Introduction
Colorectal cancer (CRC) is one of the most common
types of cancer in men and women, with ~1.5 million new cases and
~0.5 million mortalities having been reported in 2013 in the United
States (1). In recent decades, the
mortality caused by CRC has decreased dramatically owing to the
great improvement in early diagnosis and treatment (2). However, CRC remains a prominent global
health problem that may be attributed to the lack of comprehensive
and systemic understanding of the underlying molecular mechanisms
of carcinogenesis.
The accumulation of specific genetic and epigenetic
changes is considered to be the main molecular mechanism of
tumorigenesis, as it can provide a selective growth advantage of
tumor cells over neighboring normal cells (3). Among the epigenetic changes, the
abnormal methylation of promoter CpG islands leading to the
transcriptional inactivation of tumor suppressors is considered to
be a common mechanism in several human malignancies including CRC
(4). Epigenetic masking may
participate in the cancerous transformation of colorectal
epithelium by affecting the expression of tumor suppressor genes
(4). Recent progress in CRC
epigenetics studies indicated DNA methylation occurs in the early
phase of cancer formation and in the premalignant phase of the
adenoma-carcinoma sequence (5). Thus,
identifying the epigenetic alterations would be of great value in
the early detection of cancers and cancer relapse, as well as in
monitoring the response of cancers to therapies (6).
Epigenetically silenced genes by hypermethylation
can be reactivated by 5-aza-2′-deoxycytidine (5-aza-dC), which is
able to inhibit DNA methylation (7).
In addition, the re-expression of silenced genes caused by 5-aza-dC
has been demonstrated in various types of tumors in a dose- and
duration-dependent manner (7). The
application of 5-aza-dC in expression microarray analysis is
considered to be a useful approach for identifying
cancer-associated methylated genes (8).
In order to elucidate silenced genes with abnormal
methylation in CRC, Khamas et al (9) performed a genome-wide expression
screening in 5 CRC cell lines prior and subsequent to 5-aza-dC
treatment, and subsequently combined the data with CRC-specific
gene expression profiling array. The gene expression data set
established by Khamas et al (9) was submitted to the Gene Expression
Omnibus (GEO) with the accession number GSE32323. In the present
study, the microarray was downloaded and analyzed to identify
potential targets for 5-aza-dC by oligonucleotide microarray
analysis. A co-expression network of CRC-specific gene expression
profile was constructed using the context likelihood of relatedness
(CLR) algorithm to identify the signaling pathways in which these
targets were involved, thus revealing the function of the selected
identified genes.
Materials and methods
Affymetrix microarray data
Transcriptional profile of GSE32323 (9) was extracted from the GEO database
(http://www.ncbi.nlm.nih.gov/geo/), which
was based on the platform of Affymetrix Human Genome U133 Plus 2.0
Array. A total of 44 chips were available for further analysis,
including 17 pairs of cancer and non-cancerous tissues from CRC
patients, and expression profiles of 5 CRC cell lines.
Data preprocessing
The raw probe-level data in CEL files were initially
converted into expression measures. Robust multiarray average
background correction, quantile normalization and probe
summarization were subsequently performed in the R (version: 3.0.3,
March, 2014) affy package (http://www.bioconductor.org/packages/release/bioc/html/affy.html)
(10), and the processed expression
matrixes were acquired. For each sample, the expression values of
all probes for a given gene were expressed as a single value by
taking an average of the values.
Differentially expressed genes (DEGs)
analysis
The limma (11)
package (http://www.bioconductor.org/packages/2.9/bioc/html/limma.html)
in R was used to identify DEGs in the present study. The following
thresholds were set for filtering DEGs: |log2
fold-change (FC)|>1.0 and P-value<0.05. The original P-values
were adjusted using Benjamini-Hochberg procedure to correct for
multiple comparisons. For CRC cell lines, gene differential
expression was calculated from each sample prior and subsequent to
5-aza-dC treatment. Only DEGs with co-upregulated or
co-downregulated expression in ≥3 cell lines were selected and
grouped as ‘DEG1’. For CRC tissues, DEGs in CRC tissue samples
compared to non-cancerous tissue were identified and grouped as
‘DEG2’. A comparison was subsequently performed between ‘DEG1’ and
‘DEG2’. The DEGs that simultaneously upregulated in ‘DEG1’ and
downregulated in ‘DEG2’, or simultaneously downregulated in ‘DEG1’
and upregulated in ‘DEG2’ were defined as reverse-overlapped DEGs,
and were screened for further analysis.
Co-expression network inference and
analysis
To identify interactions between genes, the CLR
algorithm was used to construct the co-expression network
(DEG2.CEN) in the CRC tissue samples. The CLR threshold was set as
2.5. The sub-network (roDEG.CEN) that associated with
reverse-overlapped DEGs was selected from DEG2.CEN by employing the
package MINET (http://www.bioconductor.org/packages/3.4/bioc/html/minet.html)
(12) implemented in R/Bioconductor
(version: 3.4; http://www.bioconductor.org/) and subsequently
visualized using Cytoscape (version 3.4.0; http://www.cytoscape.org/) (13).
The CLR algorithm (14) is an extension of the relevance network
approach, which increases the contrast between physical
interactions and indirect associations and takes into account the
context of each interaction and association. Links are assigned
based on the mutual information (MI) that can accommodate
non-linear associations between pair-wise gene expression patterns.
The most probable interactions are those whose MI scores stand
significantly above the background distribution of MI scores. The
MI for two discrete random variables X and Y is defined as:
MI=I(X;Y)=∑i,jP(xi,yj)logP(xi,yj)P(xi)P(yj)
where xi, yj represent ith and
jth expression level of X and Y, respectively. P(xi) and P(yj) are
the marginal probability distributions. P(xi,
yj) is the joint distribution that the expression levels
of X and Y are xi and yj, respectively
(14).
Pathway enrichment analysis
The Database for Annotation Visualization and
Integrated Discovery (15) provides a
comprehensive set of functional annotation tools to elucidate
biological meaning behind large lists of genes or proteins. Kyoto
Encyclopedia of Genes and Genomes (KEGG) analysis was performed in
the present study for functional pathway enrichment of
reverse-overlapped DEGs with P<0.05 selected as a cut-off
criterion. In addition, the enriched functional pathways of
reverse-overlapped DEGs and roDEG.CEN were integrated. Thus, a
reverse-overlappedDEG would be correlated with a particular
enriched functional pathway if the neighboring genes of this
particular reverse-overlapped DEG were involved.
Results
Identification of DEGs
For database GSE32323 (|log2 FC|>1.0;
P<0.05), a total of 59 DEG1 s in five CRC cell lines prior and
subsequent to 5-aza-dC treatment, including 48 upregulated and 11
downregulated genes, were identified. A total of 1,341 DEG2 s with
675 upregulated and 666 downregulated genes were selected when CRC
and normal tissue samples were compared. Following comparing
between the ‘DEG1’ and ‘DEG2’ groups, 10 reverse-overlapped DEGs
were selected (Table I). Among the
identified reverse-overlapped DEGs, 6 genes [amine oxidase, copper
containing 3, fibulin-2 (FBLN2), uridine phosphorylase 1,
cysteine-rich protein 1, protein phosphatase 1, regulatory
inhibitor subunit 14A (PPP1R14A; CPI-17) and heat shock 70 kDa
protein 2] were downregulated in CRC tissue sample, but upregulated
in CRC cell lines following treatment with 5-aza-dC.
 | Table I.The characteristics of identified
reverse-overlapped differentially expressed genes. |
Table I.
The characteristics of identified
reverse-overlapped differentially expressed genes.
| Gene | EntrezID | Name | aDE_State | Degree |
|---|
| AOC3 |
8,639 | Amine oxidase, copper
containing 3 | down | 76 |
| FBLN2 |
2,199 | Fibulin 2 | down | 66 |
| UPP1 |
7,378 | Uridine
phosphorylase 1 | down | 64 |
| MIPEP |
4,285 | Mitochondrial
intermediate peptidase | up | 63 |
| RRM2 |
6,241 | Ribonucleotide
reductase M2 | up | 59 |
| KIF11 |
3,832 | Kinesin family
member 11 | up | 54 |
| PPP1R14A | 94,274 | Protein phosphatase
1, regulatory inhibitor subunit 14A | down | 54 |
| HSPA2 |
3,306 | Heat shock 70 kDa
protein 2 | down | 47 |
| SLC12A2 |
6,558 | Solute carrier
family 12 (sodium/potassium/chloride transporter), member 2 | up | 45 |
| CRIP1 |
1,396 | Cysteine-rich
protein 1 (intestinal) | down | 39 |
Co-expression network of DEGs
The co-expression network of DEGs (DEG2.CEN) was
constructed by employing the CLR algorithm. The co-expression
network was based on the DEG2 expression profile in the CRC tissue
samples and the sub-network (roDEG.CEN) that correlated with the
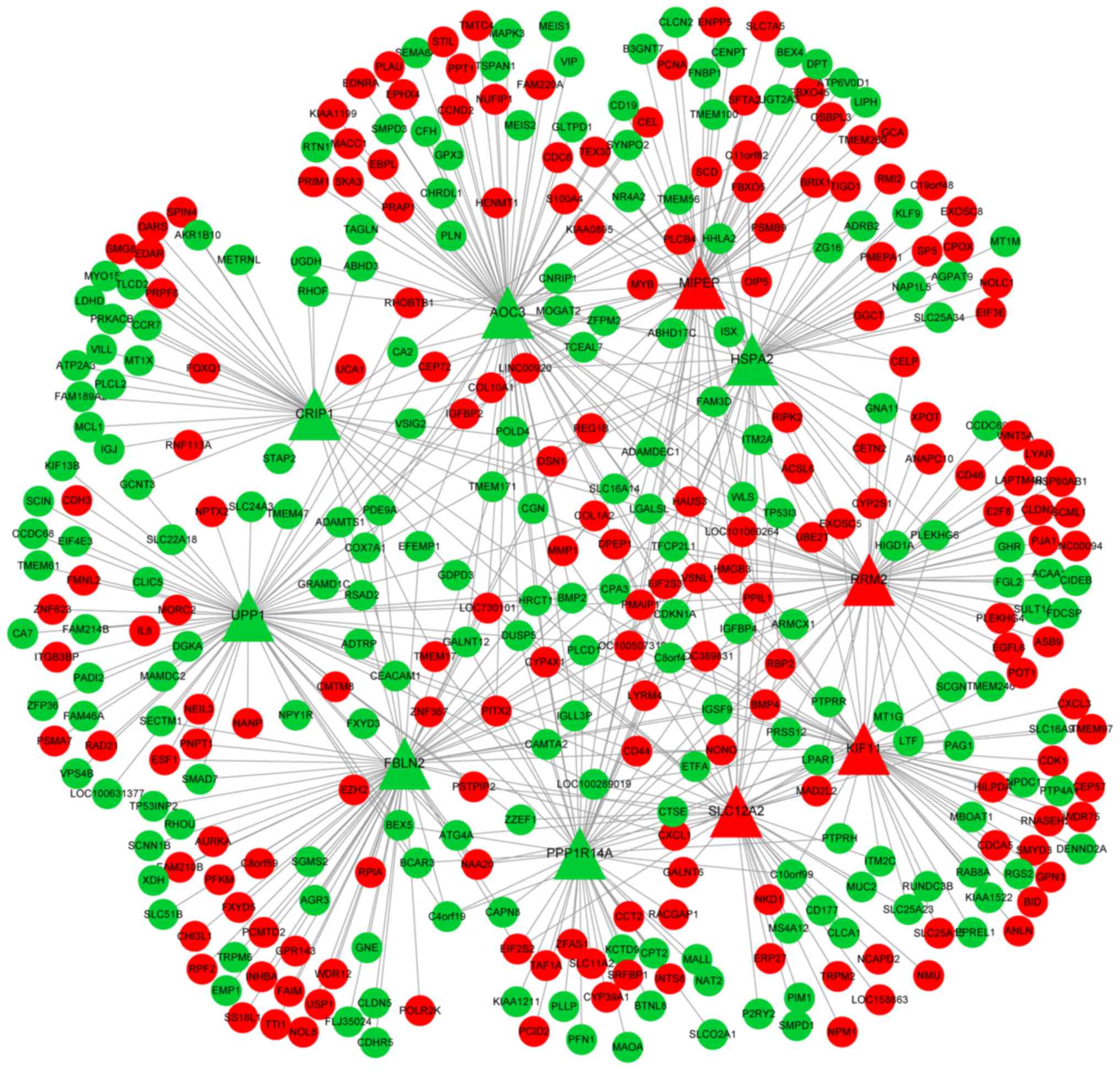
identified reverse-overlapped DEGs (Fig.
1). There were 374 nodes and 567 edges in roDEG.CEN. The number
of edges emerging from a node was set as the degree of a DEG, as
shown in Table I.
Functional pathway analysis of
network
Following integrating the roDEG.CEN with the
enriched functional pathway, the downregulated genes were enriched
in the drug metabolism pathway, while the upregulated genes were
enriched in the cellular tumor antigen p53, cell cycle, oocyte
meiosis and nucleotide-binding oligomerization domain-like
receptors (NLR) signaling pathways (Fig.
2).
 | Figure 2.Sub-network of associated Kyoto
Encyclopedia of Genes and Genomes signaling pathways in colorectal
cancer. Red triangle nodes represent upregulated genes and green
triangle nodes represent downregulated genes. Red rectangles
represent upregulated gene-enriched pathways and green rectangles
represent downregulated gene-enriched pathways. AOC3, amine
oxidase, copper containing 3; FBLN2, fibulin 2; KIF11, kinesin
family member 11; HSPA2, heat shock 70 kDa protein 2; MIPEP,
mitochondrial intermediate peptidase; PPP1R14A, protein phosphatase
1, regulatory inhibitor subunit 14A; RRM2, ribonucleotide reductase
M2; SLC12A2, solute carrier family 12 (sodium/potassium/chloride
transporter), member 2; UPP1, uridine phosphorylase 1. |
Discussion
Significant progress has been achieved in the
diagnosis and treatment of CRC. However, CRC remains the third most
common cancer worldwide (16). In the
present study, the mRNA expression profile of GSE32323 was
downloaded and DEGs were analyzed. The DEGs in 5 CRC cell lines
prior and subsequent to 5-aza-dC treatment were combined with the
CRC-specific gene expression profiling array. A total of 6
reverse-overlapped DEGs were obtained. These reverse-overlapped
DEGs were downregulated in the CRC tissue samples but upregulated
in CRC cell lines following 5-aza-dC treatment. The CLR algorithm
was employed to construct a co-expression network of CRC-specific
gene expression profile and the sub-network that correlated with
reverse-overlapped DEGs was selected. Furthermore, functional
pathway analysis identified the reverse-overlapped DEGs enriched in
a number of critical cellular pathways, including p53, cell cycle
and the NLR signaling pathway.
The 6 reverse-overlapped DEGs identified in the
present study were involved in a variety of cellular functions.
Among them, two genes (FBLN2 and PPP1R14A) have been previously
reported to be hypermethylated (17,18).
FBLN2, an extracellular matrix (ECM) protein, is recognized as a
multifunctional binding protein (19). Due to its ability to mediate
interactions between diverse ECM components, FBLN2 plays an
important role in the maintenance of extracellular structures such
as the basement membranes, as well as contacts between cells and
ECM (20,21). FBLN2 has also been reported to have
the opposite effects in pathological conditions including cancer.
The pro-tumor effects of FBLN2 were demonstrated in pancreatic
cancer cells (22); however, an
increasing number of studies indicate that FBLN2 may act as an
anti-angiogenic factor in various types of cancer, including
nasopharyngeal carcinoma (18,23,24),
as well as an anti-tumor factor in breast cancer cells (25). In addition, FBLN2 has been previously
demonstrated to be epigenetically silenced in B cell acute
lymphoblastic leukemia (26) and
methylated in breast and other epithelial cancer types (27). In the present study, FBLN2 was
downregulated in the CRC tissue sample but upregulated in CRC cell
lines following treatment with 5-aza-dC.
PPP1R14A was also identified to be methylated in
CRC. Following treatment with 5-aza-dC, the expression of PPP1R14A
increased significantly. PPP1R14A is a phosphorylation-dependent
inhibitory protein of smooth muscle myosin phosphatase activity
(28), which has been reported to be
an epigenetic biomarker in CRC (29).
The PPP1R14A gene has also been reported to be associated with
growth arrest and DNA damage (30).
Following treatment with anti-cancer drugs, including Fluorouracil,
PPP1R14A is upregulated (31). In
addition, a previous study has demonstrated that PPP1R14A is
aberrantly methylated in human esophageal squamous cell carcinoma
(18) and various types of B-cell
non-Hodgkin lymphoma (32).
A co-expression network based on the data of the
CRC-specific gene expression profile was constructed using the CLR
algorithm and the sub-network corresponding to reverse-overlapped
DEGs was selected. Bias from uneven conditions of sampling,
upstream regulation and inter-laboratory variations in microarray
can make it difficult to infer network between genes (14). CLR algorithm increases the contrast
between the physical interactions and the indirect associations by
taking the context of each interaction and association into
consideration, thus minimizing the bias from these factors
(14). Therefore, the CLR algorithm
is an attractive method to use for the identification of indirect
links and for uncovering associations between genes within
co-regulated communities. The CLR algorithm estimates a likelihood
of the MI score for a particular pair of genes by comparing the MI
values for that particular pair of genes to a background
distribution of MI values. The most probable interactions are those
whose MI scores are significantly above the background distribution
of MI scores (11). Following KEGG
pathway enrichment, the reverse-overlapped DEGs identified in the
present study were demonstrated to be enriched in several pathways,
including p53, cell cycle and NLR signaling pathways. As previously
reported, p53, cell cycle and NLR receptor signaling pathways are
closely associated with tumorigenesis and metastasis (33–35). The
results of co-expression network analysis indicated the identified
reverse-overlapped DEGs may be important tumor suppressors and are
inactivated by methylation in CRC.
Compared to the previous study published by Khamas
et al (9), the criteria used
for selecting DEGs in the present study were different. Khamas
et al (9) selected probe sets
from cell lines using a combination of two criteria: Upregulation
of gene expression in ≥4 CRC cell lines and FC>1.6 in at least
one cell line. In the present study, DEGs were selected if they
were co-upregulated or co-downregulated in ≥3 cell lines and at the
same time FC>2 in at least one cell line. Due to the difference
in threshold selection for DEGs, the identified genes in the
present study were different from the previously published report
(9). Furthermore, a co-expression
network was constructed using the CLR algorithm and the sub-network
correlated with the identified genes was selected. Pathway
enrichment analysis was performed to reveal the function of
identified genes.
There were a number of limitations in the present
study. The expression of the identified targets (FBLN2 and
PPP1R14A), as well as the association of the methylation status of
these genes with the development of CRC, remains to be confirmed by
future investigations.
In the present study, two silenced genes FBLN2 and
PPP1R14A with abnormal methylation in CRC were identified.
Furthermore, the co-expression network of identified DEGs in the
CRC tissue samples was constructed by employing the CLR algorithm
and a sub-network of reverse-overlapped DEGs was selected.
Functional pathway analysis indicated that the identified
reverse-overlapped DEGs were enriched in a number of pathways,
including p53, cell cycle and NLR signaling pathway. The results of
the present study may provide novel targets for the treatment of
CRC.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Ward E and Thun M: Declining
death rates reflect progress against cancer. PLoS One. 5:e95842010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bender CM, Pao MM and Jones PA: Inhibition
of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth
of human tumor cell lines. Cancer Res. 58:95–101. 1998.PubMed/NCBI
|
|
4
|
Kim MS, Lee J and Sidransky D: DNA
methylation markers in colorectal cancer. Cancer Metastasis Rev.
29:181–206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S,
Carter KT, Morris SM, Wang J, Willis JE, Makar KW, Ulrich CM, et
al: Differences in DNA methylation signatures reveal multiple
pathways of progression from adenoma to colorectal cancer.
Gastroenterology. 147:418–429.e8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zitt M, Zitt M and Müller HM: DNA
methylation in colorectal cancer-impact on screening and therapy
monitoring modalities? Dis Markers. 23:51–71. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takai N and Narahara H: Array-based
approaches for the identification of epigenetic silenced tumor
suppressor genes. Curr Genomics. 9:22–24. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carmona FJ and Esteller M: Epigenomics of
human colon cancer. Mutat Res. 693:53–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khamas A, Ishikawa T, Shimokawa K, Mogushi
K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H and Sugihara
K: Screening for epigenetically masked genes in colorectal cancer
using 5-Aza-2′-deoxycytidine, microarray and gene expression
profile. Cancer Genomics Proteomics. 9:67–75. 2012.PubMed/NCBI
|
|
10
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy - analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and computational biology solutions
using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
12
|
Meyer PE, Lafitte F and Bontempi G: Minet:
AR/Bioconductor package for inferring large transcriptional
networks using mutual information. BMC Bioinformatics. 9:4612008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Treviño S III, Sun Y, Cooper TF and
Bassler KE: Robust detection of hierarchical communities from
Escherichia coli gene expression data. PLoS Comput Biol.
8:e10023912012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
da W Huang, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009.PubMed/NCBI
|
|
16
|
Benson AB III: Epidemiology, disease
progression, and economic burden of colorectal cancer. J Manag Care
Pharm. 13 6 Suppl C:S5–S18. 2007.PubMed/NCBI
|
|
17
|
Hill VK: Identification of DNA Methylation
Changes in Sporadic Breast and Other Cancers. University of
Birmingham; 2011
|
|
18
|
Jung N, Won JK, Kim BH, Suh KS, Jang JJ
and Kang GH: Pharmacological unmasking microarray approach-based
discovery of novel DNA methylation markers for hepatocellular
carcinoma. J Korean Med Sci. 27:594–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Olin AI, Mörgelin M, Sasaki T, Timpl R,
Heinegård D and Aspberg A: The proteoglycans aggrecan and Versican
form networks with fibulin-2 through their lectin domain binding. J
Biol Chem. 276:1253–1261. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Argraves WS, Greene LM, Cooley MA and
Gallagher WM: Fibulins: Physiological and disease perspectives.
EMBO Rep. 4:1127–1131. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Vega S, Iwamoto T and Yamada Y:
Fibulins: Multiple roles in matrix structures and tissue functions.
Cell Mol Life Sci. 66:1890–1902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Senapati S, Gnanapragassam VS, Moniaux N,
Momi N and Batra SK: Role of MUC4-NIDO domain in the MUC4-mediated
metastasis of pancreatic cancer cells. Oncogene. 31:3346–3356.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Law EW, Cheung AK, Kashuba VI, Pavlova TV,
Zabarovsky ER, Lung HL, Cheng Y, Chua D, Lai-Wan Kwong D, Tsao SW,
et al: Anti-angiogenic and tumor-suppressive roles of candidate
tumor-suppressor gene, Fibulin-2, in nasopharyngeal carcinoma.
Oncogene. 31:728–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shuen WH and Lung ML: Fibulin-2 suppresses
tumor growth and angiogenesis through the inhibition of Erk1/2 and
p65 pathways in nasopharyngeal carcinoma. Proceedings of the AACR
104th Annual Meeting 2013. AACR. Washington, DC. pp. 43092013;
|
|
25
|
Fontanil T, Rúa S, Llamazares M,
Moncada-Pazos A, Quirós PM, García-Suárez O, Vega JA, Sasaki T,
Mohamedi Y, Esteban MM, et al: Interaction between the ADAMTS-12
metalloprotease and fibulin-2 induces tumor-suppressive effects in
breast cancer cells. Oncotarget. 5:1253–1264. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dunwell TL, Hesson LB, Pavlova T,
Zabarovska V, Kashuba V, Catchpoole D, Chiaramonte R, Brini AT,
Griffiths M, Maher ER, et al: Epigenetic analysis of childhood
acute lymphoblastic leukemia. Epigenetics. 4:185–193. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hill VK, Hesson LB, Dansranjavin T, Dallol
A, Bieche I, Vacher S, Tommasi S, Dobbins T, Gentle D, Euhus D, et
al: Identification of 5 novel genes methylated in breast and other
epithelial cancers. Mol Cancer. 9:512010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eto M, Ohmori T, Suzuki M, Furuya K and
Morita F: A novel protein phosphatase-1 inhibitory protein
potentiated by protein kinase C. Isolation from porcine aorta media
and characterization. J Biochem. 118:1104–1107. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ali D, Honne H, Danielsen S, Cekaite L,
Meling G, Rognum T, Lothe R and Lind G: 694 Identification of novel
epigenetic biomarkers in colorectal cancer, GLDC and PPP1R14A. Eur
J Cancer Suppl. 8:1752010. View Article : Google Scholar
|
|
30
|
Hollander MC, Zhan Q, Bae I and Fornace AJ
Jr: Mammalian GADD34, an apoptosis- and DNA damage-inducible gene.
J Biol Chem. 272:13731–13737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park JS, Yoon S Young, Kim JM, Yeom YI,
Kim YS and Kim NS: Identification of novel genes associated with
the response to 5-FU treatment in gastric cancer cell lines using a
cDNA microarray. Cancer Lett. 214:19–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bethge N, Honne H, Hilden V, Trøen G,
Eknæs M, Liestøl K, Holte H, Delabie J, Smeland EB and Lind GE:
Identification of highly methylated genes across various types of
B-cell non-hodgkin lymphoma. PLoS One. 8:e796022013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin WW and Karin M: A cytokine-mediated
link between innate immunity, inflammation, and cancer. J Clin
Invest. 117:1175–1183. 2007. View
Article : Google Scholar : PubMed/NCBI
|