Introduction
Subarachnoid hemorrhage (SAH), typically caused by
the rupture of an intracranial aneurysm, is a fatal disease with
high associated morbidity and mortality rates (1). The annual incidence of SAH is ~22.5
cases per 100,000 population, according to a World Health
Organization study (2). SAH has a
mortality rate of 40–50%, and the prognosis of survivors is poor
(3,4).
This is associated with a notable economic burden as 50% of SAH
patients are <55 years old (5–7).
Experimental evidence supports the hypothesis that neurological
deterioration and a poor outcome are a response to cerebral
vasospasm subsequent to SAH (8).
However, a number of patients undergo neurological deterioation
without an accompanying vasospasm and preventing vasospasm may not
always improve the clinical outcome (9). There is research to suggest that delayed
neuronal and astrocytic apoposis is an important contributor to a
poor outcome in patients with SAH (10,11). The
present study aimed to assess the factors responsible for this
process and outline the associated future possibilities for the
improvement of SAH treatment.
MSK (mitogen- and stress-activated protein kinase)
proteins are a particularly interesting family of mitogen-activated
protein kinases. They were originally identified through their
homology with the N-terminal ribosomal S6 kinase domain (12). MSK1 is a nuclear protein kinase with
two kinase domains, including a C-terminal kinase domain related to
the Ca2+/calmodulin-dependent protein kinase family and
an N-terminal kinase domain related to the AGC kinase family
(13,14). It can be activated downstream of the
mitogen-activated protein kinase (MAPK) 2/1 or MAPK 11/14 cascades
with the phosphorylation of Thr581 in the C-terminal kinase domain
(15). Once activated, the N-terminal
domain phosphorylates a variety of substrates, including nuclear
factor κB (NFκB), cAMP responsive element binding protein (CREB)
(16), histone subunit H3, and
high-mobility group nucleosome binding domain 1 (HMGN1). The major
role of MSKs in the CNS is to regulate the immediate early (IE)
genes, plasticate neuronal synapses and promote cytokine
production. The number of identified MSK1 targets continues to
increase (17). It was previously
established that MSKs are a novel type of pro-survival gene that
enhances the phosphorylation of Bcl-2-associated death promoter
(Bad) (18). Phosphorylation of Bad
at Ser112 in response to growth factors or cytokines is a common
mechanism for the promotion of cell survival; MSK1 knockdown was
previously demonstrated to suppress Bad phosphorylation after
calcium ionophore A23187 treatment in neuronal cells (18).
However, the function and expression of MSK1 in the
central nervous system (CNS) have yet to be well-characterized. It
is established that MSK1 is highly expressed in the nervous system
(19), but its function is not well
understood. In the present study, the expression and distribution
of MSK1 in the brain were examined following the experimental
induction of SAH in rats. The study aimed to identify the
physiological functions of MSK1 and the molecular mechanisms
underlying lesion and repair in the CNS.
Materials and methods
Animals and surgical procedures
A total of 48 3-months old male Sprague-Dawley rats
(280–320 g) were used. Rats were kept in the laboratory's
temperature of 20°C and the humidity was 60%. The rats were raised
with a 12-h light/dark cycle and free access to water and food. The
animals were anesthetized with 10% chloral hydrate and positioned
in a stereotactic frame with their heads tilted ~30° downwards. A
midline scalp incision was made in the neck and the
atlanto-occipital membrane was exposed subsequent to separating the
muscle layers. The atlanto-occipital membrane was punctured with a
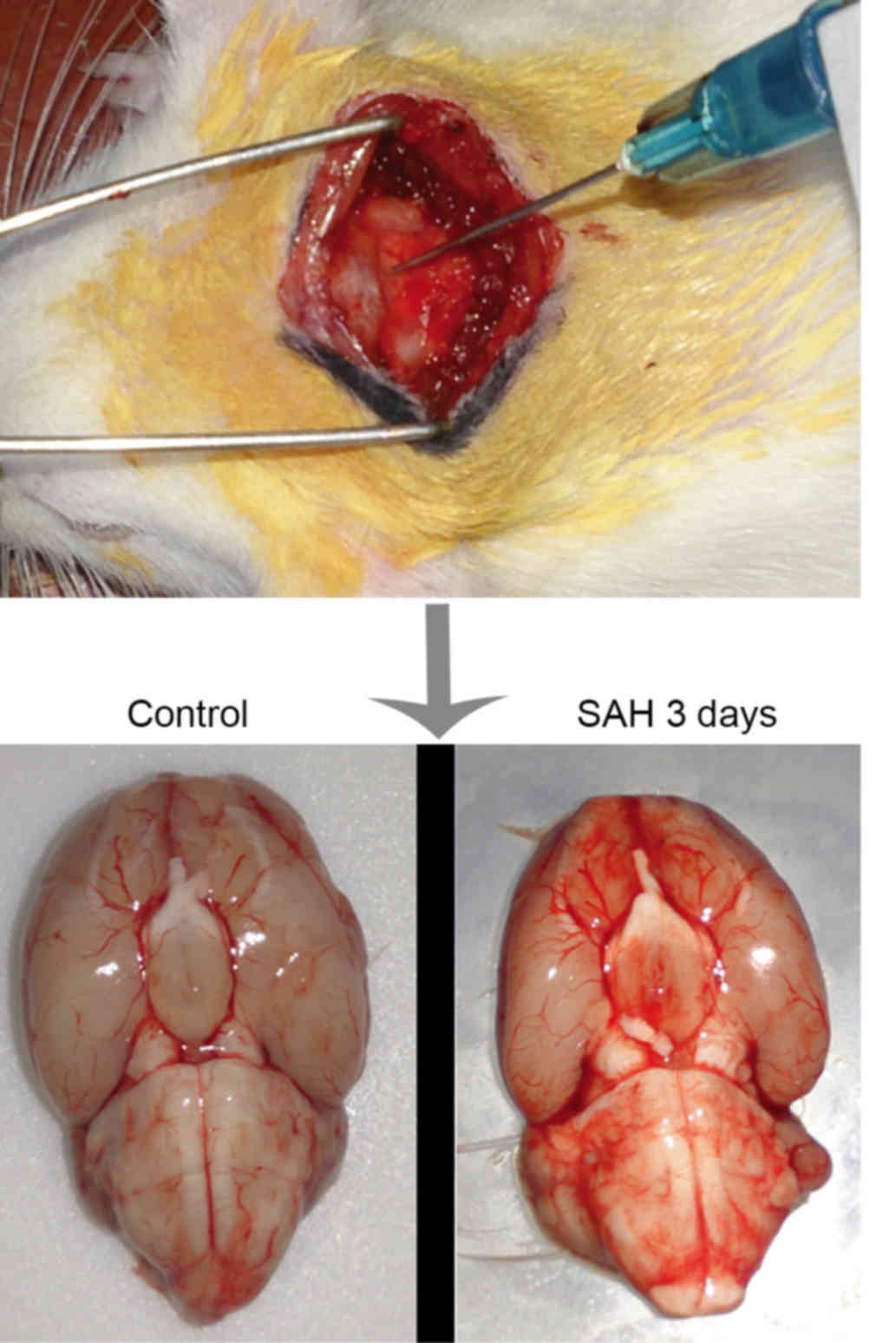
needle (Fig. 1), and 0.3 ml
autologous arterial blood was injected into the cisterna magna with
a squirt pump within 10 min. A second injection of blood was
performed with the same method following a days recovery. The
control group were injected with 0.3 ml sterile saline. Rats had
free access to water and food during recovery from anesthesia. The
animals with induced SAH were randomly divided into five sub-groups
and sacrificed by decapitation on day 1, 3, 5, 7 or 14 post-SAH,
(n=6; Fig. 1). A further 6 rats with
SAH were sacrificed with ventricle perfusion for
immunohistochemical and immunofluorescence studies on day 3. An
additional control group, sham animals (n=6), experienced the same
surgery process without the injection into the cisterna magna. Sham
group animals were then sacrificed 24 h after the sham operation.
All rats were supplied by Taishan Medical University (Taian, China)
and all surgical interventions and postoperative animal care were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals (National Research Council, 1996, USA) and were
approved by the Ethics Committee of Animal Experiments of Taishan
Medical University. All surgery was performed under 10% chloral
hydrate and every effort was made to minimize suffering.
Western blotting
For western blot analysis, brain tissues were
homogenized in lysis buffer (1% sodium deoxycholate, 50 mmol/l
Tris, 1% NP-40, 1% Triton X-100, 5 mmol/l EDTA, 1% SDS, 1 mmol/l
phenylmethane sulfonyl fluoride, 10 µg/ml aprotinin and 1 µg/ml
leupeptin) and clarified by centrifuging at 14,000 × g for 15 min
at 4°C, from which the supernatant was collected. Then a BCA kit
(Beyotime Institute of Biotechnology, Haimen, China) was used to
determinate the protein concentration. Samples (80 µg/lane) were
subjected to 10% SDS-PAGE for 40 min at 70 V followed by 90 min at
120V and then transferred onto a PVDF membrane by a transfer
apparatus at 180 mA for 2.5 h. The membrane was blocked with 5%
skimmed milk for 2 h at 20°C and then incubated with the
appropriate primary antibodies (MSK1, ab81294, 1:200, Abcam,
Cambridge, UK; active caspase-3, ab49822, 1:200, Abcam; neuronal
nuclear antigen, ab177487, 1:100, Abcam; glial fibrillary acidic
protein, sc-71143, 1:100, Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) at 4°C overnight. The membrane was washed three times in
TBST and incubated with goat-anti-rabbit (sc-2030, 1:500, Santa
Cruz Biotechnology, USA) or goat-anti-mouse IgG (sc-2031, 1:500,
Santa Cruz Biotechnology, USA) conjugated to horseradish peroxidase
antibody for 2 h at room temperature. The blotted protein bands
were developed with enhanced chemiluminescence reagent (Thermo
Fisher Scientific, Inc.) exposed on X-ray film. The relative
density of each band compared with GADPH was estimated with Scion
Image software version 4.0.3.2 (Scion Corporation, Frederick, MD,
USA).
Double immunofluorescence
staining
Brain tissue was fixed with 4% paraformaldehyde for
3 h, then 20% saccharose solution for 2 days, then 30% sucrose
solution for 2 days to remove water from the sample, all these
procedures were performed at 4°C. Sections of 8 µm thickness were
prepared and blocked with 5% normal fetal bovine serumin in PBS
containing 0.1% Triton X-100 for 2 h. The slices were then
incubated overnight at 4°C with primary antibodies against MSK1
(cat. no., ab81294, 1:200, Abcam); active caspase-3 (cat. no.,
ab49822, 1:200); neuronal nuclear antigen (cat. no., ab177487,
1:100, Abcam); glial fibrillary acidic protein (cat. no.,
sc-71,143, 1:100, Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Subsequently, secondary antibodies goat-anti-rabbit IgG (cat. no.,
sc-2030, 1:500, Santa Cruz Biotechnology, Inc.) were added and
incubated for 2 h at room temperature in the dark. Following 3
washes in PBS, the slides were observed under a fluorescence
microscope (Leica Microsystems GmbH, Wetzlar, Germany). Negative
controls omitted the primary antibodies.
Immunohistochemistry
Frozen cross-sections (8 µm) were prepared and
blocked with 5% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) in PBS for 2 h at room temperature. Then each of
the sections was incubated with the anti-MSK1 antibody (cat. no.,
ab81294, 1:200, Abcam) overnight at 4°C followed by incubation in
biotinylated Goat Anti-Rat IgG Antibody (cat. no., BP-9400, 1:500,
Vector Laboratories, Inc., Burlingame, CA, USA). Staining was
visualized with 3,3′-diaminobenzidine (DAB, Vector Laboratories,
Inc.). Staining was visualized with 3,3′-diaminobenzidine. Cells
with strong or moderate brown staining were considered as
positive.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
TRIzol reagent (Takara Biotechnology Co., Ltd.,
Dalian, China) was used to isolate RNA from rat tissues and the
concentration of RNA was measured with spectrophotometric analysis
(optical density at 260/280). Total RNA was reversely transcribed
into cDNA using a 25-µl mixture [Primer Mix (12 µl), 5xRT Reaction
Buffer (5 µl), 25 mM dNTPs (1 µl), 25 U/µl RNase Inhibitor (1 µl),
200 U/µl M-MLV Rtase (1 µl), Oligo (dt) 18 (1 µl) and
ddH2O (DNase-free; 4 µl)] at 37°C for 60 min, 85°C for 5
min and 4°C for 5 min. cDNA was used as template for PCR (Prism
7300 Real-Time PCR System, Applied Biosystems; Thermo Fisher
Scientific, Inc., USA) and the mixture used for PCR included
SYBR-Green Mix (12.5 µl; Invitrogen; Thermo Fisher Scientific,
Inc.), forward primer (0.5 µl), reverse primer (0.5 µl),
ddH2O (9.5 µl) and cDNA (2 µl). PCR was performed under
the following conditions: 95°C for 10 min; 30 cycles of 95°C for 30
sec, 60°C for 30 sec and 70°C for 30 sec. Conventional PCR was
performed with 4 µl cDNA, 12.5 µl Taq MasterMix (CWBIO, Beijing,
China), 1 µl of each forward and reverse primer (10 µM), and
RNase-free water to a final 25 µl. Conventional PCR amplification
was performed with a PTC-200 Peltier Thermal Cycler (MJ Research;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). The following
primers were used: MSK1 forward, 5′-CCTCAAGACCCCATGCTTCA-3′ and
reverse, 5′-ACTTCTGTCATGGGACTGGA-3′; and GAPDH forward,
5′-GAGGCCGGTGCTGAGTATGT-3′ and reverse, 5′-GGTGGCAGTGATGGCATGGA-3′.
GAPDH was used as an endogenous control and the
2−∆∆Cq method was used to quantify relative
mRNA expression (20).
Statistical analysis
At least three replicates were performed per
condition in each experiment. All values are expressed as the mean
± standard error of the mean. SPSS version 21.0 (IBM SPSS, Armonk,
NY, USA) was used for statistical analysis of the data. The
statistical significance of differences between groups was
determined by the Kruskal-Wallis test and Dunnett's multiple
comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of MSK1 is reduced at the
protein and mRNA levels following SAH
MSK1 exhibits a high-level expression in the nervous
system (18), however the function
has not been well understood. Western blot analysis,
immunohistochemistry, and conventional and quantitative PCR were
performed in order to investigate the expression profiles of MSK1
in the cortex of rats following simulated SAH.
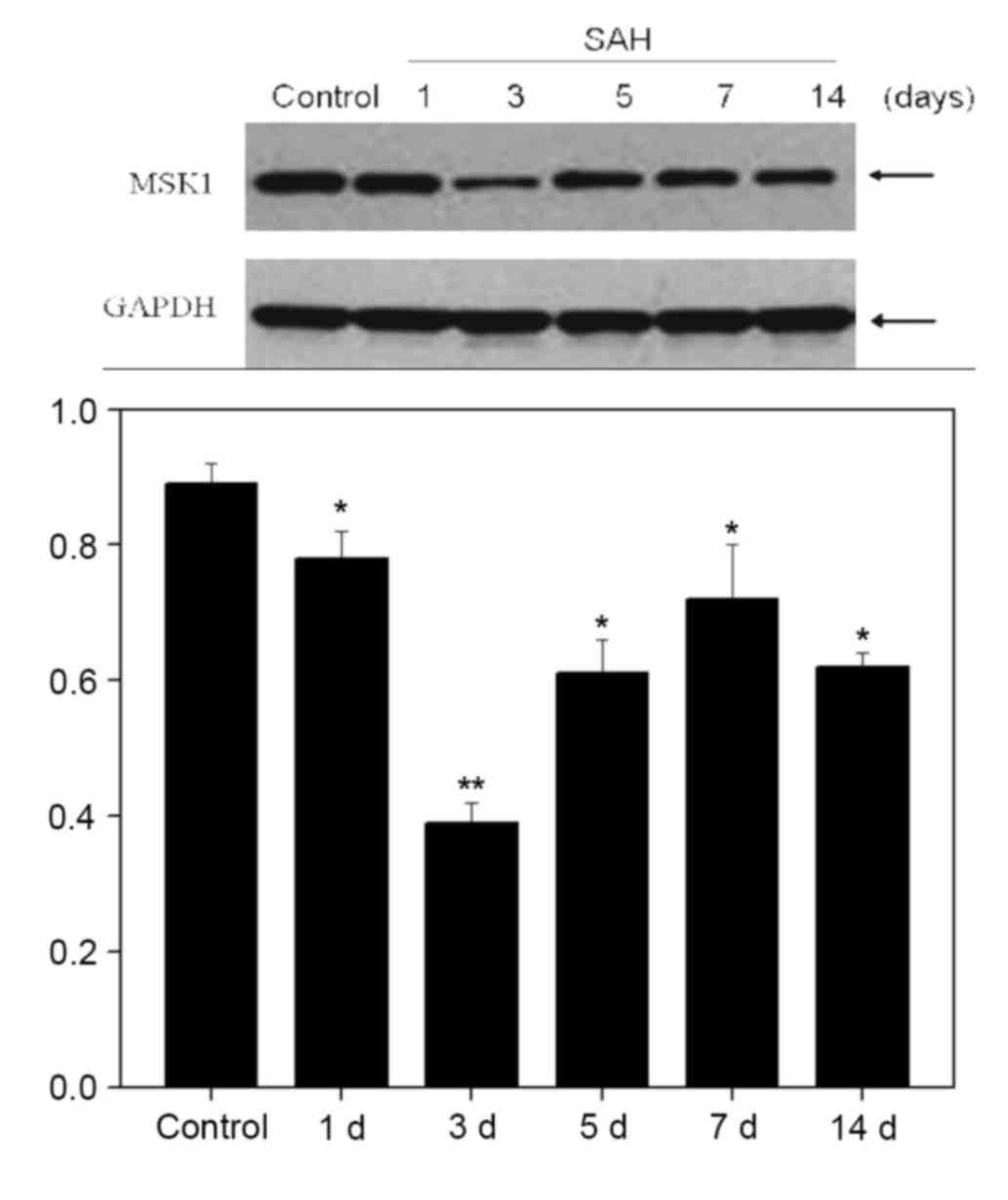
Western blot analysis at 1, 3, 5, 7 and 14 days
demonstrated that the expression of MSK1 gradually reduced to a low
point at 3 days after SAH, and recovered on day 5 onwards
(P<0.05; Fig. 2).
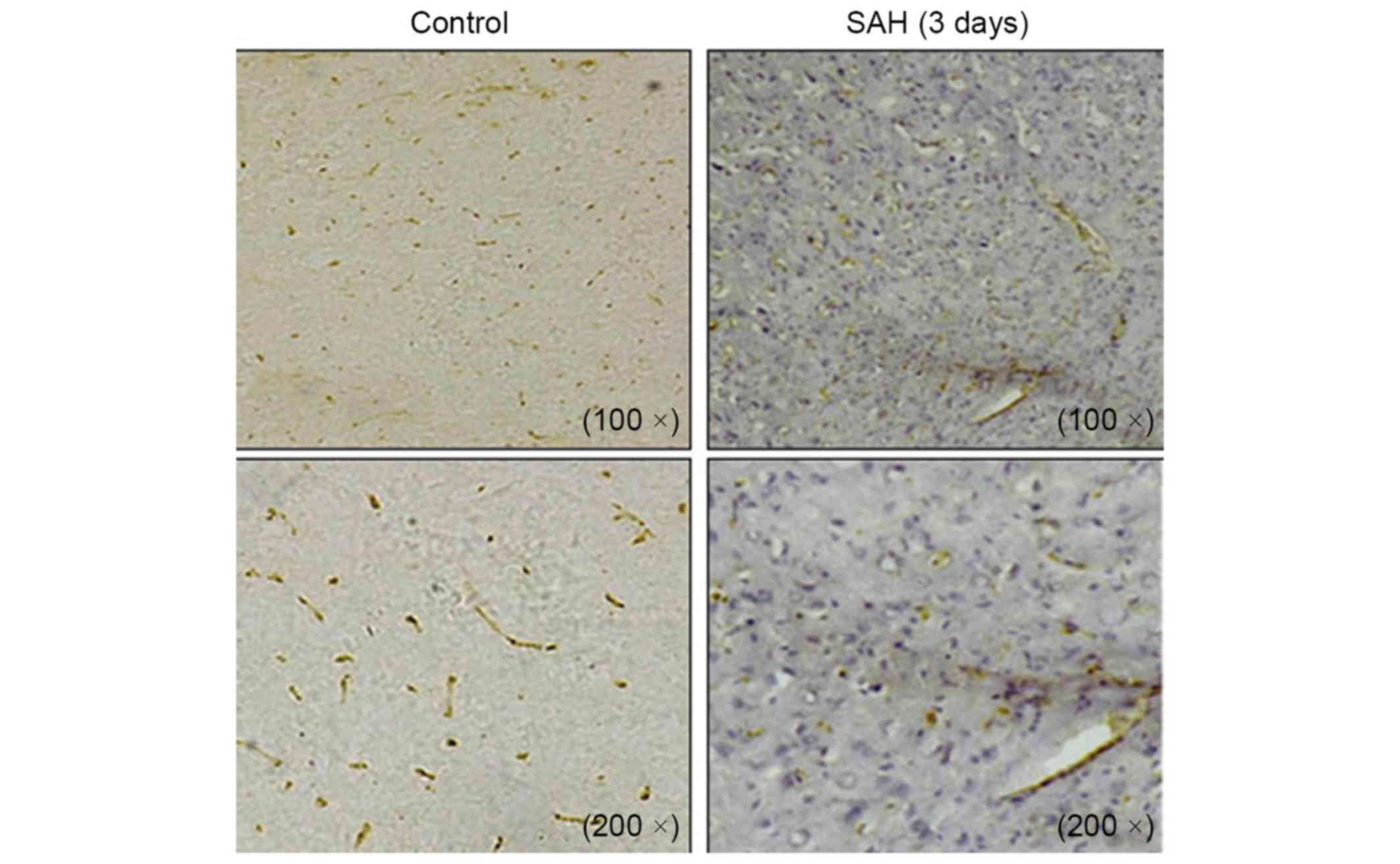
Immunohistochemical staining on cross sections of the rat brain
also demonstrated the differential expression of MSK1 between the
control and SAH groups at 3 days. Abundant MSK1-positive cells were
detected in the control group, whereas MSK1 positivity was visibly
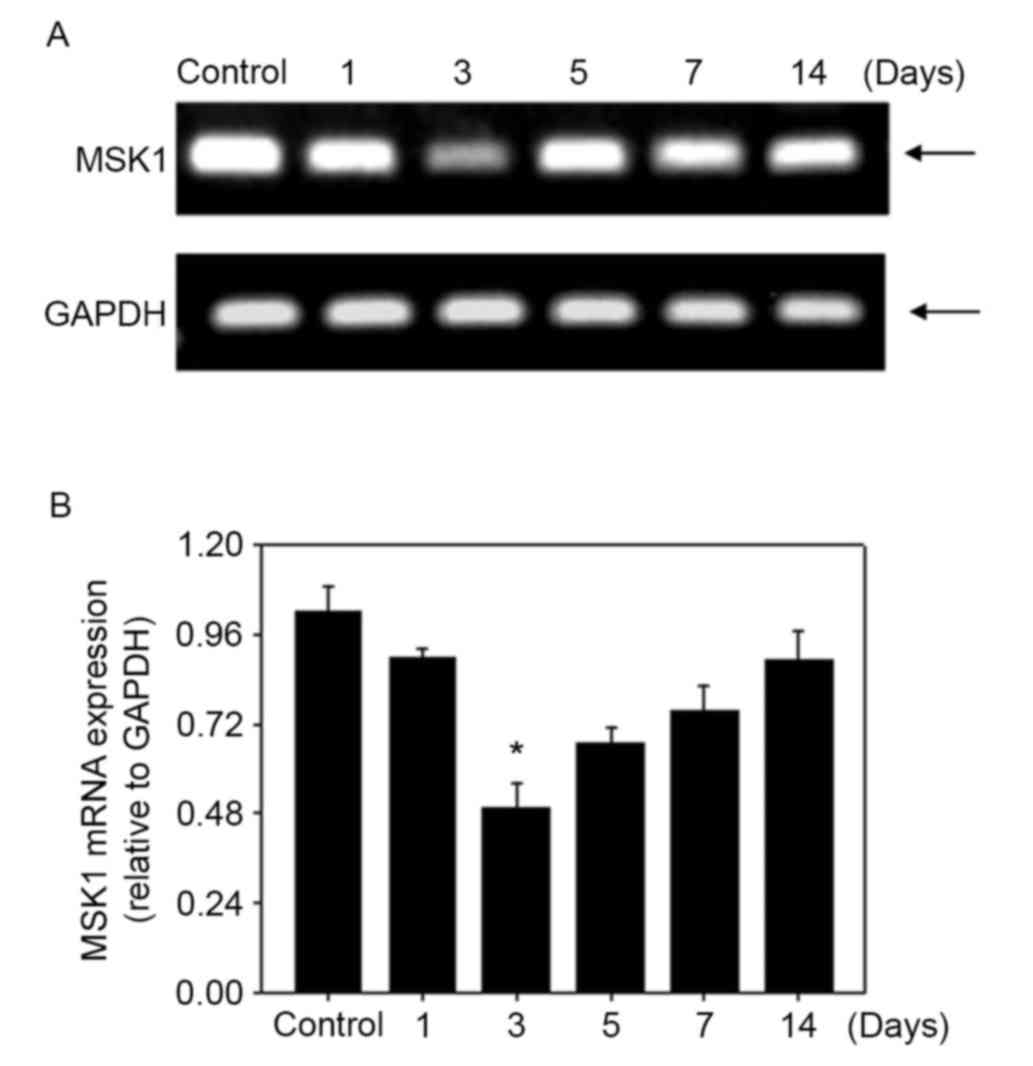
reduced in the brain at 3 days after SAH (Fig. 3). MSK1 mRNA was reverse transcribed
and the relative level was assessed with qPCR and conventional PCR.
MSK1 mRNA was expressed at a relatively high level in the control
group and was significantly decreased at 3 days in the SAH group.
The expression level reached the lowest point at 3 days after SAH,
and recovered over the following days, similar to the western blot
result (Fig. 4). The results
indicated that MSK1 may be associated with SAH-induced brain
damage.
Detection of neuron and astrocyte
apoptosis following SAH
Apoptosis serves a vital function in the control of
cell numbers and removal of damaged cells. A critical step in the
apoptosis process is the activation of caspases. The effect of MSK1
on the apoptosis of neurons and astrocytes in the CNS following SAH
remains unclear.
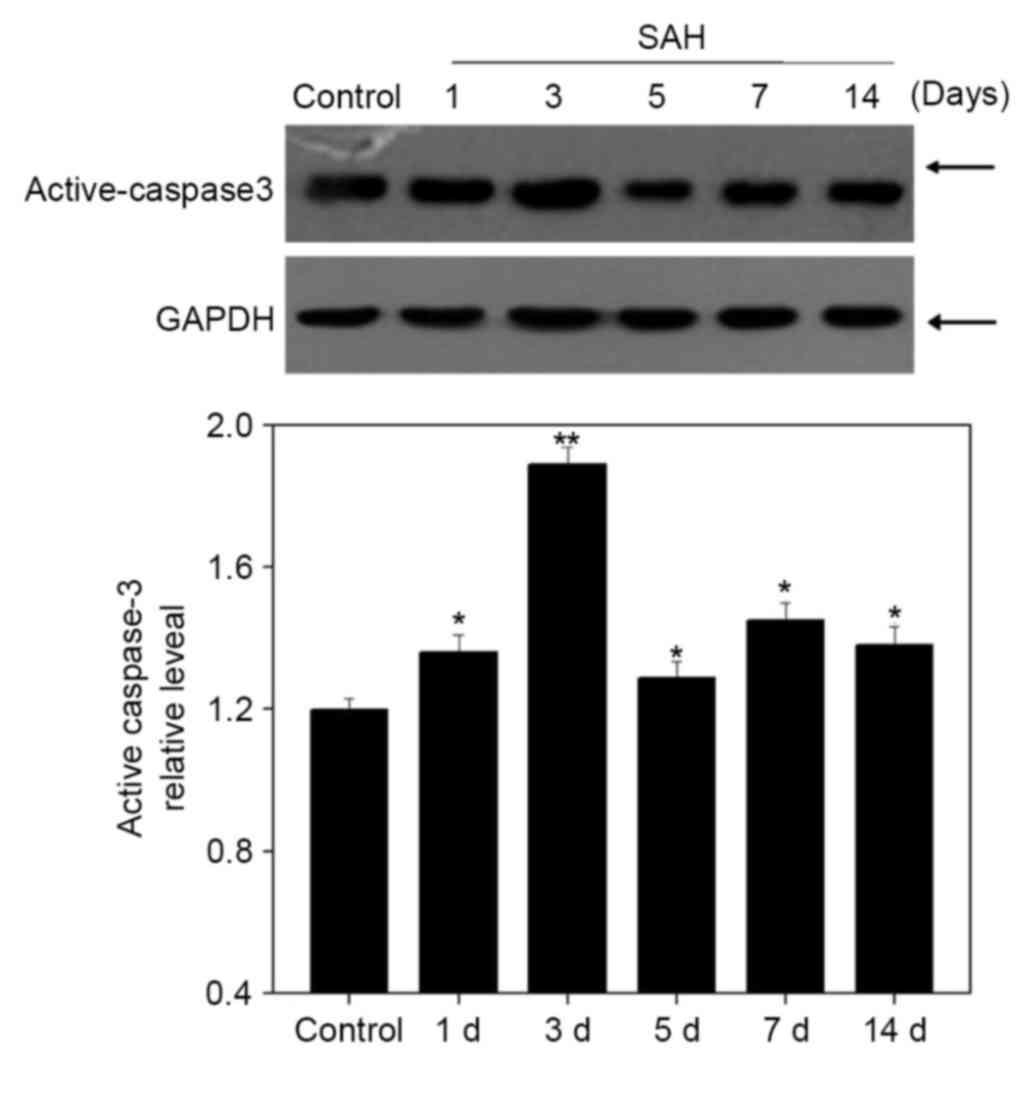
A western blot analysis was performed to examine the
expression of active caspase-3 (Fig.
5). Its expression was gradually elevated after SAH to a peak
at 3 days (P<0.05), which was negatively associated with MSK1
expression (Fig. 2).
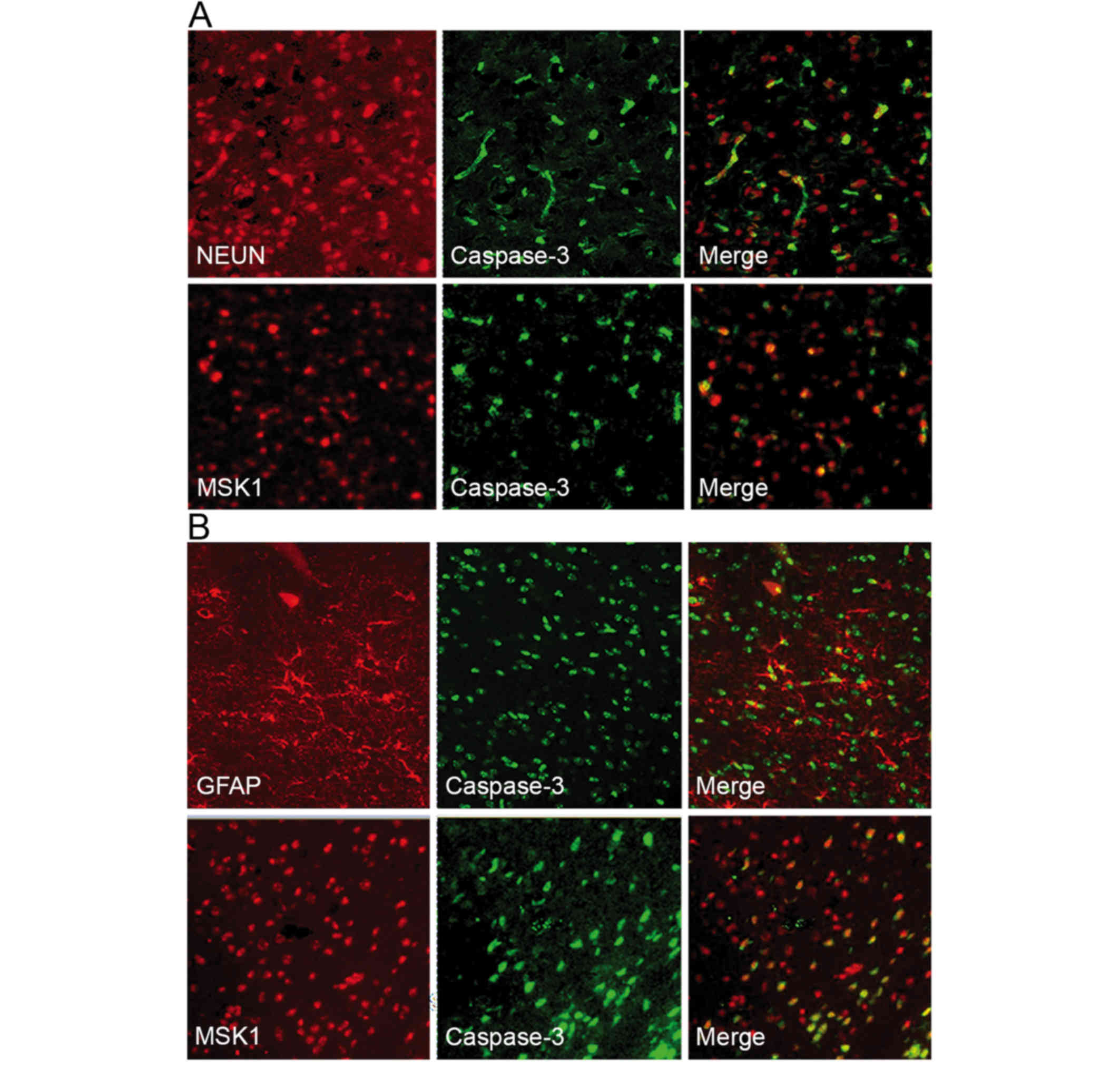
In order to examine the association between MSK1 and
apoptotic neurons at 3 days following SAH, double
immunofluorescence staining for active caspase-3 and MSK1 or NeuN
was performed. Active caspase-3 and NeuN were observed to
co-localize, suggesting that neurons were undergoing apoptosis.
Furthermore, when MSK1 and active caspase-3 fluorescence images
were merged, MSK1 and active caspase-3 appeared to frequently
co-localize (Fig. 6A).
To assess the association between MSK1 and apoptotic
astrocytes, double immunofluorescence staining for GFAP or MSK1 and
active caspase-3 was performed. GFAP and active caspase-3
immunoreactivity were observed in the cortex at day 3 post-SAH.
Double labeling revealed the co-localization of active caspase-3
with GFAP, suggesting that there were apoptotic astrocytes.
Additionally, MSK1 expression was observed in a number of apoptotic
astrocytes as evaluated by active caspase-3 staining. MSK1
reactivity coincided with astrocyte apoptosis in adjacent serial
sections (Fig. 6B). These data
indicated that MSK1 may serve a role in the apoposis of neurons and
astrocytes following SAH.
Discussion
SAH is accompanied by a number of complicated
molecular mechanisms in the brain which may result in ongoing
cellular damage, the formation of scar tissue and neurological
dysfunction. Several events, including increased intracranial
pressure, transient global ischemia and blood clots obstructing the
cerebral vasculature, may be responsible for the development of
brain damage following SAH (21,22).
Experimental evidence supports that vasospasms induce neurological
deterioration in the processes of secondary brain dysfunction
(23,24). However, clinical evidence suggests
that certain patients deteriorate neurologically without vasospasms
subsequent to SAH and preventing vasospasms does not always improve
the patient outcome (9). A possible
reason is that other processes injure neurons following SAH.
Previous reseach has revealed that the delayed apoposis of neurons
and astrocytes may be an important reason for a poor outcome in
patients with SAH (10,11). The purpose of the present study was to
identify if MSK1, a protein from a particularly interesting family
of MAPKs, is associated with this process, and to outline future
possibilities of for improving the treatment of patients subsequent
to SAH.
MSK1 contains two kinase domains and can be
activated in vivo downstream of the MAPK2/mitogen-activated
protein kinase 1 or MAPK11/14 cascades by the phosphorylation of
Thr581, located within the C-terminal kinase domain (15). Once activated, the N-terminal domain
phosphorylates substrates including NFκB, CREB, histone subunit H3,
and HMGN1. In the present study, the dynamic changes to MSK1
expression following SAH were investigated with a model of
autologous blood injections. Double immunofluorescence staining
revealed that MSK1 expression occurred in neurons and astrocytes at
3 days following SAH. These data correspond with the hypothesis
that MSK1 is associated with the pathophysiology of the CNS
following SAH. Furthermore, it can be concluded that MSK1 might
perform an important role the molecular mechanisms of brain damage
subsequent to SAH.
MSKs regulate the IE genes, plasticate neuronal
synapses and accommodate cytokine production. It was previously
established that MSK1 represents a novel type of anti-cell death
gene, which may enhance the phosphorylation of Bcl2-associated
agonist of cell death (Bad) (25).
The phosphorylation of Bad at Ser112 in response to growth factors
or cytokines is a common mechanism for cell survival. The knockdown
of MSK1 suppressed Bad phosphorylation subsequent to calcium
ionophore A23187 treatment in neuronal cells in a previous study
(18). In the present study, the
expression of active caspase-3, which can initiate and effect the
process of apoptosis, was negatively correlated with MSK1. Double
immunofluorescence staining demonstrated that active caspase-3 and
NeuN were co-localized in the rat brain at 3 days subsequent to
SAH. Furthermore, MSK1 fluorescence overlapped with active
caspase-3 fluorescence. These data suggested that MSK1 may be
associated with neuronal apoptosis subsequent to SAH. The detailed
mechanisms for this require further study.
Astrocytes are one of the main types of cell that
constitute the normal CNS parenchyma. CNS regeneration requires a
largely astrocytic environment (26).
However, the role of the astrocyte is under debate. Although
astrocytes secrete important growth factors for neurons and prevent
damage signals from spreading throughout the brain, the role
following CNS injury appears detrimental to neuronal survival,
axonal outgrowth and remyelination, preventing repair processes
(27–29). The data of the present study revealed
the co-localization of MSK1/active caspase-3 and GFAP/active
caspase-3 in the brains at 3 days subsequent to SAH. These data
indicated that MSK1 may serve a function in the procedure of
astrocytic apoposis subsequent to SAH. Though the same comments as
the previous paragraph regarding neuronal death apply, the cause of
astrocyte apoposis following SAH remains unknown.
In summary, the present study revealed, for the
first time, the expression of MSK1 following SAH. The
co-localization and correlating changes in expression of
MSK1/active caspase-3 at neurons and astrocytes indicated that MSK1
downregulation may contribute to SAH-induced apoptosis. To further
understand the effect of MSK1 in the diversity of responses that
may occur subsequent to SAH is a challenge for future
investigations.
References
|
1
|
Cahill J, Calvert JW and Zhang JH:
Mechanisms of early brain injury after subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 26:1341–1353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schuette AJ and Barrow DL: Epidemiology
and long-term mortality in subarachnoid hemorrhage. World Neurosur.
80:264–265. 2013. View Article : Google Scholar
|
|
3
|
Ingall TJ and Whisnant JP: Epidemiology of
Subarachnoid HemorrhageYanagihara T, Piepgras DG and Atkinson JLD:
Subarachnoid Hemorrhage. Medical and Surgical Management. Marcel
Dekker; New York: pp. 63–78. 1998
|
|
4
|
Taylor TN, Davis PH, Torner JC, Holmes J,
Meyer JW and Jacobson MF: Lifetime cost of stroke in the United
States. Stroke. 27:1459–1466. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Epidemiology of aneurysmal subarachnoid
hemorrhage in Australia and New Zealand: Incidence and case
fatality from the australasian cooperative research on subarachnoid
hemorrhage study (ACROSS). Stroke. 31:1843–1850. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van Gijn J, Kerr RS and Rinkel GJ:
Subarachnoid haemorrhage. Lancet. 369:306–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kooijman E, Nijboer CH, van Velthoven CT,
Mol W, Dijkhuizen RM, Kesecioglu J and Heijnen CJ: Long-term
functional consequences and ongoing cerebral inflammation after
subarachnoid hemorrhage in the rat. PLoS One. 9:e905842014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nolan CP and Macdonald RL: Can
angiographic vasospasm be used as a surrogate marker in evaluating
therapeutic interventions for cerebral vasospasm? Neurosurg Focus.
21:E12006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Macdonald RL, Kakarieka A, Mayer SA,
Pasqualin A, Rufenacht DA, Schmiedek P and Kassell NF: Prevention
of cerebral vasospasm after aneurysmal subarachnoid hemorrhage with
clazosentan, an endothelin receptor antagonist. Neurosurgery.
59:4532006. View Article : Google Scholar
|
|
10
|
Hansen-Schwartz J, Vajkoczy P, Macdonald
RL, Pluta RM and Zhang JH: Cerebral vasospasm: Looking beyond
vasoconstriction. Trends Pharmacol Sci. 28:252–256. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Macdonald RL, Pluta RM and Zhang JH:
Cerebral vasospasm after subarachnoid hemorrhage: The emerging
revolution. Nat Clin Pract Neurol. 3:256–263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deak M, Clifton AD, Lucocq LM and Alessi
DR: Mitogen- and stress-activated protein kinase-1 (MSK1) is
directly activated by MAPK and SAPK2/p38, and maymediate activation
of CREB. EMBO J. 17:4426–4441. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pierrat B, Correia JS, Mary JL,
Tomás-Zuber M and Lesslauer W: RSK-B, a novel ribosomal S6
kinasefamily member, is a CREB kinase under dominant control of
p38amitogen-activated protein kinase (p38aMAPK). J Biol Chem.
273:29661–29671. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Drobic B, Espino PS and Davie JR: Mitogen-
and stress-activated protein kinase 1activity and histone h3
phosphorylation in oncogene-transformed mouse fibroblasts. Cancer
Res. 64:9076–9079. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arthur JS: MSK activation and
physiological roles. Front Biosci. 13:5866–5879. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johansen KM and Johansen J: Regulation of
chromatin structure by histone H3S10 phosphorylation. Chromosome
Res. 14:393–404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deak M, Clifton AD, Lucocq LM and Alessi
DR: Mitogen- and stress-activated protein kinase-1 (MSK1) is
directly activated by MAPK and SAPK2/p38, and may mediate
activation of CREB. EMBO J. 17:4426–4441. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clark CJ, McDade DM, O'Shaughnessy CT and
Morris BJ: Contrasting roles of neuronal Msk1 and Rsk2 in Bad
phosphorylation and feedback regulation of Erk signalling. J
Neurochem. 102:1024–1034. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ning B, Li Z, Zhu N, Hou G and Pang Q:
Traumatic brain injury induces a downregulation of MSK1 in rat
brain cortex. J Mol Neurosci. 49:380–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prunell GF, Svendgaard NA, Alkass K and
Mathiesen T: Delayed cell death related to acute cerebral blood
flow changes following subarachnoid hemorrhage in the rat brain. J
Neurosurg. 102:1046–1054. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeon H, Ai J, Sabri M, Tariq A and
Macdonald RL: Learning deficits after experimental subarachnoid
hemorrhage in rats. Neuroscience. 169:1805–1814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dziurdzik P, Krawczyk L, Jalowiecki P,
Kondera-Anasz Z and Menon L: Serum interleukin-10 in ICU patients
with severe acute central nervous system injuries. Inflamm Res.
53:338–334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ostrowski RP, Colohan AR and Zhang JH:
Molecular mechanisms of early brain injury after subarachnoid
hemorrhage. Neurol Res. 28:399–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
She QB, Ma WY, Zhong S and Dong Z:
Activation of JNK1, RSK2, and MSK1 is involved in serine 112
phosphorylation of Bad by ultraviolet B radiation. J Biol Chem.
277:24039–24048. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fawcett JW and Asher RA: The glial scar
and central nervoussystem repair. Brain Res Bull. 49:377–391. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Franklin RJM and Ffrench-Constant C:
Remyelination in the CNS: From biology to therapy. Nat Rev
Neurosci. 9:839–855. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Segovia KN, McClure M, Moravec M, Luo NL,
Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS and Back SA:
Arrested oligodendrocyte lineage maturation in chronic perinatal
white matter injury. Ann Neurol. 63:520–530. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fawcett JW: Astrocytic and neuronal
factors affecting axon regeneration in the damaged central nervous
system. Cell Tissue Res. 290:371–377. 1997. View Article : Google Scholar : PubMed/NCBI
|