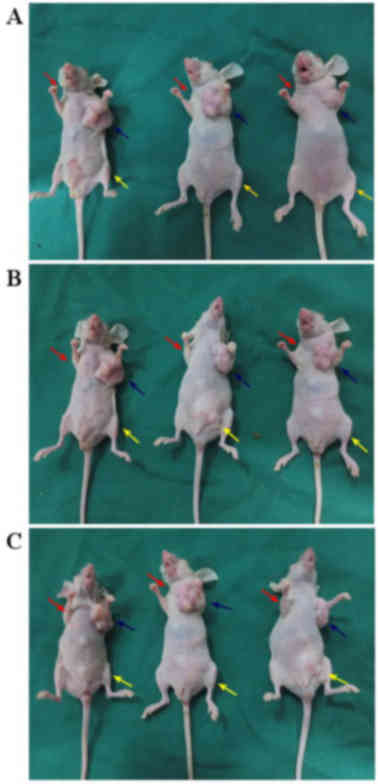
Following injection with cancer cells or normal
saline, the tumor growth of the nude mice was observed and recorded
daily (Fig. 3). No visible tumors
were observed in the nude mice injected with the CD133−
cells (Fig. 3A-a, B-a and C-a) or
normal saline. In the group injected with CD133+ cells,
visible tumors were observed in the left axilla of all nude mice at
15, 20 and 30 days post-injection with 1,000 (Fig. 3A-b), 10,000 (Fig. 3B-b) and 100,000 cells (Fig. 3C-b). In the group injected with the
COLO 205 cells, only two visible tumors were observed in the left
groin of the nine nude mice 35 days post-injection with 10,000
cells (Fig. 3B-c and. C-c).
Therefore, the tumorigenicity of the CD133+ cells is
significantly higher, compared with the CD133− cells
(P<0.01) or COLO 205 cells (P<0.01), and no significant
difference in tumorigenicity was observed between the
CD133− cells and the COLO 205 cells (P=0.47).
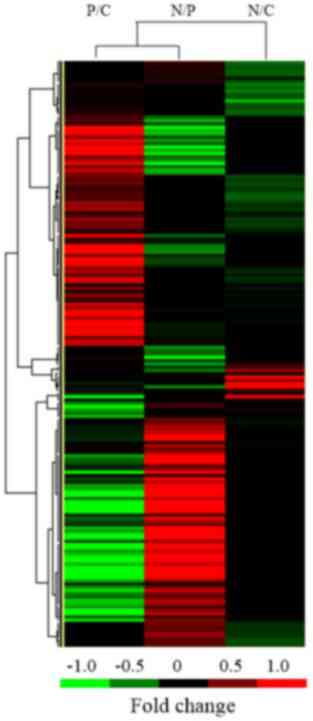
Gene expression profiling of DNA
damage repair genes
To detect changes in the mRNA expression levels of
specific DNA damage repair genes in the colon cancer cells, data
for the 180 genes involved in DNA repair were extracted from the
results of expression profile sequencing of the isolated
CD133+, CD133− and COLO 205 colon cancer
cells (Fig. 4).
Of these 43 genes which mRNA expression levels were
altered, 30 (8 upregulated and 22 downregulated) were
differentially expressed in the COLO 205 cells, as compared with
the CD133− cells, and 6 genes (all downregulated) were
differentially expressed in the COLO 205 cells, as compared with
the CD133+ cells. A total of 18 genes (10 upregulated
and 8 downregulated) were differentially expressed in the
CD133− cells, compared with the CD133+ cells.
By contrast, only six genes were downregulated and none were
upregulated in the CD133+ cells, as compared with the
COLO 205 cells. Differential expression of DNA damage repair
genes. The results of KEGG pathway analysis revealed 14
signaling pathways that were implicated among the 40 DNA damage
repair genes that were observed to be differentially expressed
during the present study (Table
II).
| Table II.Expression levels of differentially
expressed genes. |
Table II.
Expression levels of differentially
expressed genes.
|
| Fold-change |
|
|
|---|
|
|
|
|
|
|---|
| Gene | Pe/Ce | Ne/Pe | Ne/Ce | Accession no. | Signaling pathway
or function |
|---|
| UNG | 0.763 | 0.470, |
0.359,
0.19 | NM_080911 | Base excision
repair and break joining factors |
| NTHL1 | 1.576, | 0.428, | 0.675, | NM_002528 | Base excision
repair and break joining factors |
| MPG | 0.840, | 0.553, | 0.464, | NM_002434 | Base excision
repair and break joining factors |
| NEIL3 | 0.604, | 0.554, | 0.335, | NM_018248 | Base excision
repair and break joining factors |
| APEX1 | 0.494, | 0.600, | 0.297, | NM_001641 | Base excision
repair and break joining factors |
| PARP3 | 0.803, | 0.447, | 0.359, | NM_001003931 | Base excision
repair and break joining factors |
| MGMT | 0.875, | 0.490, | 0.428, | NM_002412 | Direct reversal of
damage/DNA-topoisomerase crosslinks |
|
|
|
|
|
| DNA-topoisomerase
crosslinks DNA-topoisomerase crosslinks/DNA-topoisomerase
crosslinks |
| ALKBH3 | 0.562, | 0.529, | 0.298, | NM_139178 | Direct reversal of
damage/DNA-topoisomerase crosslinks |
| PMS2L3 | 0.508, | 2.305, | 1.170, | NM_005395 | Mismatch excision
repair genes |
| RPA1 | 0.666, | 0.744, | 0.496, | NM_002945 | NER associated
genes |
| RPA2 | 0.783, | 0.595, | 0.465, | NM_002946 | NER associated
genes |
| GTF2H4 | 0.812, | 0.584, | 0.474, | NM_001517 | NER associated
genes |
| MNAT1 | 0.677, | 0.529, | 0.358, | NM_002431 | NER associated
genes |
| ERCC4 | 1.338, | 1.835, | 2.455, | NM_005236 | NER associated
genes |
| DMC1 | 1.927, | 1.921, | 3.702, | NM_007068 | HR genes |
| SHFM1 | 1.095, | 0.409, | 0.448, | NM_006304 | HR genes |
| MRE11A | 0.269, | 1.317, | 0.355, | NM_005590 | HR genes |
| GEN1 | 0.448, | 2.649, | 1.187, | NM_001014999 | HR genes |
| BRIP1 | 0.396, | 2.075, | 0.822, | NM_032043 | Genes involved in
Fanconi anemia |
| FAAP20 | 1.318 | 0.410 | 0.540 | NM_182533.2 | Genes involved in
Fanconi anemia |
| FAAP24 | 0.747 | 0.627 | 0.469 | NM_152266 | Genes involved in
Fanconi anemia |
| XRCC6 | 0.591 | 0.778 | 0.460 | NM_001469 | Non-homologous
end-joining genes |
| NUDT1 | 1.037 | 0.399 | 0.414 | NM_002452 | Genes involved in
modulation of nucleotide pools |
| RRM2B | 0.470 | 2.019 | 0.948 | NM_015713 | Genes involved in
modulation of nucleotide pools |
| POLN | 1.471 | 1.531 | 2.251 | NM_181808 | DNA polymerases
(cofactor or catalytic subunits) |
| FEN1 | 0.589 | 0.849 | 0.500 | NM_004111 | Editing and
processing nucleases |
| TREX2 | 0.758 | 0.473 | 0.358 | NM_007205 | Editing and
processing nucleases |
| RAD18 | 0.593 | 2.696 | 1.598 | NM_020165 | Genes involved in
ubiquitination and modification |
| HLTF | 1.987 | 1.740 | 3.457 | NM_003071 | Genes involved in
ubiquitination and modification |
| H2AFX | 0.971 | 0.508 | 0.493 | NM_002105 | Genes involved in
chromatin structure and modification |
| SETMAR | 0.778 | 0.631 | 0.491 | NM_006515 | Genes involved in
chromatin structure and modification |
| BLM | 0.637 | 2.037 | 1.296 | NM_000057 | Diseases associated
with sensitivity to DNA damaging agents |
| RPA4 | 0.766 | 0.557 | 0.426 | NM_013347 | Identified genes
with DNA repair functions |
| PRPF19 | 0.451 | 1.637 | 0.739 | NM_014502 | Identified genes
with DNA repair functions |
| RDM1 | 1.546 | 0.656 | 0.424 | NM_145654 | Identified genes
with DNA repair functions |
| NABP2 | 0.492 | 0.567 | 0.279 | NM_024068 | Identified genes
with DNA repair functions |
| ATR | 1.392 | 1.768 | 2.462 | NM_001184 | Conserved DNA
damage response genes |
| MDC1 | 0.618 | 6.529 | 4.035 | NM_014641 | Conserved DNA
damage response genes |
| TP53 | 1.087 | 0.424 | 0.461 | NM_000546 | Conserved DNA
damage response genes |
| RIF1 |
0.601
0.0000 | 2.072 | 1.245 | NM_001177665 | Conserved DNA
damage response genes |
Expression analysis of differential
genes
Changes in the mRNA expression levels of three
genes, the conserved DNA damage response gene TP53, B-cell
lymphoma/leukemia 11A (BCL-11a) and zinc-finger protein 1 (ZF1), in
various cells were compared in order to examine the reliability of
the results obtained from expression profile sequencing. The data
were concordant between the results of the two methods of
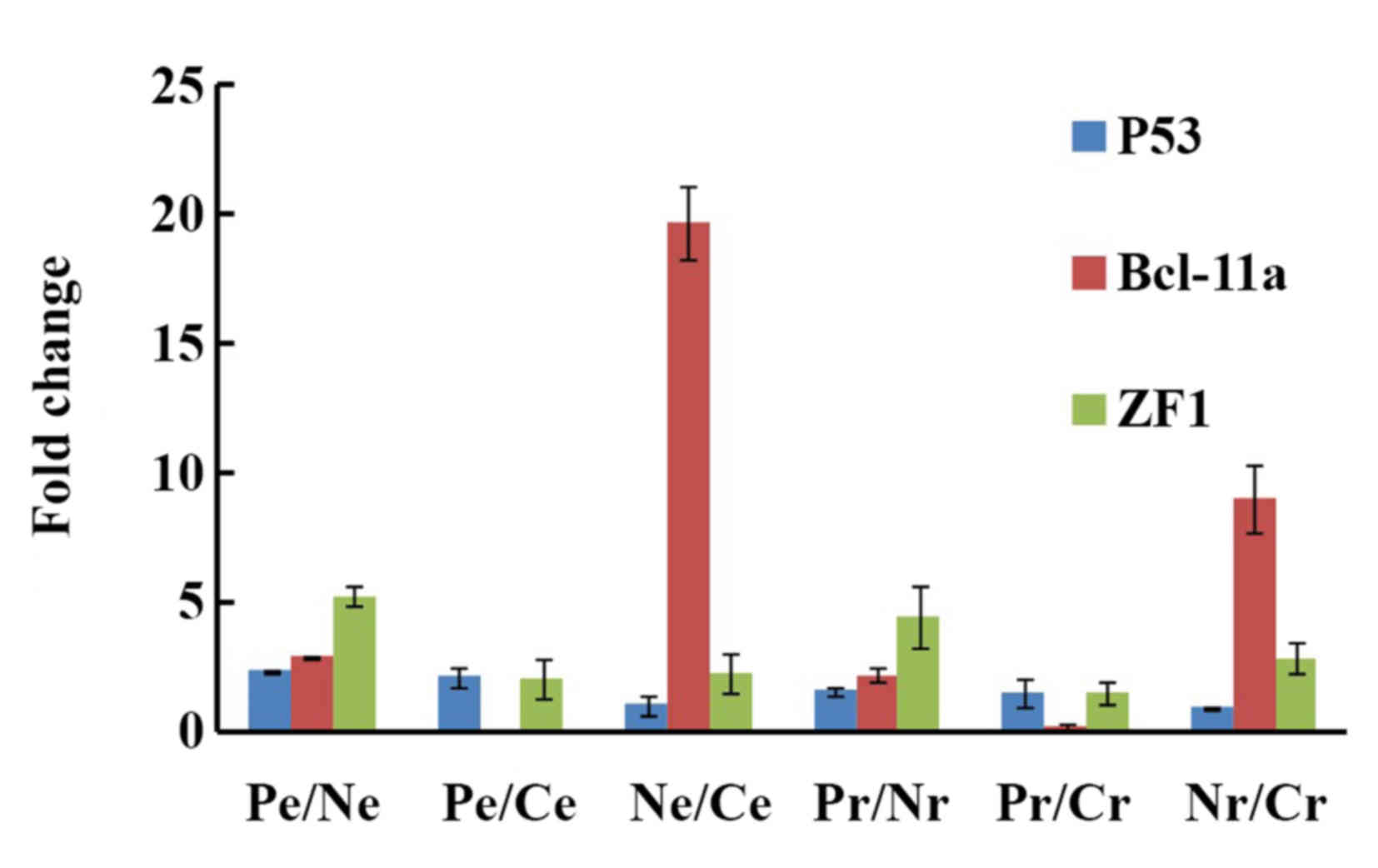
expression profile sequencing and RT-qPCR (Fig. 5). The change in the p53 mRNA
expression levels in the CD133+/CD133−,
CD133+/COLO 205 and CD133−/COLO 205 cells was
2.37, 2.17 and 1.09 folds, respectively, from expression profile
sequencing and 1.61, 1.60 and 1.00 fold, respectively, from
RT-qPCR. Similar trends in expression levels were observed in the
CD133+/CD133−, CD133+/COLO 20 and
CD133−/COLO 205 cells for BCL-11a (expression profile
sequencing, 2.95, 0.15 and 19.67 fold; RT-qPCR, 2.25, 0.25 and 9
fold, respectively) and ZF1 (expression profile sequencing, 5.27,
2.12 and 2.36 fold; RT-qPCR, 4.53, 1.58 and 2.87 fold,
respectively).
Discussion
Of the 12 genes involved in base excision repair and
break joining, no variation in gene expression levels was observed
in the CD133− cells, as compared with the
CD133+ cells; however, the expression levels of four
genes were downregulated in the CD133− cells, compared
with the COLO 205 cells, and the expression levels of three genes
were downregulated in the CD133− cells, compared with
the CD133+ cells. Of these genes, uracil-DNA glycosylase
and poly (adenosine diphosphate-ribose) polymerase 3 were
downregulated in the COLO 205 cells and the CD133+
cells. In CD133− cells, nth endonuclease III-like 1,
N-methylpurine DNA glycosylase, nei endonuclease VIII-like 1 and
apurinic/apyrimidinic endodeoxyribonuclease 1 were downregulated,
compared with the CD133+ cells. As those genes that were
downregulated in expression are all involved in base excision
repair (31) or break joining
(32), their downregulation may have
negative effects on the base excision repair capacity towards
damaged DNA in CD133− cells.
A total of five human genes are involved in the
direct reversal of DNA damage (33),
including two tyrosyl-DNA phosphodiesterases, two
alpha-ketoglutarate-dependent dioxygenases, and an
O6-methylguanine-DNA methyltransferase (MGMT). No
changes in the expression levels of these five genes were observed
in the CD133+ cells, compared with the COLO 205 cells.
By contrast, MGMT gene expression was upregulated in the
COLO 205 cells, compared with the CD133− cells.
Similarly, alkylation repair homolog 3 expression was downregulated
in the CD133− cells compared with the COLO 205
cells.
By contrast, no marked changes in the expression
levels of the ten genes involved in the mismatch excision repair
signaling pathway were detected in all three types of cells, with
the exception of postmeiotic segregation increased 2-like protein
3, which was upregulated in the CD133− cells, compared
with the CD133+ cells.
Nucleotide excision repair (NER) is a complex
biological process, involving more than 29 genes that are able to
correct DNA damage through nuclease cleavage of the damaged base,
removal of the damaged oligonuclotide and resynthesis using the
undamaged strand as the template (34). No significant changes in gene
expression levels were observed in the CD133+ cells
compared with the COLO 205 cells and the CD133− cells.
By contrast, five genes were differentially expressed, with the
expression of four genes downregulated and upregulated for one gene
in the CD133− cells, compared with COLO 205 cells.
Replication protein A1 (RPA1) and replication
protein A2 (RPA2) are components of the alternative RPA complex,
which is essential for the binding and stabilizing of
single-stranded DNA intermediates, preventing the reannealing of
complementary DNA (35). Therefore,
downregulation of the expression of these two genes may reduce the
NER capacity of CD133− cells. As components of the seven
subunits, as well as kinase subunits of general transcription
factor IIH, downregulation of general transcription factor IIH
subunit 4 and CDK-activating kinase assembly factor MAT1
expression levels may also reduce the NER capacity (36) of CD133− cells. As excision
repair cross-complementing rodent repair deficiency complementation
group 4 (ERCC4) forms a complex with ERCC1 and is involved in the
5′ incision made during NER (37),
upregulating ERCC4 expression may affect the nucleotide
excision repair capacity of CD133− cells.
More than 19 genes are involved in homologous
recombination repair (38) and, in
the present study, only 4 of these genes were differentially
expressed. As a component of the Mre11-Rad50-Nbs1 (MRN) complex,
meiotic recombination 11 homolog A (MRE11A) exhibits single-strand
endonuclease activity and double-strand 3′-5′ exonuclease activity
specific to the MRN complex, which is critical to the processes of
double-strand break (DSB) repair, DNA recombination, maintenance of
telomere integrity and meiosis (39).
The expression levels of the MRE11A gene were downregulated
in the CD133+ cells and the CD133− cells, as
compared with the COLO 205 cells. The Holliday junction 5′ flap
endonuclease, which possesses Holliday junction resolvase activity
in vitro and is considered to function in homology-driven
repair of DNA DSBs (40), exhibited
upregulated expression levels in the CD133− and COLO 205
cells, as compared with the CD133+ cells. Upregulated
expression of the BRCA2-associated split hand/foot malformation
(ectrodactyly) type 1 gene was observed in the CD133−
cells, compared with the CD133+ and COLO 205 cells. DNA
meiotic recombinase 1, which assembles at the sites of programmed
DNA DSBs and searching for allelic DNA sequences located on
homologous chromatids during homologous recombination in the
process of meiosis, exhibited upregulated expression in the
CD133− cells, compared with the COLO 205 cells.
Of the 17 genes involved in Fanconi anemia, only
three genes exhibited changes in expression levels during the
present study. These were as follows: Breast cancer 1
(BRCA1)-interacting protein 1 (BRIP1), involved in the repair of
DNA DSBs by BRCA1-dependent homologous recombination (41); the 20 kDa Fanconi anemia-associated
protein (FAAP20), a component of the Fanconi anemia complex that is
required to recruit the complex to DNA interstrand cross-links to
promote repair; the 24 kDa Fanconi anemia-associated protein
(FAAP24), which regulates Fanconi anemia group D2 protein
monoubiquitination upon DNA damage (42). Seven genes have been reported to be
involved in non-homologous end-joining (43), among them, X-ray repair cross
complementing 6 (XRCC6) (44), the 70
kDa subunit of the single-stranded DNA-dependent ATP-dependent
helicase II was the only one which gene expression level was
changed in CD133+ cells compared with CD133−
cells and COLO 205 cells. Downregulation of XRCC6 expression levels
(2.173 folds) in CD133− cells, compared with COLO 205
cells, may have a negative effect on the non-homologous end joining
capacity of CD133− cells.
Three genes are involved in the modulation of
nucleotide pools during the process of DNA damage repair in human
cells (45). As an antimutagen,
nucleoside diphosphate linked moiety X-type motif 1 (NUDT1)
hydrolyzes oxidized purine nucleoside triphosphates, preventing the
misincorporation of nucleotides (46). Downregulation of NUDT1
expression levels in CD133− cells may reduce their
capacity to sanitize oxidized nucleotide pools and increase the
risk of misincorporation. In addition, the small subunit 2 of the
p53-inducible ribonucleotide reductase catalyzes the conversion of
ribonucleoside diphosphates to deoxyribonucleoside diphosphates
(47). Therefore, the downregulated
expression of the ribonucleotide reductase regulatory TP53
inducible subunit M2B in the CD133+ cells, as compared
with the CD133− and COLO 205 cells, may increase cell
survival by promoting p53-dependent damage repair and supplying
deoxyribonucleotides for DNA damage repair in cells arrested at the
G1 or G2 phases of the cell cycle (48).
Although 17 DNA polymerases are involved in DNA
damage repair as cofactors or catalytic subunits, only DNA
polymerase N (POLN) expression was upregulated in the
CD133− cells, as compared with the COLO 205 cells. As
POLN is involved in the repair of DNA crosslinks (49), a change in its expression levels may
affect the DNA repair capacity of CD133− cells.
Of the seven editing and processing nucleases
involved in DNA damage repair (50),
the expression levels of flap structure-specific endonuclease 1
(FEN1) and three prime repair exonuclease 2 (TREX2)
were downregulated by 2 fold in the CD133− cells, as
compared with the COLO 205 cells, and the expression of
TREX2 was downregulated in the CD133− cells,
compared with the CD133+ cells (2.11 folds) and the COLO
205 cells (2.79 folds). Since FEN1 and TREX2 are involved in DNA
replication and mismatch repair, possessing a structure-specific
nuclease activity preference for double-stranded DNA with
mismatched 3′termini and 5′-flap structures (51). Therefore, downregulation of the mRNA
expression levels of the two enzymes may affect the fidelity of DNA
replication in human cells, which can affect the viability of
cells.
Among the 11 genes involved in ubiquitination and
post-translational protein modification (52) in human cells, only two genes
(RAD18 and HLTF) exhibited upregulated expression
levels in the CD133− cells, as compared with the
CD133+ and COLO 205 cells. E3 ubiquitin-protein ligase
RAD18, which is involved in the post-replication repair of
UV-damaged DNA, and helicase-like transcription factor, which
functions in the error-free post-replication repair of damaged DNA
and the maintenance of genomic stability (53). HLTF is a SWI2/SNF2-family
ATP-dependent chromatin remodeling enzyme that acts in the
error-free branch of DNA damage tolerance (DDT), a cellular
mechanism that enables replication of damaged DNA while leaving
damage repair for a later time (54).
Alterations in the expression levels of these genes may affect the
cellular DNA damage repair capacity.
Three genes are known involved in chromatin
structure and modification including chromatin assembly factor 1
subunit A (CHAF1A), H2A histone family member X
(H2AFX) as well as SET domain and mariner transposase fusion
gene (SETMAR). In CD133− cells, the SETMAR
and H2AX genes showing the same downregulated trends in mRNA
expression levels compared with the COLO 205 cells and
CD133+ cells. Because of H2AX has previously been
demonstrated to interact with several types of cancer-associated
genes, including mediator of DNA damage checkpoint protein 1
(MDC1), nibrin, tumor protein p53 binding protein 1, bloom syndrome
protein, BRCA1, BRCA1-associated RING domain protein 1 and
γ-H2AX (phosphorylated on serine 139) (55,56), and
is, therefore, a sensitive target for locating DSBs in cells.
SETMAR specifically methylates histone H3 at lysines 4 and 36 and,
therefore, has a role in DNA repair activities, including
non-homologous end joining and DSB repair (57). Downregulation of the expression of
SETMAR and H2AX may, therefore, serve a role in DNA
damage though chromatin remodeling.
The five genes including Bloom syndrome RecQ like
helicase (BLM), Werner syndrome RecQ like helicase
(WRN), RecQ like helicase 4 (RECQL4), ATM
serine/threonine kinase (ATM) as well as M-Phase specific
PLK1 interacting protein (MPLKIP) that are defective in
diseases associated with a sensitivity to DNA damaging agents
(58,59), only the Bloom syndrome helicase gene,
involved in the 5′ end resection of DNA during DSB repair,
exhibited upregulated expression levels in the CD133−
cells, as compared with the CD133+ cells.
Of the nine genes that have established or suspected
DNA repair functions such as DNA cross-link repair 1A
(DCLRE1A), DNA cross-link repair 1B (DCLRE1B),
pre-mRNA processing factor 19 (PRPF19), replication protein
A 30 kDa subunit (RPA4), RecQ like helicase (RECQL),
RecQ like helicase 5 (RECQL5), Helicase, POLQ-like
(HELQ), RAD52 motif containing 1 (RDM1) as well as
nucleic acid binding protein 2 (NABP2), the expression of
three genes were changed. The mRNA expression levels of RPA4
and RDM1 were downregulated in CD133− cells
compared with the COLO 205 cells and CD133+ cells while
PRPF19 and NABP2 were downregulated in
CD133+ cells compared with the COLO 205 cells and
CD133− cells.
Of the 15 conserved DNA damage response genes
(60) including ATR serine/threonine
kinase (ATR), ATR interacting protein (ATRIP),
mediator of DNA damage checkpoint 1 (MDC1), RAD1 checkpoint
DNA exonuclease (RAD1), RAD9 checkpoint clamp component A
(RAD9A), HUS1 checkpoint clamp component (HUS1),
RAD17 checkpoint clamp loader component (RAD17), checkpoint
kinase 1 (CHEK1), checkpoint kinase 2 (CHEK2), tumor
protein P53 (TP53), tumor protein P53 binding protein 1
(TP53BP1), replication timing regulatory factor 1
(RIF1), CDC like kinase 2 (CLK2) and PER1
(period circadian clock 1), the expression of ATR and
MDC1 were upregulated and TP53 expression was
downregulated in the CD133− cells, compared with the
COLO 205 cells, whereas RIF1 and MDC1 expression
levels were upregulated; TP53 exhibited downregulated expression
levels in the CD133− cells, compared with the
CD133+ cells.
In conclusion, the present study demonstrated that
the mRNA expression levels of 43 genes varied in all three types of
colon cancer cells. Heterogeneity in the gene expression profiles
of DNA damage repair genes was present in the COLO 205 cells, and
in the COLO 205-derived CD133− cells and
CD133+ cells. The results of the present study may
provide a reference for molecular diagnosis, therapeutic target
selection, determination of treatment and prognostic judgment in
colorectal cancer (61).
Acknowledgements
This study was supported by the Foundation of
Guizhou Province Social Development of Science and Technology
Research Projects [grant no. (2009) 3068)] and the Foundation of
Guizhou Province Commission of Health and Family Planning [grant
no. gzwjkj2015-1-045].
References
|
1
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gasser S and Raulet D: The DNA damage
response, immunity and cancer. Semin Cancer Biol. 16:344–347. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mallette FA and Ferbeyre G: The DNA damage
signaling pathway connects oncogenic stress to cellular senescence.
Cell Cycle. 6:1831–1836. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liang Y, Lin SY, Brunicardi FC, Goss J and
Li K: DNA damage response pathways in tumor suppression and cancer
treatment. World J Surg. 33:661–666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgan MA and Lawrence TS: Molecular
pathways: Overcoming radiation resistance by targeting DNA damage
response pathways. Clin Cancer Res. 21:2898–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimizu I, Yoshida Y, Suda M and Minamino
T: DNA damage response and metabolic disease. Cell Metab.
20:967–977. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rajewsky MF, Engelbergs J, Thomale J and
Schweer T: DNA repair: Counteragent in mutagenesis and
carcinogenesis- accomplice in cancer therapy resistance. Mutat Res.
462:101–105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goldstein M and Kastan MB: The DNA damage
response: Implications for tumor responses to radiation and
chemotherapy. Annu Rev Med. 66:129–143. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Annovazzi L, Caldera V, Mellai M, Riganti
C, Battaglia L, Chirio D, Melcarne A and Schiffer D: The DNA
damage/repair cascade in glioblastoma cell lines after
chemotherapeutic agent treatment. Int J Oncol. 46:2299–2308.
2015.PubMed/NCBI
|
|
10
|
Paull TT: Mechanisms of ATM activation.
Annu Rev Biochem. 84:711–738. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suvorova II, Kozhukharova IV, Nikol'skiĭ
NN and Pospelov VA: ATM/ATR signaling pathway activation in human
embryonic stem cells after DNA damage. Tsitologiia. 55:841–851.
2013.(In Russian). PubMed/NCBI
|
|
12
|
Kühne C, Tjörnhammar ML, Pongor S, Banks L
and Simoncsits A: Repair of a minimal DNA double-strand break by
NHEJ requires DNA-PKcs and is controlled by the ATM/ATR checkpoint.
Nucleic Acids Res. 31:7227–7737. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Knittel G, Liedgens P and Reinhardt HC:
Targeting ATM-deficient CLL through interference with DNA repair
pathways. Front Genet. 6:2072015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakazawa K, Dashzeveg N and Yoshida K:
Tumor suppressor p53 induces miR-1915 processing to inhibit Bcl-2
in the apoptotic response to DNA damage. FEBS J. 281:2937–2944.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mlynarczyk C and Fåhraeus R: Endoplasmic
reticulum stress sensitizes cells to DNA damage-induced apoptosis
through p53-dependent suppression of p21 (CDKN1A). Nat Commun.
5:50672014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yajima N, Wada R, Matsuzaki Y and
Yagihashi S: DNA damage response and its clinicopathological
relationship in appendiceal tumors. Int J Colorectal Dis.
29:1349–1354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun T and Cui J: A plausible model for
bimodal p53 switch in DNA damage response. FEBS Lett. 588:815–821.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Broustas CG and Lieberman HB: DNA damage
response genes and the development of cancer metastasis. Radiat
Res. 181:111–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kelley MR, Logsdon D and Fishel ML:
Targeting DNA repair pathways for cancer treatment: what's new?
Future Oncol. 10:1215–1237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khanna A: DNA damage in cancer
therapeutics: A boon or a curse? Cancer Res. 75:2133–2138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Korwek Z and Alster O: The role of the DNA
damage response in apoptosis and cell senescence. Postepy Biochem.
60:248–262. 2014.(In Polish). PubMed/NCBI
|
|
23
|
Prakash A and Doublié S: Base excision
repair in the mitochondria. J Cell Biochem. 116:1490–1499. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian H, Gao Z, Li H, Zhang B, Wang G,
Zhang Q, Pei D and Zheng J: DNA damage response-a double-edged
sword in cancer prevention and cancer therapy. Cancer Lett.
358:8–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wallace NA and Galloway DA: Manipulation
of cellular DNA damage repair machinery facilitates propagation of
human papillomaviruses. Semin Cancer Biol. 26:30–42. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou L, Wei X, Cheng L, Tian J and Jiang
JJ: CD133, one of the markers of cancer stem cells in Hep-2 cell
line. Laryngoscope. 117:455–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gussin HA, Sharma AK and Elias S: Culture
of endothelial cells isolated from maternal blood using anti-CD105
and CD133. Prenat Diagn. 24:189–193. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu SC, Ping YF, Yi L, Zhou ZH, Chen JH,
Yao XH, Gao L, Wang JM and Bian XW: Isolation and characterization
of cancer stem cells from a human glioblastoma cell line U87.
Cancer Lett. 265:124–134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li HZ, Yi TB and Wu ZY: Suspension culture
combined with chemotherapeutic agents for sorting of breast cancer
stem cells. BMC Cancer. 8:1352008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ballester M, Castelló A, Ibáñez E, Sánchez
A and Folch JM: Real-time quantitative PCR-based system for
determining transgene copy number in transgenic animals.
Biotechniques. 37:610–613. 2004.PubMed/NCBI
|
|
31
|
Krokan HE and Bjørås M: Base excision
repair. Cold Spring Harb Perspect Biol. 5:a0125832013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lieber MR: The mechanism of human
nonhomologous DNA end joining. J Biol Chem. 283:1–5. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yi C and He C: DNA repair by reversal of
DNA damage. Cold Spring Harb Perspect Biol. 5:a0125752013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bowden NA: Nucleotide excision repair: Why
is it not used to predict response to platinum-based chemotherapy?
Cancer Lett. 346:163–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deng SK, Chen H and Symington LS:
Replication protein A prevents promiscuous annealing between short
sequence homologies: Implications for genome integrity. Bioessays.
37:305–313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu Q, Wani G, Sharma N and Wani A: Lack
of CAK complex accumulation at DNA damage sites in XP-B and XP-B/CS
fibroblasts reveals differential regulation of CAK anchoring to
core TFIIH by XPB and XPD helicases during nucleotide excision
repair. DNA Repair (Amst). 11:942–950. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McDaniel LD and Schultz RA: XPF/ERCC4 and
ERCC1: Their products and biological roles. Adv Exp Med Biol.
637:65–82. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Prakash R, Zhang Y, Feng W and Jasin M:
Homologous recombination and human health: The roles of BRCA1,
BRCA2, and associated proteins. Cold Spring Harb Perspect Biol.
7:a0166002015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Borde V and Cobb J: Double functions for
the Mre11 complex during DNA double-strand break repair and
replication. Int J Biochem Cell Biol. 41:1249–1253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sarbajna S and West SC: Holliday junction
processing enzymes as guardians of genome stability. Trends Biochem
Sci. 39:409–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li S, Ting NS, Zheng L, Chen PL, Ziv Y,
Shiloh Y, Lee EY and Lee WH: Functional link of BRCA1 and ataxia
telangiectasia gene product in DNA damage response. Nature.
406:210–215. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Koczorowska AM, Białkowska A, Kluzek K and
Zdzienicka MZ: The role of the Fanconi anemia pathway in DNA repair
and maintenance of genome stability. Postepy Hig Med Dosw (Online).
68:459–472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Radhakrishnan SK, Jette N and Lees-Miller
SP: Non-homologous end joining: Emerging themes and unanswered
questions. DNA Repair (Amst). 17:2–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fell VL and Schild-Poulter C: The Ku
heterodimer: Function in DNA repair and beyond. Mutat Res Rev Mutat
Res. 763:15–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Garre P, Briceño V, Xicola RM, Doyle BJ,
de la Hoya M, Sanz J, Llovet P, Pescador P, Puente J, Díaz-Rubio E,
et al: Analysis of the oxidative damage repair genes NUDT1, OGG1,
and MUTYH in patients from mismatch repair proficient HNPCC
families (MSS-HNPCC). Clin Cancer Res. 17:1701–1712. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mishima M, Sakai Y, Itoh N, Kamiya H,
Furuichi M, Takahashi M, Yamagata Y, Iwai S, Nakabeppu Y and
Shirakawa M: Structure of human MTH1, a Nudix family hydrolase that
selectively degrades oxidized purine nucleoside triphosphates. J
Biol Chem. 279:33806–33815. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pontarin G, Ferraro P, Håkansson P,
Thelander L, Reichard P and Bianchi V: p53R2-dependent
ribonucleotide reduction provides deoxyribonucleotides in quiescent
human fibroblasts in the absence of induced DNA damage. J Biol
Chem. 282:16820–16828. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Link PA, Baer MR, James SR, Jones DA and
Karpf AR: p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a
DNA hypomethylation-independent decitabine gene target that
correlates with clinical response in myelodysplastic syndrome/acute
myelogenous leukemia. Cancer Res. 68:9358–9366. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Takata K, Tomida J, Reh S, Swanhart LM,
Takata M, Hukriede NA and Wood RD: Conserved overlapping gene
arrangement, restricted expression, and biochemical activities of
DNA polymerase ν (POLN). J Biol Chem. 290:24278–24293. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zheng L and Shen B: Okazaki fragment
maturation: Nucleases take centre stage. J Mol Cell Biol. 3:23–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mason PA and Cox LS: The role of DNA
exonucleases in protecting genome stability and their impact on
ageing. Age (Dordr). 34:1317–1340. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dianov GL, Meisenberg C and Parsons JL:
Regulation of DNA repair by ubiquitylation. Biochemistry (Mosc).
76:69–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hedglin M and Benkovic SJ: Regulation of
Rad6/Rad18 activity during DNA damage tolerance. Annu Rev Biophys.
44:207–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hishiki A, Hara K, Ikegaya Y, Yokoyama H,
Shimizu T, Sato M and Hashimoto H: Structure of a novel DNA-binding
domain of helicase-like transcription factor (HLTF) and its
functional implication in DNA damage tolerance. J Biol Chem.
290:13215–13223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cook PJ, Ju BG, Telese F, Wang X, Glass CK
and Rosenfeld MG: Tyrosine dephosphorylation of H2AX modulates
apoptosis and survival decisions. Nature. 458:591–596. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Turinetto V and Giachino C: Multiple
facets of histone variant H2AX: A DNA double-strand-break marker
with several biological functions. Nucleic Acids Res. 43:2489–2498.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fnu S, Williamson EA, De Haro LP,
Brenneman M, Wray J, Shaheen M, Radhakrishnan K, Lee SH, Nickoloff
JA and Hromas R: Methylation of histone H3 lysine 36 enhances DNA
repair by nonhomologous end-joining. Proc Natl Acad Sci USA.
108:pp. 540–545. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Frank B, Hoffmeister M, Klopp N, Illig T,
Chang-Claude J and Brenner H: Colorectal cancer and polymorphisms
in DNA repair genes WRN, RMI1 and BLM. Carcinogenesis. 31:442–445.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nimonkar AV, Genschel J, Kinoshita E,
Polaczek P, Campbell JL, Wyman C, Modrich P and Kowalczykowski SC:
BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end
resection machineries for human DNA break repair. Genes Dev.
25:350–362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Strzyz P: DNA damage response: Cell
thriving despite DNA damage. Nat Rev Mol Cell Biol. 17:3962016.
View Article : Google Scholar
|
|
61
|
Lord CJ and Ashworth A: The DNA damage
response and cancer therapy. Nature. 481:287–294. 2012. View Article : Google Scholar : PubMed/NCBI
|