Introduction
The epidermal growth factor receptor (EGFR)
signaling pathway is importantly implicated in tumor cell growth,
invasion, angiogenesis and metastasis (1). Molecular aberrations on the EGFR pathway
are the most commonly studied predictive biomarkers of response to
targeted agents in lung cancer. Non-small cell lung cancers
(NSCLCs) harboring somatic gain-of-function mutations in the EGFR
tyrosine kinase domain respond well to treatment with the EGFR
tyrosine kinase inhibitors (TKIs), gefitinib and erlotinib
(2,3).
These small molecule TKIs compete with ATP to bind the kinase
domain of their targets. Several distinct activating mutations of
the EGFR gene have been described in NSCLC, including
in-frame deletions in exon 19 (delE746-A750) and a
leucine-to-arginine substitution at position 858 (L858R) in exon 21
(2,3).
All patients who experience a marked improvement
with these drugs eventually develop progression of disease
subsequent to a median of 12 months due to the acquisition of drug
resistance (4). Approximately half of
the cases with acquired resistance to EGFR TKIs can be accounted
for by a second-site mutation in exon 20 of the EGFR kinase domain,
which results in the substitution of methionine for threonine at
position 790 (T790M) (5,6). The bulkier methionine residue at
position 790 sterically hinders binding of either gefitinib or
erlotinib to the ATP-binding pocket. An additional study has
indicated that T790M also increases the affinity of EGFR for ATP,
thereby out-competing ATP-competitive TKIs and restoring enzymatic
activity in their presence (7).
However, the changes of gene expression involved in EGFR TKI
resistance due to the T790M mutation remain poorly defined.
Amplification of MET, a gene encoding a different membrane
bound receptor tyrosine kinase, is a separate mechanism of acquired
resistance to EGFR TKIs (8). Less
frequent forms of acquired resistance include histological
transformation to small cell lung cancer (9,10),
PIK3CA mutation (9,11) and epithelial to mesenchymal transition
(9,12). However, the exact frequencies of these
mechanisms remain to be established.
Previously, gene expression profiling of human
cancers has proved valuable in studies into cancer, providing
insights into mechanisms and targets involved in carcinogenesis and
drug response in several different types of cancer (13,14).
Analysis using mRNA microarrays allows simultaneous assessment of
the expression of thousands of genes and this approach provides a
valuable means to identify novel molecular targets for therapeutic
intervention. Additionally, it may be used to identify genes whose
expression is changed in cells with acquired drug resistance by
comparing gene expression in drug-resistant cells to that in
parental cells that are sensitive to treatment with, for example,
EGFR TKIs.
To additionally investigate resistance to EGFR TKIs,
the present study has established cell lines that are resistant to
either gefitinib or erlotinib. Using mRNA microarrays, genome-wide
analysis of gene expression profiles has established a clear
division between parental and resistant cells, with altered
expression of genes involved in the regulation of cellular
proliferation, apoptosis and the cell cycle in the EGFR
TKI-resistant cells. The present study may provide key insights
into gene expression profiles involved in conferring resistance to
EGFR TKI therapy in lung cancer cells.
Materials and methods
Cell culture and establishment of the
gefitinib- and erlotinib-resistant cell lines
The EGFR-mutated NSCLC cell line, PC-9, was
used. PC-9 cells are known to contain a deletion in exon 19
(delE746-A750) of EGFR and be highly sensitive to gefitinib
and erlotinib (15). Gefitinib- or
erlotinib-resistant sublines of PC-9 were established as described
previously (16) and these resistant
cells were >100 times more resistant to gefitinib and erlotinib
compared with parental cells. Briefly, parental PC-9 cells were
exposed to 10 nmol/l of gefitinib or erlotinib for 48 h in medium
containing 10% fetal bovine serum. Subsequently, cells were washed
and cultured in drug-free medium until surviving cells were 80%
confluent. These cells were then re-exposed to increasing
concentrations of gefitinib or erlotinib. Six months after initial
exposure, cells were able to grow in 1 µmol/l of gefitinib or
erlotinib. The established drug-resistant cell lines were then
maintained in medium containing 1 µmol/l of gefitinib or erlotinib.
Gefitinib- or erlotinib-resistant cells are referred to as PC-9G
and PC-9E, respectively. PC-9G and PC-9E cells were confirmed to
contain the EGFR T790M mutation by DNA sequencing (16). All cells were maintained as monolayer
cultures in RPMI 1640 medium supplemented with 10% fetal bovine
serum, 1% penicillin-streptomycin and 1% sodium pyruvate at 37°C in
a humidified incubator in an atmosphere of 5% CO2. All
cell culture materials were obtained from HyClone (GE Healthcare,
Logan, UT, USA).
mRNA microarray analysis
Duplicated total RNA sample of each cell lines was
prepared using TRIzol® Reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. The quantity and integrity of extracted
RNA was confirmed using a 2100 Bioanalyzer (Agilent Technologies,
Inc., Santa Clara, CA, USA) prior to samples being deemed suitable
for microarray analysis. Fluorescent complimentary RNA (cRNA)
probes were generated and purified using the Agilent's Low RNA
Input Linear Amplification kit (Agilent Technologies, Inc.)
according to the manufacturer's protocol. Labeled cRNA target was
quantified using a NanoDrop-1000 spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). mRNA microarray
hybridization was performed for the samples using the Agilent Whole
Human Genome Oligo Microarray (44 K) according to manufacturer's
protocol. Briefly, 600 ng Cy3-labeled fragmented cRNA was
hybridized overnight to an Agilent Whole Human Genome Oligo
Microarray, washed twice, blocked, and scanned using Agilent's
microarray scanner.
Data acquisition and statistical
analysis
The hybridized images were quantified using the
Feature Extraction Software (Agilent Technologies, Inc.). All data
normalization and the selection of fold-changed genes were
performed using the GeneSpring GX 7.3 (Agilent Technologies, Inc.).
The averages of normalized ratios were calculated by dividing the
average of normalized signal channel intensity by the average of
normalized control channel intensity. Functional annotation of
genes was performed according to the Gene Ontology™ Consortium
(http://www.geneontology.org/index.shtml) using the
GeneSpring GX 7.3. Gene functional classification was based on
searches carried out using the Database for Annotation,
Visualization, and Integrated Discovery (DAVID) bioinformatics
resources (http://david.abcc.ncifcrf.gov/).
Results
Distinctive gene expression patterns
are revealed by mRNA microarray analysis in EGFR TKI-resistant lung
cancer cells with the EGFR T790M mutation
To detect expression changes in the EGFR
TKI-resistant cells, an mRNA microarray analysis of PC-9G and PC-9E
cells in comparison with drug-naïve parental PC-9 cells was
performed. In total, >20,000 genes were reliably analyzed
following quantile normalization, and those showing fold changes of
>1.5 were defined as being differentially expressed between the
parental and either the PC-9G or PC-9E cells. Among the genes whose
expression was significantly altered, the present study focused on
those genes whose expression was altered in PC-9G and PC-9E cells
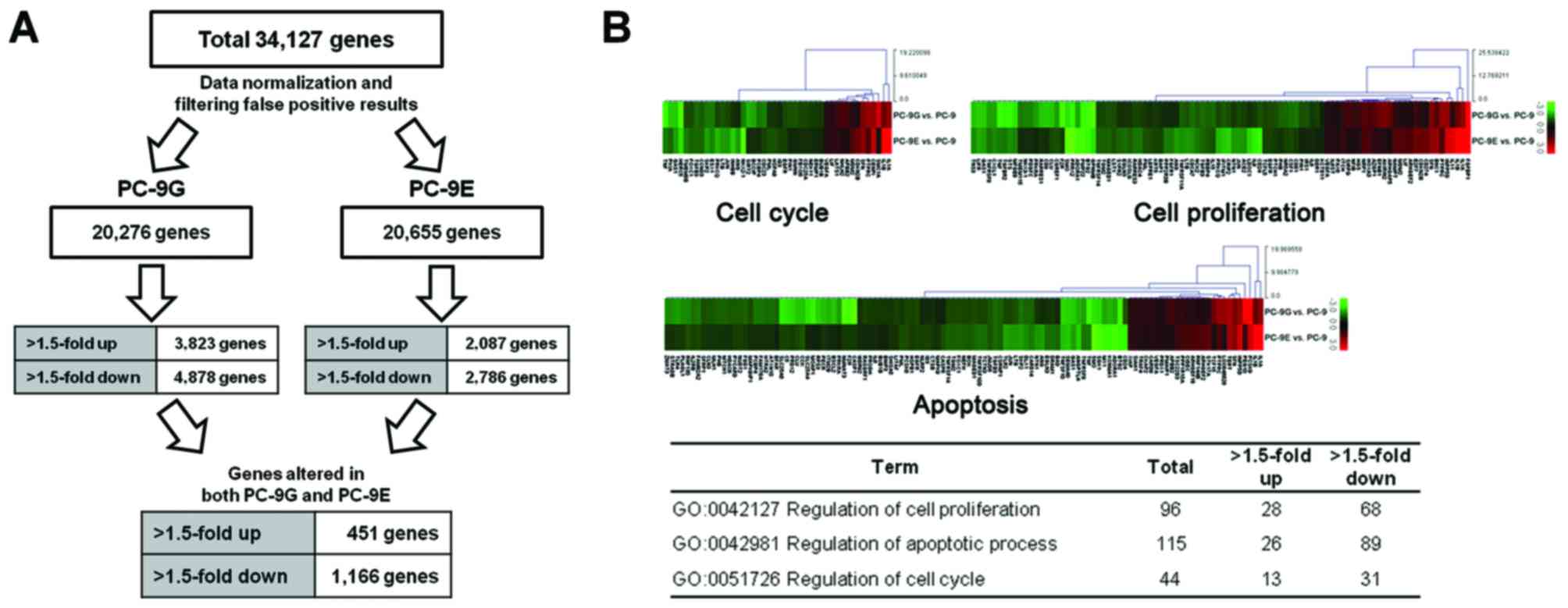
(Fig. 1A). A total of 1,617 genes
were identified as being differentially expressed in PC-9G and
PC-9E cells. Cluster analysis of these differentially expressed
genes between PC-9G/PC-9E and parental PC-9 cells identified 451
mRNAs with significantly increased expression in PC-9G and PC-9E
compared with in PC-9 cells, and 1,166 mRNAs with significantly
reduced expression in PC-9G and PC-9E compared with in PC-9
cells.
Gene ontology analyses of microarray
data reveals functional pathways associated with altered mRNA
expression in PC-9G and PC-9E cells
Genes were functionally categorized with regard to
their associated biological process. Gene ontology analysis for the
>1.5-fold up- or downregulated genes in PC-9G and PC-9E cells
was performed using the DAVID tools. According to the DAVID gene
ontology program, 1,104 (68%) of the 1,617 genes are associated
with a known annotated biological process, 288 (64%) of the 451
upregulated genes and 816 (70%) of the 1,166 downregulated genes.
In particular, 96, 115 and 44 genes are associated with the
regulation of cellular proliferation, apoptosis and the cell cycle,
respectively (Fig. 1B).
Altered expression of genes involved
in the regulation of cellular proliferation, apoptosis and the cell
cycle
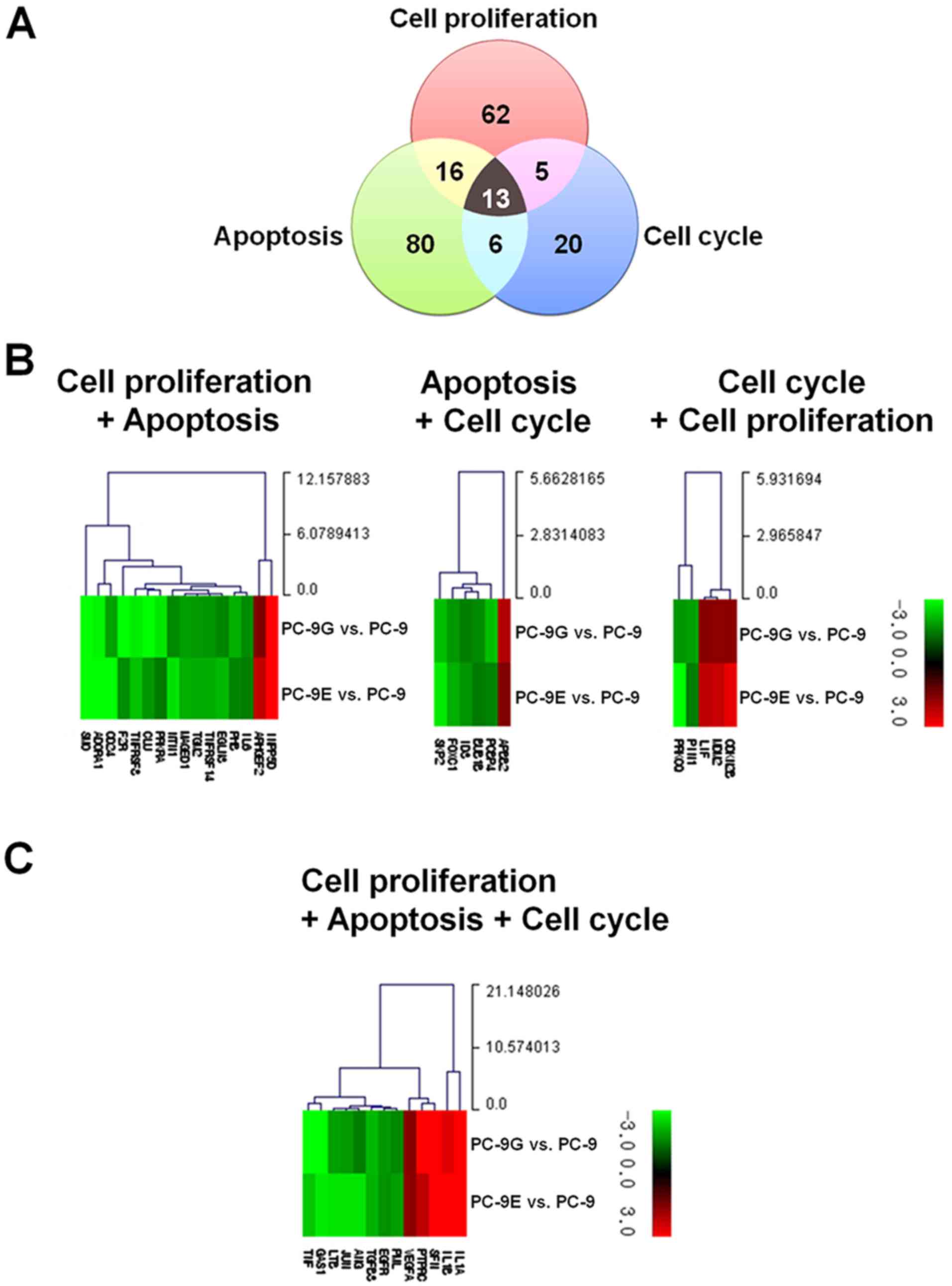
The present study subsequently investigated the
genes whose expression was significantly altered in PC-9G and PC-9E
cells and that were common in any two or all three ontological
clusters: Regulation of cellular proliferation, apoptosis and the
cell cycle (Fig. 2A). In total, 40
genes are involved in any two or all three ontological clusters.
With the exception of genes involved in all of three ontological
clusters, 16 genes are involved in cellular proliferation and
apoptosis (Fig. 2B, Table I), six in apoptosis and the cell cycle
(Fig. 2B, Table II), and five in the cell cycle and
cellular proliferation (Fig. 2B,
Table III). A total of 13 genes
were identified to be involved in cellular proliferation, apoptosis
and the cell cycle (Fig. 2C, Table IV).
 | Table I.Altered expression of genes involved
in cellular proliferation and apoptosis. |
Table I.
Altered expression of genes involved
in cellular proliferation and apoptosis.
|
|
|
| Fold change (vs.
PC-9) |
|---|
|
|
|
|
|
|---|
| Gene symbol | Description | Genbank | PC-9G | PC-9E |
|---|
| Upregulated |
|
ARHGEF2 | Rho/Rac guanine
nucleotide exchange factor (GEF) 2 | NM_004723 | 1.52 | 2.29 |
|
INPP5D | Inositol
polyphosphate-5-phosphatase, 145 kDa | NM_001017915 | 3.49 | 5.22 |
| Downregulated |
|
ADORA1 | Adenosine A1
receptor | NM_000674 | −2.90 | −5.15 |
|
CD24 | CD24 molecule | NM_013230 | −1.91 | −4.39 |
|
CLU | Clusterin | NM_001831 | −3.24 | −1.79 |
|
EGLN3 | Egl-9 family
hypoxia-inducible factor 3 | NM_022073 | −1.59 | −2.18 |
|
F2R | Coagulation factor
II (thrombin) receptor | BC016059 | −5.15 | −1.68 |
|
IL6 | Interleukin 6
(interferon, β2) | NM_000600 | −1.65 | −1.49 |
|
MAGED1 | Melanoma antigen
family D, 1 | NM_001005333 | −1.86 | −2.08 |
|
NTN1 | Netrin 1 | NM_004822 | −1.63 | −2.68 |
|
PHB | Prohibitin | NM_002634 | −2.07 | −1.58 |
|
PRKRA | Protein kinase,
interferon-inducible double stranded RNA dependent activator | NM_003690 | −2.74 | −1.43 |
|
SMO | Smoothened frizzled
class receptor | NM_005631 | −3.72 | −10.07 |
|
TGM2 | Transglutaminase 2
(C polypeptide, protein-glutamine-gamma-glutamyltransferase) | NM_198951 | −1.81 | −2.02 |
|
TNFRSF14 | Tumor necrosis
factor receptor superfamily, member 14 | AB208808 | −1.76 | −2.05 |
|
TNFRSF8 | Tumor necrosis
factor receptor superfamily, member 8 | NM_001243 | −2.78 | −2.27 |
| Table II.Altered expression of genes involved
in apoptosis and the cell cycle. |
Table II.
Altered expression of genes involved
in apoptosis and the cell cycle.
|
|
|
| Fold change (vs.
PC-9) |
|---|
|
|
|
|
|
|---|
| Gene symbol | Description | Genbank | PC-9G | PC-9E |
|---|
| Upregulated |
|
|
|
|
|
APBB2 | Amyloid β (A4)
precursor protein-binding, family B, member 2 | NM_004307 | 2.09 | 1.55 |
| Downregulated |
|
|
|
|
|
BUB1B | BUB1 mitotic
checkpoint serine/threonine kinase B | NM_001211 | −1.65 | −1.45 |
|
FOXC1 | Forkhead box
C1 | NM_001453 | −1.79 | −2.06 |
|
ID3 | Inhibitor of DNA
binding 3, dominant negative helix-loop-helix protein | NM_002167 | −1.51 | −1.75 |
|
PCBP4 | Poly(rC) binding
protein 4 | NM_033010 | −2.24 | −1.54 |
|
SKP2 | S-phase
kinase-associated protein 2, E3 ubiquitin protein ligase | NM_032637 | −2.25 | −2.77 |
| Table III.Altered expression of genes involved
in the cell cycle and cellular proliferation. |
Table III.
Altered expression of genes involved
in the cell cycle and cellular proliferation.
|
|
|
| Fold change (vs.
PC-9) |
|---|
|
|
|
|
|
|---|
| Gene symbol | Description | Genbank | PC-9G | PC-9E |
|---|
| Upregulated |
|
|
|
|
|
CDKN2B | Cyclin-dependent
kinase inhibitor 2B (p15, inhibits CDK4) | NM_004936 | 1.64 | 2.74 |
|
LIF | Leukemia inhibitory
factor | NM_002309 | 1.59 | 2.24 |
|
MDM2 | MDM2
proto-oncogene, E3 ubiquitin protein ligase | NM_002392 | 1.66 | 2.35 |
| Downregulated |
|
|
|
|
|
PIN1 | Peptidylprolyl
cis/trans isomerase, NIMA-interacting 1 | NM_006221 | −1.87 | −1.47 |
|
PRKCQ | Protein kinase C,
theta | NM_006257 | −1.78 | −3.08 |
| Table IV.Altered expression of genes involved
in cellular proliferation, apoptosis and also in the cell
cycle. |
Table IV.
Altered expression of genes involved
in cellular proliferation, apoptosis and also in the cell
cycle.
|
|
|
| Fold change (vs.
PC-9) |
|---|
|
|
|
|
|
|---|
| Gene symbol | Description | Genbank | PC-9G | PC-9E |
|---|
| Upregulated |
|
|
|
|
|
IL1A | Interleukin 1,
α | NM_000575 | 3.47 | 23.92 |
|
IL1B | Interleukin 1,
β | NM_000576 | 2.68 | 17.35 |
|
PTPRC | Protein tyrosine
phosphatase, receptor type, C | NM_001267798 | 3.61 |
2.25 |
|
SFN | Stratifin | NM_006142 | 3.10 |
3.39 |
|
VEGFA | Vascular
endothelial growth factor A | NM_001025370 | 1.61 |
1.68 |
| Downregulated |
|
|
|
|
|
ANG | Angiogenin,
ribonuclease, RNase A family, 5 | NM_001145 | −1.50 | −2.70 |
|
EGFR | Epidermal growth
factor receptor | NM_201283 | −1.79 | −1.72 |
|
GAS1 | Growth
arrest-specific 1 | NM_002048 | −3.38 | −2.77 |
|
JUN | Jun
proto-oncogene | NM_002228 | −1.81 | −2.70 |
|
LTB | Lymphotoxin β (TNF
superfamily, member 3) | NM_002341 | −1.90 | −2.70 |
|
PML | Promyelocytic
leukemia | NM_033238 | −1.62 | −1.87 |
|
TGFB3 | Transforming growth
factor, β3 | NM_003239 | −2.18 | −2.13 |
|
TNF | Tumor necrosis
factor | NM_000594 | −4.62 | −2.35 |
Genes listed in Table
I–IV were sorted according to
their functions, and the genes whose altered expression could
promote tumor progression or reduce tumor suppression, favoring
tumor growth were selected for further analysis (Table V). Upregulation of the gene encoding
amyloid βA4 precursor protein-binding family B member 2, which has
been reported as an apoptosis inhibitor in aneuploid fibrosarcoma
(17), was detected. The gene
encoding Rho/Rac guanine nucleotide exchange factor 2, which is
reported as an oncogenic protein in pancreatic cancer (18) and associated with the invasion and
metastasis of breast cancer (19),
was also upregulated. Previously, interleukin 1α (IL1A) and
interleukin 1β (IL1B) have been reported as being associated with
the growth of lung cancer cells (20). Significant upregulation of the
IL1A and IL2B genes was observed in the present
study, which implied that the tumor microenvironment of resistant
cells was altered. Leukemia inhibitory factor (encoded by the
LIF gene) has been known to promote tumor progression and
radioresistance in nasopharyngeal carcinoma (21). Among the upregulated genes, mouse
double minute 2 homolog is a well-known oncogene that can promote
tumor formation by targeting tumor suppressor proteins, including
p53, for proteasomal degradation (22). The protein tyrosine phosphatase
receptor type C gene, which is associated with the recurrence of
colorectal cancer (23), was also
upregulated. The stratifin gene, a gene which was upregulated in
the present study, is associated with resistance to chemotherapy
and radiotherapy in pancreatic cancer (24) and to tumor invasion in lung cancer
(25). The vascular endothelial
growth factor A gene, which is associated with angiogenesis in
cancer (26) and the migration and
invasion of lung cancer (27), was
also upregulated.
| Table V.Genes whose expression was altered
towards enhanced tumor progression or reduced tumor
suppression. |
Table V.
Genes whose expression was altered
towards enhanced tumor progression or reduced tumor
suppression.
|
|
| Fold change (vs.
PC-9) |
|---|
|
|
|
|
|---|
| Gene symbol | Associated
process | PC-9G | PC-9E |
|---|
| Tumor
progression |
|
|
|
|
APBB2 | Inhibition of tumor
cell apoptosis | 2.09 | 1.55 |
|
ARHGEF2 | Oncogenic
signaling, invasion and metastasis | 1.52 | 2.29 |
|
IL1A | Growth of tumor
cells | 3.47 | 23.92 |
|
IL1B | Growth of tumor
cells | 2.68 | 17.35 |
|
LIF | Tumor progression
and radioresistance | 1.59 | 2.24 |
|
MDM2 | Tumor
formation | 1.66 | 2.35 |
|
PTPRC | Cancer
recurrence | 3.61 | 2.25 |
|
SFN | Resistance to
chemotherapy and radiotherapy, invasion | 3.1 | 3.39 |
|
VEGFA | Angiogenesis,
migration and invasion | 1.61 | 1.68 |
| Tumor
suppression |
|
|
|
|
GAS1 | Inhibition of
cellular proliferation and induction of apoptosis | −3.38 | −2.77 |
|
PCBP4 | Suppression of
cellular proliferation | −2.24 | −1.54 |
|
TNF | Apoptosis of tumors
cells | −4.62 | −2.35 |
Among the downregulated genes, the growth
arrest-specific 1 gene, which is known to be a tumor suppressor
gene (28), was detected. The poly
(rC) binding protein 4 gene, which is induced by the p53 tumor
suppressor and whose product suppresses cellular proliferation by
inducing apoptosis and cell cycle arrest in G2/M
(29), was also downregulated.
Decreased expression of the tumor necrosis factor gene encoding
tumor necrosis factor, an inducer of apoptosis (30), was observed.
Discussion
Gefitinib and erlotinib, commonly considered as the
standard treatment for patients with NSCLC who harbor EGFR
activating mutations, are two oral drugs that bind the ATP-binding
site in the tyrosine kinase domain of the EGFR protein. Somatic
mutations occurring in the tyrosine kinase domain of the
EGFR gene and responsible for ligand-independent activation
of the EGFR pathway have been reported; exon 19 deletions and L858R
substitution in exon 21 are the most common, accounting for 90% of
all EGFR activating mutations in NSCLC (2,3). These
mutations lead to increased growth signaling, thus conferring
susceptibility to treatment with TKIs. Despite the initial clear
responses to EGFR TKIs and substantial increase in progression free
survival observed in various clinical trials, the majority of
patients with NSCLC with an EGFR activating mutation develop
acquired resistance subsequent to a median of ~12 months from
treatment initiation (4). Although
significant advances have been made, in ≤30% of cases the acquired
resistance mechanisms remain unexplained. Therefore, there is an
urgent need to identify the underlying mechanisms of resistance in
order to develop effective targeted therapies for cancer
progression subsequent to EGFR TKI treatment.
The present study established two NSCLC cell lines
resistant to the commonly used EGFR TKIs, gefitinib or erlotinib.
These cell lines harbor the EGFR T790M mutation, which is
the most frequently reported mechanism of acquired resistance to
EGFR TKI treatment. The present study aimed to analyze possible
gene expression changes associated with EGFR TKI resistance using
PC-9G and PC-9E cell lines. Among the genes whose expression was
significantly altered, those whose expression was altered in PC-9G
and PC-9E cells were focused on, in order to increase the
reliability of the results.
It is known that when binding to one of its several
ligands, EGFR forms homodimers or heterodimers with other members
of the ERBB family receptor tyrosine kinases, and activates
downstream pathways, including the phosphatidylinositol
3-kinase/protein kinase B, Raf/mitogen activated protein
kinase/extracellular signal-regulated kinases, and janus
kinase/signal transducers and activators of transcription signaling
pathways, initiating a cascade of signaling events that trigger
anti-apoptotic signaling, cellular proliferation, angiogenesis and
tumor invasion and metastasis. Treatment with EGFR TKIs will induce
blockage of these pathways. The present data indicated that EGFR
TKI-resistant cells are likely to exhibit altered expression of
genes that are associated with apoptosis, cellular proliferation
and the cell cycle.
Genes, that are associated with at least two of the
processes of cellular proliferation, apoptosis, and the cell cycle,
and whose expression was significantly altered in EGFR
TKI-resistant cells to favor tumor progression, are listed in
Table V. As shown in Table V, there was increased expression of
genes whose functions are known to promote tumor growth, and
decreased expression of genes whose functions are known to suppress
tumor growth, in PC-9G and PC-9E cells. It will be necessary to
determine the action of these genes in the development of EGFR TKI
resistance in NSCLC cells.
Two previous studies were identified that had used
mRNA microarray analysis to examine changes of gene expression in
lung cancer cells with acquired resistance to either gefitinib or
erlotinib (12,31). However, these studies only examined
one cell line resistant to either gefitinib or erlotinib. The
present study examined two cell lines with acquired resistance-one
to gefitinib and one to erlotinib. Furthermore, genes whose
expression was altered in the gefitinib- and the
erlotinib-resistant cell line were investigated and the changes of
gene expression in terms of major survival pathways were
analyzed.
To the best of our knowledge, this is the first
study to identify a common mRNA expression profile in cells with
acquired resistance to either gefitinib or erlotinib, and can
provide key insights into gene expression profiles involved in
conferring resistance to EGFR TKI therapy in lung cancer cells. In
conclusion, the present study observed distinctive gene expression
patterns via mRNA microarray analysis of EGFR TKI-resistant lung
cancer cells harboring the EGFR T790M mutation.
Acknowledgements
The present study was supported by the Korea
Institute of Radiological and Medical Sciences Research Fund (grant
no. 50452-2014).
References
|
1
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–344. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pao W and Chmielecki J: Rational,
biologically based treatment of EGFR-mutant non-small-cell lung
cancer. Nat Rev Cancer. 10:760–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cataldo VD, Gibbons DL, Pérez-Soler R and
Quintás-Cardama A: Treatment of non-small-cell lung cancer with
erlotinib or gefitinib. N Engl J Med. 364:947–955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors-impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:pp. 2070–2075. 2008; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S
and Heist RS: Genotypic and histological evolution of lung cancers
acquiring resistance to EGFR inhibitors. Sci Transl Med.
3:75ra262011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zakowski MF, Ladanyi M and Kris MG;
Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome
Group, : EGFR mutations in small-cell lung cancers in patients who
have never smoked. N Engl J Med. 355:213–215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suda K, Tomizawa K, Fujii M, Murakami H,
Osada H, Maehara Y, Yatabe Y, Sekido Y and Mitsudomi T: Epithelial
to mesenchymal transition in an epidermal growth factor
receptor-mutant lung cancer cell line with acquired resistance to
erlotinib. J Thorac Oncol. 6:1152–1161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
van 't Veer LJ, Dai H, van de Vijver MJ,
He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ,
Witteveen AT, et al: Gene expression profiling predicts clinical
outcome of breast cancer. Nature. 415:530–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruzzo A, Graziano F, Loupakis F, Rulli E,
Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P and
Bisonni R: Pharmacogenetic profiling in patients with advanced
colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J
Clin Oncol. 25:1247–1254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ono M, Hirata A, Kometani T, Miyagawa M,
Ueda S, Kinoshita H, Fujii T and Kuwano M: Sensitivity to gefitinib
(Iressa, ZD1839) in non-small cell lung cancer cell lines
correlates with dependence on the epidermal growth factor (EGF)
receptor/extracellular signal-regulated kinase 1/2 and EGF
receptor/Akt pathway for proliferation. Mol Cancer Ther. 3:465–472.
2004.PubMed/NCBI
|
|
16
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Lee SS, Kim CH, Yoo YD and Lee JC: The role of MET
activation in determining the sensitivity to epidermal growth
factor receptor tyrosine kinase inhibitors. Mol Cancer Res.
7:1736–1743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lange A, Thon L, Mathieu S and Adam D: The
apoptosis inhibitory domain of FE65-like protein 1 regulates both
apoptotic and caspase-independent programmed cell death mediated by
tumor necrosis factor. Biochem Biophys Res Commun. 335:575–583.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cullis J, Meiri D, Sandi MJ, Radulovich N,
Kent OA, Medrano M, Mokady D, Normand J, Larose J, Marcotte R, et
al: The RhoGEF GEF-H1 is required for oncogenic RAS signaling via
KSR-1. Cancer Cell. 25:181–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liao YC, Ruan JW, Lua I, Li MH, Chen WL,
Wang JR, Kao RH and Chen JH: Overexpressed hPTTG1 promotes breast
cancer cell invasion and metastasis by regulating GEF-H1/RhoA
signalling. Oncogene. 31:3086–3097. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Engels EA, Wu X, Gu J, Dong Q, Liu J and
Spitz MR: Systematic evaluation of genetic variants in the
inflammation pathway and risk of lung cancer. Cancer Res.
67:6520–6527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu SC, Tsang NM, Chiang WC, Chang KP,
Hsueh C, Liang Y, Juang JL, Chow KP and Chang YS: Leukemia
inhibitory factor promotes nasopharyngeal carcinoma progression and
radioresistance. J Clin Invest. 123:5269–5283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Momand J, Zambetti GP, Olson DC, George D
and Levine AJ: The mdm-2 oncogene product forms a complex with the
p53 protein and inhibits p53-mediated transactivation. Cell.
69:1237–1245. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chew A, Salama P, Robbshaw A, Klopcic B,
Zeps N, Platell C and Lawrance IC: SPARC, FOXP3, CD8 and CD45
correlation with disease recurrence and long-term disease-free
survival in colorectal cancer. PLoS One. 6:e220472011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Z, Dong Z, Myer D, Yip-Schneider M, Liu
J, Cui P, Schmidt CM and Zhang JT: Role of 14-3-3σ in poor
prognosis and in radiation and drug resistance of human pancreatic
cancers. BMC Cancer. 10:5982010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shiba-Ishii A and Noguchi M: Aberrant
stratifin overexpression is regulated by tumor-associated CpG
demethylation in lung adenocarcinoma. Am J Pathol. 180:1653–1662.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carmeliet P: VEGF as a key mediator of
angiogenesis in cancer. Oncology. 69 Suppl 3:S4–S10. 2005.
View Article : Google Scholar
|
|
27
|
Chen CH, Lai JM, Chou TY, Chen CY, Su LJ,
Lee YC, Cheng TS, Hong YR, Chou CK, Whang-Peng J, et al: VEGFA
upregulates FLJ10540 and modulates migration and invasion of lung
cancer via PI3K/AKT pathway. PLoS One. 4:e50522009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Domínguez-Monzón G, Benítez JA, Vergara P,
Lorenzana R and Segovia J: Gas1 inhibits cell proliferation and
induces apoptosis of human primary gliomas in the absence of Shh.
Int J Dev Neurosci. 27:305–313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu J and Chen X: MCG10, a novel p53
target gene that encodes a KH domain RNA-binding protein, is
capable of inducing apoptosis and cell cycle arrest in G(2)-M. Mol.
Cell Biol. 20:5602–5618. 2000.
|
|
30
|
Carswell EA, Old LJ, Kassel RL, Green S,
Fiore N and Williamson B: An endotoxin-induced serum factor that
causes necrosis of tumors. Proc Natl Acad Sci USA. 72:pp.
3666–3670. 1975; View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Terai H, Soejima K, Yasuda H, Sato T,
Naoki K, Ikemura S, Arai D, Ohgino K, Ishioka K, Hamamoto J, et al:
Long-term exposure to gefitinib induces acquired resistance through
DNA methylation changes in the EGFR-mutant PC9 lung cancer cell
line. Int J Oncol. 46:430–436. 2015.PubMed/NCBI
|