Introduction
Liver cancer in males is the fifth most frequently
diagnosed type of cancer worldwide; however, it is the second most
frequent cause of cancer mortality. In females, it is the seventh
most commonly diagnosed type of cancer and the sixth leading cause
of cancer mortality (1). An estimated
748,300 cases of liver cancer were newly diagnosed and 695,900
cancer mortalities occurred worldwide in 2008, with 50% of these
cases and mortalities estimated to have occurred in China (2). The majority of patients with hepatopathy
in China suffer as a result of hepatitis B cirrhosis; furthermore,
the disease is typically diagnosed late, opportunities for excision
are limited and sensitivity to radiotherapy is poor, therefore the
majority of cases are treated using traditional chemotherapy
(3). However, liver cancer exhibits
poor sensitivity to chemotherapeutic drugs, and the side effects
are not ideal; in particular, resistance to doxorubicin [Adriamycin
(ADM)] in the treatment of liver cancer is <80% (4). The poor sensitivity to chemotherapeutic
drugs is associated with natural drug resistance and acquired
multidrug resistance (MDR) in liver cancer (4). A number of drugs exist that are able to
reverse the MDR of liver cancer; however, they exhibit increased
toxicity and a limited effect. Therefore, the development of a
novel drug to reverse MDR is of key importance for the treatment of
liver cancer.
MDR describes the property of malignant cells
exhibiting resistance to a number of chemotherapeutic drugs of
distinct structure and mechanism of action. MDR is a key mechanism
used by tumor cells to resist chemotherapeutic drugs. A number of
factors may result in MDR, including the drug-efflux pump mechanism
of drug resistance proteins, including P-glycoprotein (P-gp),
MDR-associated protein (MRP) and lung resistance-related protein
(LRP), mutations in DNA topoisomerase and DNA repair abnormality
(5). The drug efflux pump mediated by
drug resistance proteins, including P-gp and MRP, is the primary
molecular mechanism of MDR in tumor cells (6).
Cellular signal transduction pathways serve a role
in malignant tumors, in which abnormalities in the
mitogen-activated protein kinase (MAPK) signaling pathway activated
by growth factors such as epidermal growth factor have been
associated with the growth, proliferation and invasion of malignant
tumors, and resistance to chemotherapeutic drugs (7,8). Mammals
possess three classical MAPK signaling pathways, in which the Raf
proto-oncogene serine/threonine protein kinase
(Raf)/MAPK/extracellular-signal-regulated kinase (ERK) kinase
(MEK)/ERK signal transduction pathway is the most well-researched
(9,10). It has been demonstrated previously
that ADM in lymphocytoma B is able to induce the expression of P-gp
to generate drug resistance by activating MAPK/ERK (11). Abrams et al (12) demonstrated that a targeted drug aimed
at Raf/MEK/ERK signal transduction pathway was able to reverse the
drug resistance of leukemia drug resistance cells and enhance the
sensitivity of tumor-resistance chemotherapeutic drugs. Katayama
et al (13) demonstrated that
the MEK inhibitor U0126 was able to downregulate the expression of
endogenous P-gp of SW620-14 cells and the expression of exogenous
P-gp of MCF-7/MDR and MDA-MB-231/MDR to enhance the anti-tumor
activity. However, the association between MAPK and MDR in primary
liver cancer remains unclear.
The aim of the present study was to elucidate the
interaction between the MAPK signaling pathway and ATP-binding
cassette (ABC) protein expression in hepatocellular carcinoma
(HCC). A selective inhibitor of MEK activity (U0126) was used to
investigate the effects on P-gp and MRP1 protein expression.
Materials and methods
Cell lines and reagents
The ADM, VCR, 5-FU and MMC were purchased from
Zhejiang Hisun Pharmaceutical Co., Ltd (Taizhou City, Zhejiang
Province). The HepG2 and HepG2/ADM cell lines were purchased from
Beijing North Carolina Souren Biotechnology Research Institute,
(Beijing, China). U0126 was purchased from (Selleck Chemicals,
Houston, TX, USA), and the RPMI 1640 mediun and fetal bovine serum
were purchased from (Hyclone; GE Healthcare, Logan, UT, USA).
Cell culture
The ADM-resistant human HCC cell line HepG2/ADM was
cultured in RPMI-1640 medium containing 10% fetal bovine serum, 100
units/ml penicillin and 100 mg/ml streptomycin (Hyclone; GE
Healthcare, Logan, UT, USA) at 37°C in an incubator with 5%
CO2 and 95% humidity. ADM (0.4 nmol/ml) was added to the
culture medium to maintain the drug resistance of HepG2/ADM cells.
The HepG2 cells also cultured in this way, without adding ADM.
Investigation of drug resistance in
HepG2/ADM cells and sensitivity to chemotherapeutic drugs
A total of 0.2 ml HepG2/ADM and HepG2 cells in the
exponential growth phase were inoculated in 96-well plates at a
density of 1×105/well). Following incubation at 37°C and
5% CO2 for 24 h, the supernatant was discarded and fresh
medium containing chemotherapeutic drugs (ADM, VCR, 5-FU and MMC)
at various concentrations (ADM; 0, 0.1, 1, 10, 100 and 1,000 mg/ml;
VCR, 5-FU and MMC; 0, 2, 4, 8, 16, 32 mg/l) was added into culture
plates. Following incubation for 24 h 37°C and 5% CO2,
the supernatant was discarded, 10 µl Cell Counting Kit-8 (CCK-8)
was added to each well and cells were cultured for a further 4 h
under the same conditions, in the absence of drugs. A microplate
reader was used to measure the absorbance of each well at 450 nm.
The half-maximal inhibitory concentration (IC50) was
calculated to determine the resistance indices of HepG2/ADM and
HepG2 cells to the chemotherapeutic drugs used.
Determination of the effect of U0126
at various concentrations on the apoptotic rates of HepG2/ADM
cells
HepG2/ADM cells in exponential growth phase were
seeded in 6-well plates (1×105 cells/well) and incubated
for 24 h. The supernatant was discarded, medium containing various
U0126 concentrations (0, 10, 20 and 40 µmol/l) was added into the
corresponding wells and the cells were incubated for 48 h.
EDTA-free trypsin (0.25%) was used to detach cells and cells were
divided into groups. The detached cells were washed twice using
centrifugation at 503.1 × g for 5 min at 37°C. An Annexin
V/propidium iodide (PI) apoptosis detection kit. A Cell
Proliferation and Cytotoxicity Assay Kit (Nanjing KeyGen Biotech.
Co., Ltd., Nanjing, China) was used to determine the apoptosis rate
of HepG2/ADM cells, according to the manufacturer's protocol.
Subsequently, cells were harvested and washed twice, and
resuspended in 1X binding buffer (Beyotime Institute of
Biotechnology, Haimen, China) at a concentration of
1×106 cells/ml. A 100 µl cell suspension was stained
with 5 µl Annexin V-fluorescein isothiocyanate and 5 µl PI at room
temperature for 15 min in the dark, prior to analyzing the cells
using flow cytometry.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The expression of MDR1 and MRP1 mRNA was analyzed
using RT-qPCR (14), with human
β-actin (284bp; sequence: Forward 1379: 5′-AGCGAGCATCCCCCAAAGTT-3′;
reverse, 1663: 5′-GGGCACGAAGGCTCATCATT-3′) as an internal control.
TRIzol® (Qingdao Jisskang Biotechnology Co., Ltd., Qingdao, China)
(1 ml) was added to the harvested cells in a microcentrifuge tube
and agitated until evenly mixed. Chloroform (0.2 ml) was added to
the mixture, and the microcentrifuge tube was inverted a number of
times and left to stand for 5 min at room temperature.
Subsequently, the tube was centrifuged at 4,024.8 × g for 15 min at
4°C. The upper aqueous phase (~400 µl) was transferred into a new
1.5 ml microcentrifuge tube and 400 µl propan-2-ol was added. The
contents of the tube were mixed evenly and left to stand for 10 min
at room temperature. The mixture was centrifuged at 4,024.8 × g for
10 min at 4°C. The supernatant was discarded, and the precipitate
was washed three times with 70% ice-cold ethanol and air-dried for
between 5 and 10 min. Diethylpyrocarbonate in H2O (20
µl) was added to the tube to dissolve the precipitate. The quality
and concentration of RNA was determined using a spectrophotometer.
[RNA concentration=OD260×40 µg/ml × Dilution Multiple (4)] cDNA was synthesized from total mRNA
using a PrimeScript RT reagent kit. Following first-strand cDNA
synthesis (50°C for 2 min, 95°C for 10 min, 95°C for 30 sec, then
60°C 30 sec for 40 cycles). PCR amplification using the Ex Taq™ kit
(Takara Biotechnology, Co., Ltd., Dalian, China) was conducted
using the following primers: MDR1: Forward,
5′TGATTGCATTTGGAGGACAA3′, reverse 5′CCAGAAGGCCAGAGCATAAG3′; MRP1,
forward, 5′AGGTGGACCTGTTTCGTGAC3′; reverse,
5′CCTGTGATCCACCAGAAGGT3′. DNA was amplified using the following
protocol: 2 min at 50°C and 10 min at 95°C, followed by 40 cycles
of 95°C for 30 sec and 60°C for 30 sec. Oligonucleotides and
reagents for the PCR assay were purchased from Nanjing Kingsley
Biological Technology Co., Ltd. (Nanjing, China). Data were
analyzed with Sequence Detector software (version 1.9; Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
mean Cq value for duplicate measurements was used to detect the
expression of target gene normalized to the housekeeping gene
β-actin, which was used as an internal control.
Western blot analysis
The expression of drug-resistant P-gp and MRP1 were
analyzed using western blot analysis. A total of 1.2×106
HepG2/ADM and HepG2 cells in exponential growth phase were
inoculated into 6-well plates. Cells were harvested following
treatment with U0126 and 10, 20 and 40 µmol/l ADM for 48 h at 37°C,
and subsequently washed twice with PBS. A radioimmunoprecipitation
assay lysis buffer (50 mM Tris (pH 7.4), 150mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate,
sodium fluoride, EDTA and leupeptin; Beyotime Institute of
Biotechnology) was used to homogenize cells. Following detection of
the protein concentration using a bicinchoninic acid assay
(Beyotime Institute of Biotechnology), 75 µg of each sample was
separated using SDS-PAGE (8% gel). Following separation of target
proteins from total protein determined according to size based on
that of the pre-stained markers, electrophoresis was stopped.
Following removal of the hydrogel, the target protein bands were
excised, washed with distilled water and electrotransferred onto
polyvinylidene fluoride membranes. The membrane was blocked with
TBS-5% Tween-20 (TBST) containing 5% skimmed milk powder at room
temperature for 2 h and incubated at 4°C overnight with the primary
antibodies directed against the following: MRP1 (cat. no.
GTX116046; dilution, 1:1,000; GeneTex International Corporation,
Hsinchu, Taiwan) and P-Glycoprotein (cat. no. GTX108354; dilution,
1:1,000; GeneTex International Corporation). Following washing with
TBST, the membrane was incubated at 37°C with horseradish
peroxidase-conjugated secondary antibody (cat. no. BA1054; Beyotime
Institute of Biotechnology) for 2 h. Following a further wash with
TBST, labeled proteins were visualized using an Film Development
enhanced chemiluminescence kit (cat. no. P0020; Wuhan Booute
Biotechnology Co, Ltd., Wuhan, China) on high-performance
chemiluminescence film according to the manufacturer's
protocol.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 19.0; IBM SPSS, Armonk, NY, USA). Results were
presented as the mean ± standard deviation of three replicates. A
one-way analysis of variance was applied for comparison among
multiple sets of data. The multiple comparison between the groups
was performed using the Student-Newman-Keuls method. P<0.05 was
considered to represent a statistically significant difference.
Results
HepG2/ADM cells exhibit increased
resistance to chemotherapeutic drugs compared with HepG2 cells,
which is decreased by U0126 treatment
HepG2 and HepG2/ADM cells were incubated with
increasing doses of ADM, VCR, 5-FU and MMC to determine their
sensitivities to each chemotherapeutic drug (Fig. 1). The IC50 values for each
drug were significantly increased in HepG2/ADM cells compared with
HepG2 cells (P<0.01; Table I;
Fig. 2), demonstrating that HepG2/ADM
cells exhibited drug resistance to ADM and the other
chemotherapeutic drugs.
 | Table I.Determination of IC50
values and resistance indices of various anticancer drugs. |
Table I.
Determination of IC50
values and resistance indices of various anticancer drugs.
|
| IC50 |
|
|---|
|
|
|
|
|---|
| Drug | HepG2 | HepG2/ADM | HepG2/ADM+U0126 | Resistance index |
|---|
| ADM (g/l) | 1.065±0.105 |
29.57±1.756a |
5.796±0.143b | 27.77 |
| VCR (mg/l) | 1.471±0.560 |
9.650±0.912a |
3.250±0.579b | 6.560 |
| 5-FU (mg/l) | 1.958±0.904 |
10.28±1.012a |
6.930±0.315b | 5.250 |
| MMC (mg/l) | 2.117±0.406 |
23.36±0.869a |
10.27±0.751b | 11.03 |
In order to investigate whether U0126 was able to
enhance the chemotherapeutic effects of the drugs, HepG2/ADM cells
were pre-treated with U0126 (20 µmol/l) for 24 h, followed by the
drugs for a further 48 h. Cell viability was determined using a
CCK-8 kit. Treatment with U0126 (20 µmol/l) on HepG2/ADM cells led
to a significant decrease in its IC50 values for all
four drugs compared with that of untreated HepG2/ADM cells
(P<0.01; Table I; Fig. 2), demonstrating that U0126-treated
HepG2/ADM cells exhibited increased sensitivity to ADM, VCR, 5-FU
and MMC compared with untreated HepG2/ADM cells.
U0126 induces apoptosis of HepG2/ADM
cells in a dose-dependent manner
Although U0126 was demonstrated to increase the
sensitivity of HepG2/ADM cells to the chemotherapeutic drugs ADM,
VCR, 5-FU and MMC, the underlying molecular mechanisms for the
effect of U0126 remain unclear. To investigate whether this
treatment was able to induce apoptosis in HepG2/ADM cells, flow
cytometry was used to determine the proportion of apoptotic cells
induced by treatment with U0126. The results demonstrated that
U0126 induced an increased incidence of apoptosis of HepG2/ADM
cells compared with untreated HepG2/ADM control cells (Fig. 3), and the increase in apoptosis was
identified to be dose-dependent (Fig.
4).
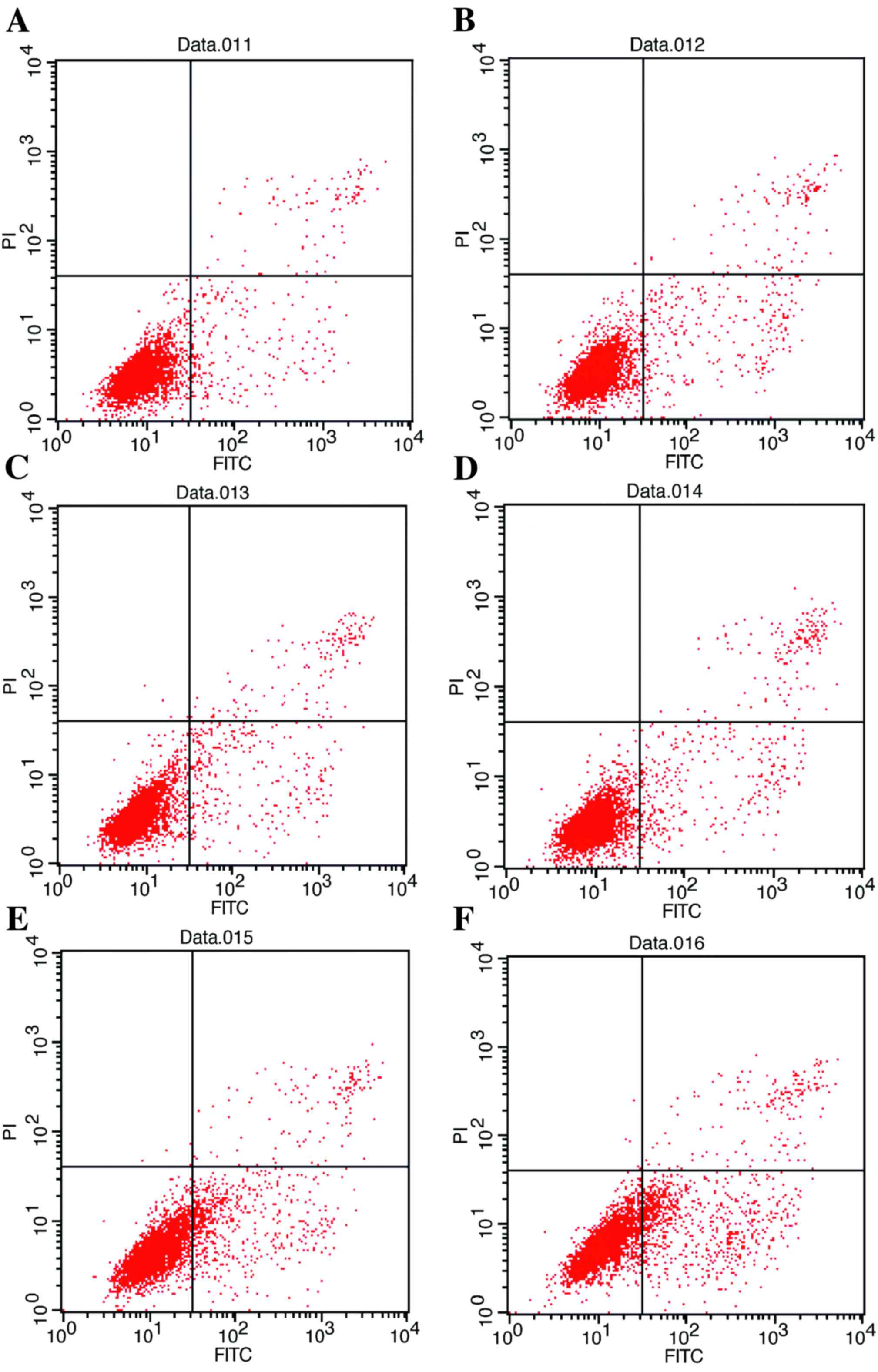 | Figure 3.Flow cytometric analysis of apoptosis
of HepG2/ADM cells. (A) Untreated HepG2/ADM cells (early apoptosis,
4.88%; late apoptosis, 1.51%). (B) HepG2/ADM cells treated with ADM
(0.4 µmol/l) after 48 h (early apoptosis, 5.49%; late apoptosis,
2.13%). (C) HepG2/ADM cells treated with U0126 (40 µmol/l) (early
apoptosis, 6.67%; late apoptosis, 3.12%). (D) HepG2/ADM cells
treated with ADM (0.4 µmol/l) and U0126 (10 µmol/l) (early
apoptosis, 10.75%; late apoptosis, 3.53%). (E) HepG2/ADM cells
treated with ADM (0.4 µmol/l) and U0126 (20 µmol/l) (early
apoptosis, 16.83%; late apoptosis, 2.93%). (F) HepG2/ADM cells
treated with ADM (0.4 µmol/l) and U0126 (40 µmol/l) (early
apoptosis, 21.15%; late apoptosis, 4.37%). ADM, Adriamycin; FITC,
fluorescein isothiocyanate; PI, propidium iodide. |
U0126 inhibits MDR1 and MRP1 mRNA
expression in HepG2/ADM cells
In order to identify the underlying molecular
mechanism for the U0126-mediated enhancement of drug sensitivity in
HepG2/ADM cells, the effects of U0126 on MDR1 and MRP1 mRNA
expression in HepG2/ADM cells were investigated using RT-qPCR.
RT-qPCR analysis demonstrated that U0126 significantly
downregulated MDR1 and MRP1 mRNA expression in a dose-dependent
manner (P<0.05; Fig. 5). MDR1 mRNA
expression in the drug-resistant HepG2/ADM cells was significantly
increased (5.37-fold) compared with that of drug-sensitive HepG2
parental cells (P<0.05; Fig. 5).
Similarly, MRP1 mRNA expression in the drug-resistant HepG2/ADM
cells was significantly increased (6.14-fold) compared with that of
drug-sensitive HepG2 parental cells (P<0.05; Fig. 5). These results indicated that a
potential underlying molecular mechanism for the U0126-mediated
enhancement of drug sensitivity in HepG2/ADM cells was the
downregulation of MDR1 and MRP1 mRNA expression.
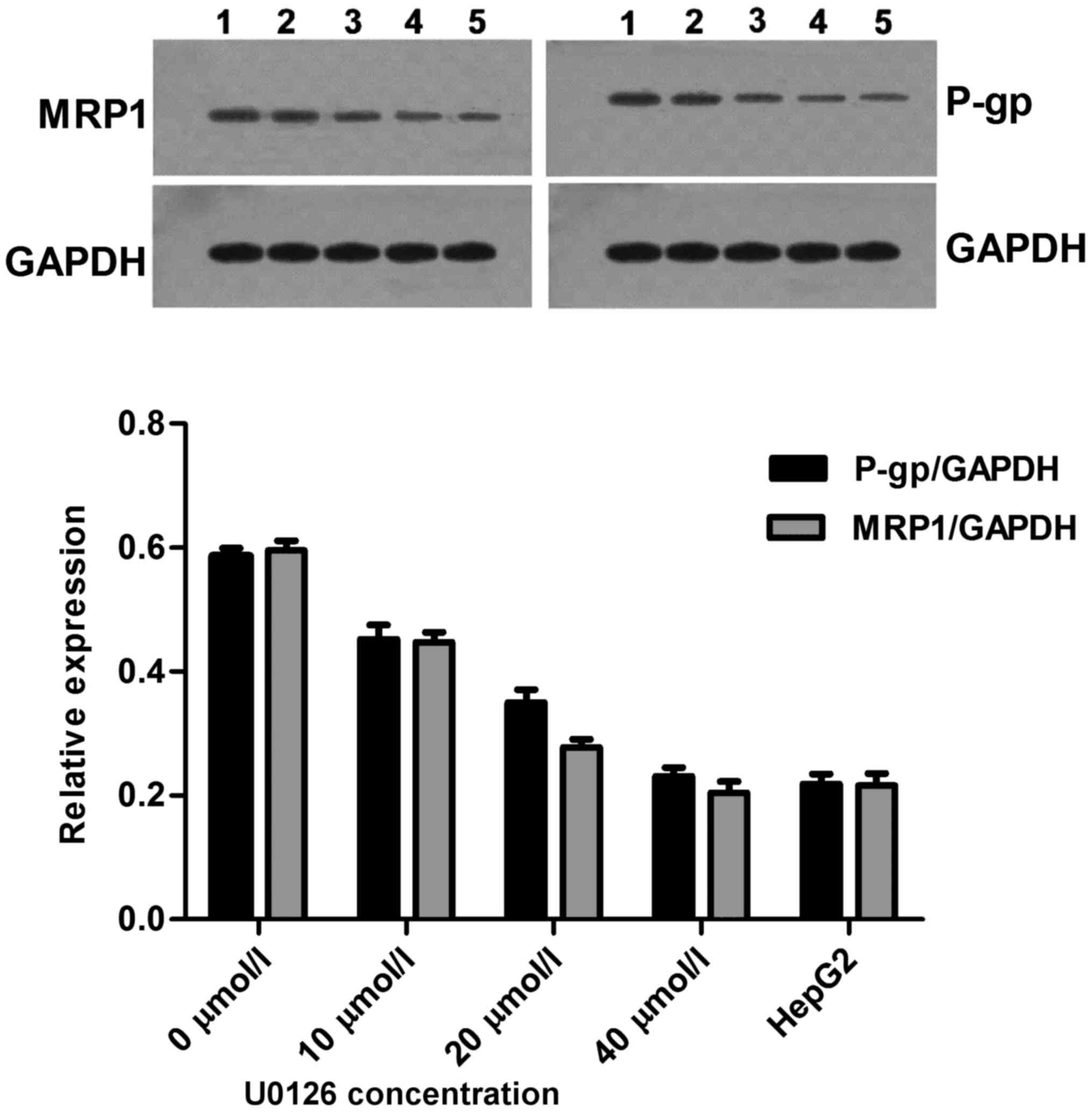
U0126 induces P-gp and MRP1 protein
downregulation
Anticancer drugs exhibit limited activity and a poor
response when used in the treatment of drug-resistant cells. P-gp
is the factor most frequently involved in MDR (15). The effects of U0126 on P-gp protein
levels in drug-sensitive HepG2 parental cells and drug-resistant
HepG2/ADM cells were investigated. Following a 24 h incubation with
U0126, a concentration-dependent decrease in P-gp levels was
detected in HepG2/ADM cells, with the decrease detectable at
concentrations of U0126 as low as 10 µmol/l. The relative
expression of P-gp protein in the drug-resistant HepG2/ADM cells
was increased 2.68-fold compared with that in drug-sensitive HepG2
parental cells (0.5857±0.0235 vs. 0.2183±0.0273, respectively;
Fig. 6). Furthermore, a number of
recent studies have demonstrated the importance of functional MRP1
in response to individual pathway inhibitors (16). For this reason, MRP1 protein
expression was determined in HepG2 and HepG2/ADM cell lines,
following 24 h treatment with increasing concentrations of U0126.
In the cell lines, a concentration-dependent decrease in MRP1
protein expression levels was demonstrated. The relative expression
of MRP1 protein in the drug-resistant HepG2/ADM cells was increased
2.76-fold compared with that of drug-sensitive HepG2 parental cells
(0.5953±0.0271 vs. 0.2153±0.0341, respectively; Fig. 6). These results indicated that U0126
enhances the sensitivity of HepG2/ADM cell to chemotherapeutic
drugs via downregulated expression of P-gp and MRP1 protein.
Discussion
MAPKs serve a critical role in the transduction of
extracellular signals into cells to regulate differentiation,
proliferation and apoptosis. Tumor cells exhibit resistance to the
chemotherapeutic drugs to which they are exposed; however, tumor
cells also exhibit resistance to chemotherapeutic drugs with a
molecular structure that has not been encountered previously as
well as to chemotherapeutic drugs that exert their effects via a
different mechanism of action (17).
HepG2/ADM cells conform to MDR standards (18) and may therefore be used to investigate
reversion of MDR.
Currently, research into the mechanism of MDR in
tumor cells has focused on the proteins P-gp, MRP and LRP, and the
enzymes glutathione transferase and DNA topoisomerase (15). In the present study, P-gp and MRP1
protein expression was demonstrated to be increased in
drug-resistant HepG2/ADM cells compared with drug-sensitive HepG2
parental cells, suggesting that typically used chemotherapeutic
drugs may lead to MDR in hepatoma cells. This is similar to the
study by Chaudhary and Roninson (19), which demonstrated that acquired
resistance may occur through the induction of short-term
chemotherapy and is retained for ~6 weeks (19). P-gp and MRP1 belong to the ABC
membrane transport protein superfamily. These proteins may mediate
the excretion of various antitumor drugs, resulting in the
concentration of the drugs being lower than the effective
concentration required to limit the proliferation of the tumor
cell; conversely, these proteins also have an effect on the
distribution of chemotherapeutic drugs in tumor cells, leading to
the acquisition of MDR by tumor cells (20). Therefore, the concentration of
chemotherapeutic drugs in tumor cells may be improved by adjusting
the expression of ABC proteins to enhance anti-tumor activity.
MAPKs are present in the majority of cell types, and
transmit extracellular stimulating signals to regulate cell growth,
differentiation, proliferation, apoptosis and other biological
effects. A number of parallel MAPK signaling pathways have been
identified, among which the ERK/MAPK signaling pathway is the
typical signal regulation and control pathway, and the c-Jun
N-terminal kinase/stress-activated protein kinase and
p38MAPK pathway frequently modulates the biological
effect under stress conditions (12).
Hu et al (21) demonstrated an
increased effect of chemotherapeutic drugs on H460 non-small cell
lung cancer cells following suppression of the ERK/MAPK signaling
pathway. Eum et al (22)
demonstrated that the Raf/MEK/ERK signaling pathway inhibitor
PLX4720 may reverse the drug resistance of NIH3T3/MDR cells by
decreasing the expression of P-gp, which is consistent with the
results of the present study. Using the MEK inhibitor U0126 in
HepG2/ADM cells, it was demonstrated that the treatment may inhibit
the proliferation of HepG2/ADM cells and promote apoptosis;
furthermore, with increasing concentrations, U0126 was able to
decrease the mRNA and protein expression of P-gp and MRP1,
suggesting that the drug resistance of HepG2/ADM cells may be
reversed. However, further research is required to identify the
specific association and underlying molecular mechanism.
The association between MAPK and MDR protein remains
unclear, and the underlying molecular mechanism of MDR regulation
by MAPK also remains unclear. Lee et al (23) reported that another signaling pathway
[the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)
signaling pathway] is associated with MDR: PI3K activation in
prostate cancer drug-resistant cell was demonstrated to generate
MDR by increasing the expression of MRP1. In leukemic
drug-resistant cells, the drug resistance of MRP1 was decreased by
inhibiting the PI3K/AKT signal transmission pathway (24); however, whether they are associated
remains unclear. The regulation and underlying molecular mechanism
of MDR is complicated, which may result from combined regulation
and control of numerous signaling pathways.
In conclusion, the HepG2/ADM cell model of MDR was
used to investigate the underlying molecular mechanism for
resistance and to identify a drug that may overcome drug
resistance. The MEK inhibitor U0126 may decrease the mRNA and
protein expression levels of drug-resistant proteins, and promote
apoptosis of drug-resistant cells, which provides a theoretical
basis for combined chemotherapy in the treatment of liver
cancer.
References
|
1
|
International Agency for Research on
Cancer (IARC), . GLOBOCAN 2008: Cancer Incidence and Mortality
Worldwide. IARC; Lyon: 2010, https://www.iarc.fr/en/media-centre/iarcnews/2010/globocan2008.phpMay
1–2010
|
|
2
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fang Y, Shang QL, Liu JY, Li D, Xu WZ,
Teng X, Zhao HW, Fu LJ, Zhang FM and Gu HX: Prevalence of occult
hepatitis B virus infection among hepatopathy patients and health
people in China. J Infect. 58:383–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Al-Rawashdeh FY, Scriven P, Cameron IC,
Vergani PV and Wyld L: Unfolded protein response activation
contributes to chemoresistance in hepatocellular carcinoma. Eur J
Gastroenterol Hepatol. 22:1099–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: Role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee CH: Reversing agents for ATP-binding
cassette drug transporters. Methods Mol Biol. 596:325–340. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anders M, Christian C, McMahon M,
McCormick F and Korn WM: Inhibition of the Raf/MEK/ERK pathway
up-regulates expression of the coxsackievirus and adenovirus
receptor in cancer cells. Cancer Res. 63:2088–2095. 2003.PubMed/NCBI
|
|
8
|
Viala E and Pouysségur J: Regulation of
tumor cell motility by ERK mitogen-activated protein kinases. Ann N
Y Acad Sci. 1030:208–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilkinson MG and Millar JB: Control of the
eukaryotic cell cycle by MAP kinase signaling pathways. FASEB J.
14:2147–2157. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shen H, Xu W, Luo W, Zhou L, Yong W, Chen
F, Wu C, Chen Q and Han X: Upregulation of mdr1 gene is related to
activation of the MARK/ERK signal transduction pathway and YB-1
nuclear translocation in B-cell lymphoma. Exp Hematol. 39:558–569.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abrams SL, Steelman LS, Shelton JG, Wong
EW, Chappell WH, Bäsecke J, Stivala F, Donia M, Nicoletti F, Libra
M, et al: The Raf/MEK/ERK pathway can govern drug resistance,
apoptosis and sensitivity to targeted therapy. Cell Cycle.
9:1781–1791. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Katayama K, Yoshioka S, Tsukahara S,
Mitsuhashi J and Sugimoto Y: Inhibition of the mitogen-activated
protein kinase pathway results in the down-regulation of
P-glycoprotein. Mol Cancer Ther. 6:2092–2102. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berger W, Setinek U, Hollaus P, Zidek T,
Steiner E, Elbling L, Cantonati H, Attems J, Gsur A and Micksche M:
Multidrug resistance markers P-glycoprotein, multidrug resistance
protein1, and lung resistance protein in non-small cell lung
cancer: Prognostic implications. J Cancer Res Clin Oncol.
131:355–363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Zhang J, Zhang L, Zhao L, Fan S,
Yang Z, Gao F, Kong Y, Xiao GG and Wang Q: Expression of P-gp, MRP,
LRP, GST-π and TopoIIα and intrinsic resistance in human lung
cancer cell lines. Oncol Rep. 26:1081–1089. 2011.PubMed/NCBI
|
|
16
|
Tivnan A, Zakaria Z, O'Leary C, Kögel D,
Pokorny JL, Sarkaria JN and Prehn JH: Inhibition of multidrug
resistance protein1 (MRP1) improves chemotherapy drug response in
primary and recurrent glioblastoma multiforme. Front Neurosci.
9:2182015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stavrovskaya AA: Cellular mechanisms of
multidrug resistance of tumor cells. Biochemistry (Mosc).
65:95–106. 2000.PubMed/NCBI
|
|
18
|
Snow K and Judd W: Characterisation of
Adriamycin- and amsacrine-resistant human leukaemic T cell lines.
Br J Cancer. 63:17–28. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chaudhary PM and Roninson IB: Induction of
multidrug resistance in human cells by transient exposure to
different chemotherapeutic drugs. J Natl Cancer Inst. 85:632–639.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang XK and Fu LW: Interaction of tyrosine
kinase inhibitors with the MDR-related ABC transporter proteins.
Curr Drug Metab. 11:618–628. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu Y, Bally M, Dragowska WH and Mayer L:
Inhibition of mitogen-activated protein kinase/extracellular
signal-regulated kinase enhances chemotherapeutic effects on H460
human non-small cell lung cancer cells through activation of
apoptosis. Mol Cancer Ther. 2:641–649. 2003.PubMed/NCBI
|
|
22
|
Eum KH, Ahn SK, Kang H and Lee M:
Differential inhibitory effects of two Raf-targeting drugs,
sorafenib and PLX4720, on the growth of multidrug-resistant cells.
Mol Cell Biochem. 372:65–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JT Jr, Steelman LS and McCubrey JA:
Phosphatidylinositol 3′-kinase activation leads to multidrug
resistance protein-1 expression and subsequent chemoresistance in
advanced prostate cancer cell. Cancer Res. 64:8397–8404. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tazzari PL, Cappellini A, Ricci F,
Evangelisti C, Papa V, Grafone T, Martinelli G, Conte R, Cocco L,
McCubrey JA and Martelli AM: Multidrug resistance-associated
protein 1 expression is under the control of the phosphoinositide 3
kinase/Akt signal transduction network in human acute myelogenous
leukemia blasts. Leukemia. 21:427–438. 2007. View Article : Google Scholar : PubMed/NCBI
|