Introduction
Ovarian cancer is the most common cause of
cancer-associated mortalities in females diagnosed with
gynecological malignancies, accounting for ~22,000 new cancer cases
(3.7% of all cancer cases in females) and ~140,000 mortalities
(4.2% of all female mortalities) annually (1,2). In spite
of advancements occurring in cytoreductive surgery and
chemotherapeutic techniques, the 5-year survival rate for ovarian
cancer remains virtually unchanged due to the high metastatic and
drug-resistant properties of the cancer. Thus, effective therapy is
required.
Wee1-like protein kinase (WEE1), an important
regulator for the cell cycle checkpoints, contributes to the
upstream regulation of the cyclin-dependent kinase (CDK) complexes
by mediating the activation of CDK1 (also known as CDC2) and CDK2
(3–5).
CDK1/2 are well-known mediators of cell division, ensuring that the
cell is healthy to undergo mitosis and subsequently driving the
cell cycle through G2/M and S phases (6,7). Previous
studies have identified the expression levels of WEE1 in a variety
of human tumor tissues (8–12). Decreased expression of WEE1 has been
demonstrated in non-small cell lung cancer (NSCLC) and increased
WEE1 expression has been identified in ovarian cancer, melanoma,
osteosarcoma, glioblastoma and breast cancer. Notably, WEE1
expression was significantly associated with poor overall survival
time in the analysis of 109 patients with ovarian cancer with
post-chemotherapy effusions (13),
suggesting that WEE1 may be a novel therapeutic target for ovarian
cancer.
The direct effect of WEE1 inhibition on ovarian
cancer cell viability requires study. MK1775, the first selective
molecular inhibitor of WEE1, has been identified as an agent that
primarily targets the G2/M checkpoint and exhibits a
toxic effect (14). Subsequent
studies were initiated to determine the synergistic antitumor role
of MK1775 in combination therapies with conventional DNA damaging
agents for ovarian, breast, prostate, pancreatic and colon cancer
(15–18). An antitumor effect was observed to be
dependent on the cytotoxicity exhibited by other combined
DNA-damaging agents or a tumor protein 53 (p53)-defect in tumor
cells (14,16,17,19). The
single-agent effect of MK1775 on tumor viability in vivo was
initially identified in NSCLC (20),
medulloblastoma (21) and sarcoma
(22). In addition, previous studies
have indicated that the cytotoxic effect of MK1775 was independent
of p53 status (10,22). However, in ovarian cancer, no
significant antitumor effect was observed when nude mice bearing
OVCAR-5 were treated with MK-1775 alone (23). The present study analyzed the
cytotoxic effect of MK1775 as a single chemotherapy agent on ID8
and SKOV3 ovarian cancer cell lines in vitro. Furthermore,
the antitumor activity of MK1775 in vivo was determined in
ID8-bearing immunocompetent mice.
Materials and methods
Animals and reagents
C57BL/6 WT mice were purchased from Jackson
Laboratory (Ben Harbour, ME, USA), while Dr Hans Schreiber
(University of Chicago, Chicago, IL, USA) provided EG7 and B16F10
cell lines. LLC1, SKOV3 and OVCAR3 cell lines were purchased from
the American Type Culture Collection (ATCC; Manassas, VA, USA).
Mouse ID8 and ID8-OVA cells were obtained as previously described
(24). The BPS-1 mouse melanoma cell
was generated by this study group (25) from a spontaneously arising tumor in
B-Raf proto-oncogene, serine/threonine kinase
[BRAF(V600E)]/phosphatase and tensin homolog (PTEN) (Tyr::CreER;
tyrosinase promoter/enhancer regions upstream of a
tamoxifen/4-hydroxytamoxifen-inducible Cre recombinase fused to a
mutated estrogen receptor ligand binding domain; BrafCA/+;
Ptenlox5/lox5, transgene for the Braf allele and the floxed
Pten allele) transgenic mice (26,27). The
animal studies were performed under the approval of the
Institutional Animal Use Committees of the Northwestern University
(Chicago, IL, USA). B16F10, ID8, ID8-OVA and BPS1 cells were
maintained in RPMI 1640 medium (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany), supplemented with 1% penicillin-streptomycin,
1% glutamine and 5% fetal bovine serum (FBS). SKOV3 and LLC1 cells
were cultured in complete medium composed of Dulbeccos modified
Eagles medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
FBS. All the antibodies for flow cytometry were obtained from
BioLegend, Inc. (San Diego, CA, USA) or eBiosience (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The annexin V apoptosis
detection kit was purchased from Biolegend, Inc., and MTT kits were
purchased from Roche Diagnostics (Basel, Switzerland).
In vitro cytotoxicity assays
An MTT assay was performed to determine the in
vitro cytotoxicity of MK1775 in distinct mouse tumor cell
lines, including B16F10, LLC1, BPS1, ID8 and EG7, and the human
ovarian cancer SKOV3 cell line. Cell density was optimized to
ensure that the cell viability for the duration of the treatment
was within the linear range. According to the manufacturers
protocol, a variety of tumor cells in the logarithmic growth phase
were plated (1,000 cells/well) in 96-well flat bottom plates and
allowed to adhere prior to treatment. Subsequently, cells were
treated in triplicates with vehicle dimethyl sulfoxide (DMSO) only,
and a variety of concentrations of MK1775 dissolved in DMSO. On the
basis of previous studies (14,28), the
following concentrations of MK1775 were used: 0.01, 0.1, 1.0 and 10
µM, and negative control cells were treated with DMSO. At 72 h
post-drug treatment, MTT was added to a final concentration of 1 mM
and the cells were incubated at 37°C in an atmosphere containing 5%
CO2. A total of 200 µl solubilization solution was
subsequently added to each well and plates were allowed to stand
overnight at 37°C. The absorbance was determined at 560 nm using a
microplate reader. The half-maximal inhibitory concentration
(IC50) of MK1775 for ID8 and SKOV3 cells was
calculated.
Flow cytometry analysis of
apoptosis
ID8 and SKOV3 cells were plated in a 75-cm2 dish
(1.5×104 cells/ml) and allowed to adhere overnight at 37°C prior to
treatment. Following treatment with the specified concentrations of
MK1775, the cell apoptosis assay was conducted using cell staining
with annexin V (2.5 µg/ml) and 7-aminoactinomycin D (7-AAD) (1
µg/ml) for 15 min at room temperature (25°C) in the dark, according
to the manufacturers protocol. The apoptosis assay was performed 24
or 48 h after drug treatment using MACS Quant Analyzer (Miltenyi
Biotec, Cologne, Germany). The data were analyzed using Flow Jo
software version 10.0.7r2 (Tree Star, Inc., Ashland, OR, USA).
Cells that stained negative for annexin V-allophycocyanin and 7-AAD
were classified as not undergoing determinable apoptosis,
double-positive cells were classified as late-state apoptosis,
single-positive cells for annexin-V were classified as early
apoptosis and single-positive cells for 7-AAD were classified as
dead or necrotic cells.
Analysis of the cell cycle
The treatment conditions were the same as in the
apoptosis assay. The treated tumor cells (1×106 cells) were
detached and fixed in ice-cold 75% ethanol for 4 h on ice. Cells
were subsequently washed with PBS and stained with 5 µg/ml
propidium iodide (PI; cat number: 421301, Biolegend, Inc.) and 50
µg/ml RNase for 30 min at 4°C. The cell cycle of ID8 cells was
determined using PI staining, whereas that of SKOV3 cells was
determined by DAPI (cat number: 422801, Biolegend, Inc.) staining
(1 µg/ml at 4°C for 30 min). The flow cytometry analysis was
performed using MACS Quant Analyzer (Miltenyi Biotec) and the
proportions of cells in G1, S, and G2/M
phases were analyzed using Flow Jo Cell Cycle software version
10.0.7r2 (Tree Star, Inc., Ashland, OR, USA).
Western blot analysis
ID8 cells were treated with DMSO, 0.5 µM MK1775 or 1
µM MK1775 for 48 h, followed by a wash with PBS. Cells were
subsequently lysed using radioimmunoprecipitation assay buffer,
including phenylmethylsulfonyl fluoride and a protease inhibitor
cocktail (Roche Diagnostics, Indianapolis, IN, USA). Protein
concentration was determined using BCA protein assay kit according
to manufacturers protocol (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The proteins were separated on SDS-PAGE (12% gel) and
subsequently transferred onto polyvinylidene fluoride membranes.
Membranes were blocked in 5% skimmed milk for 2 h at room
temperature and incubated with a primary antibody against
phosphorylated-CDC2 (residue Typ15), a target of WEE1 kinase
(1:1,000; catalog no. 4539; Cell Signaling Technology, Inc.,
Danvers, MA, USA), at 4°C overnight. Membranes were washed with
Tris-buffered saline-Tween-20 (TBST) 3 times prior to incubation at
room temperature for 2 h with the appropriate secondary antibody
conjugated to horseradish peroxidase (1:2,000; catalog no. 7074;
Cell Signaling Technology, Inc.). Subsequently, membranes were
washed 3 times with TBST and developed using enhanced
chemiluminescence (Thermo Fisher Scientific, Inc.). Blots were
visualized using C-DiGit Blot Scanner (LI-COR Biosciences, Lincoln,
NE, USA). β-actin served as a loading control.
Tumor challenges
Mice were housed in single-sex groups, and had ad
libitum access to bottled acidified water (pH 2.8 to 3.1) and
feed pellets with 6% fat (NIH 31M, Purina Mills, Richmond, IN,
USA). The number of room air changes/hour was maintained at 15±1,
temperature at 22±2°C, and relative humidity at 45±5%, with a 14:10
h light: dark cycle. ID8-OVA tumor cells (6×106) in suspension were
challenged intraperitoneally into 6–8 week-old C57BL/6 WT female
mice (20–25 g body weight) (n=8) and tumor progression was
monitored every 7 days by determining the abdominal circumferences
of the mice with the aid of a wire measured by a ruler, expressed
in millimeters. At day 30 after challenge, when abdominal swelling
was determinable, mice were randomly divided into two groups (n=4
in each). DMSO or MK1775 treatment was administered daily at 50
mg/kg. The ascites fluid from ID8-bearing mice was harvested with a
20-gauge needle for volume measurement 30 days after MK1775
treatment. The endpoint criterion was a gain of >20% of body
weight compared to the pre-study weight. Mice were euthanized by
combined high dose of CO2 gas (a flow rate of 1–3 l per
minute for a 10 l volume chamber) followed by cervical
dislocation.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 10.0; SPSS Inc., Chicago, IL, USA). Statistical
significance between two groups was assessed using an unpaired
Students two-tailed t-test and multiple comparisons between the
groups were determined using an analysis of variance
(Student-Newman-Keuls). P<0.05 was considered to indicate a
statistically significant difference.
Results
Cytotoxic effect of MK1775 on a
variety of tumor cell lines
The in vitro cytotoxic effect of MK1775 on
distinct tumor cells was assessed using an MTT assay. As presented
in Fig. 1, MK1775 treatment decreased
the viability of murine melanoma B16F10 and BPS1, lymphoma EG7,
ovarian cancer ID8, LLC1 lung cancer and human ovarian cancer SKOV3
cells in a dose-dependent manner. The responses of the tumor cells
to MK1775 at a variety of concentrations were indicative of the
relative cytotoxicity of MK1775 against distinct tumor cell lines.
Notably, the mean IC50 of MK1775 to ID8 and SKOV3 cells
were 0.75 and 0.46 µM, respectively. The results of the present
study demonstrated that ID8 and SKOV3 ovarian cancer cells were
sensitive to MK1775 treatment.
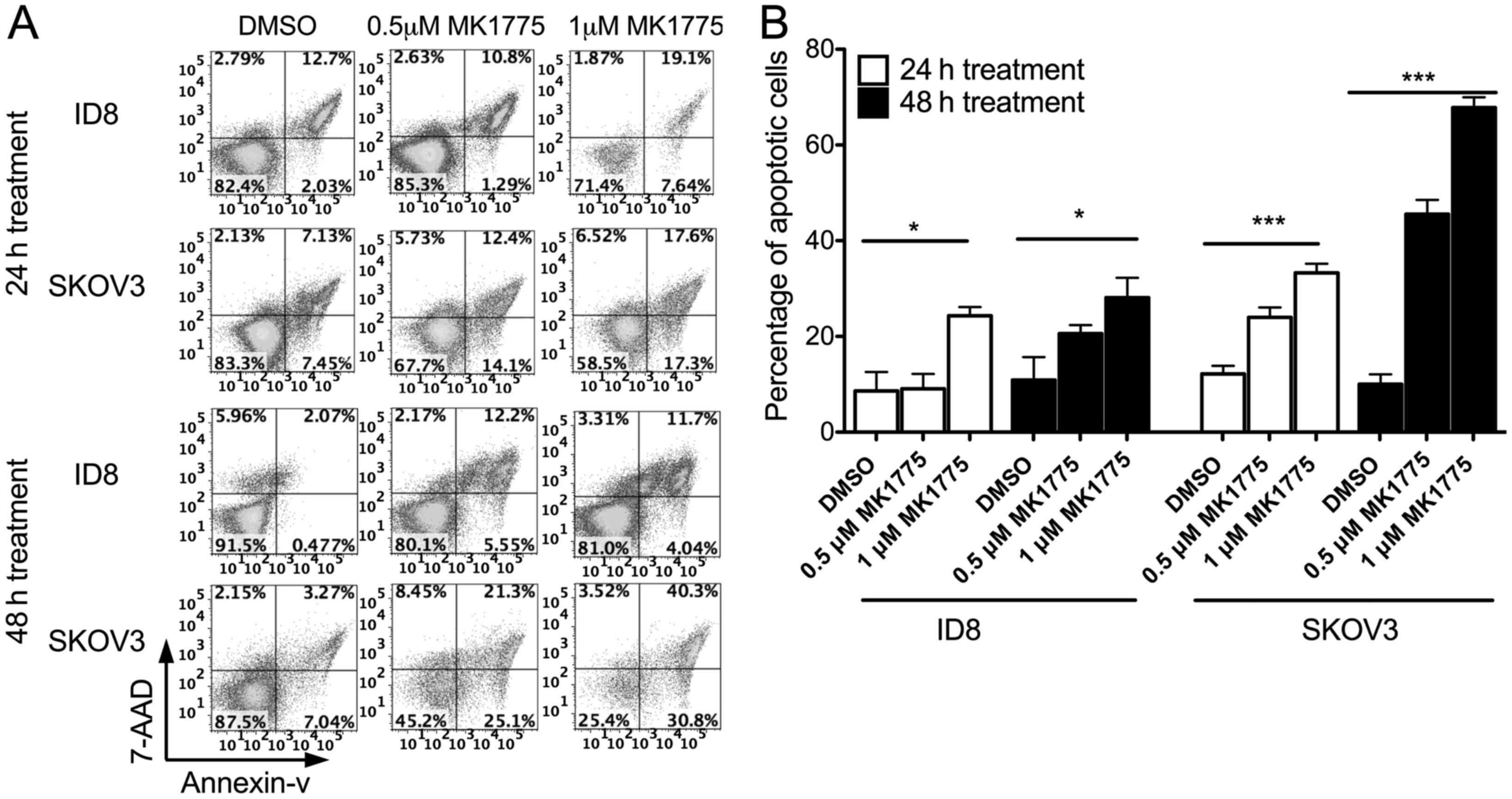
MK1775 as a single agent induces the
apoptosis of ID8 and SKOV3 ovarian cancer cells
A cell apoptosis assay was conducted in ovarian
cancer ID8 and SKOV3 cell lines to examine the cytotoxic activity
of MK1775. Flow cytometry was used to determine annexin V/7-AAD
expression and the stages of apoptosis. Previously, studies have
identified that ID8 cells express WT p53 (29), whereas SKOV3 cells have null p53
(30). Fig.
2 presents the proportion of cells undergoing early, late and
end-stage apoptosis. In ID8 and SKOV3 cells, MK1775 induced tumor
cell apoptosis. SKOV3 cells were more susceptible to MK1775-induced
apoptosis compared with ID8 cells. The results of the present study
identified the cytotoxic role of MK1775 as a single chemotherapy
agent against either WT p53 or p53-defective cell lines.
Furthermore, a p53 defect may sensitize tumor cells to MK1775
treatment.
MK1775 treatment abrogates the
G2/M checkpoint of ID8 cells through attenuating the
phosphorylation of CDK1
To determine the underlying molecular mechanism of
cell death induced by MK1775 treatment, independent of p53, cell
cycle analysis and a western blot analysis was performed on ID8
cell lines with WT p53. As presented in Fig. 3, MK1775 treatment arrested tumor cells
in S phase and abrogated the G2/M cell cycle checkpoint.
The abrogation of the G2 DNA damage checkpoint resulted
in cell death and the presence of a sub-G1 peak
indicated that MK1775 treatment induced cell apoptosis, which was
consistent with the results from the cell apoptosis assay (Fig. 2). In addition, a dose-dependent
decrease in phosphorylated-CDC2 (Tyr15) was observed in ID8 cells
following MK1775 treatment (Fig. 3C),
indicating that MK1775 may abrogate the G2 checkpoint
through the phosphorylation of CDC2. A significant increase in
cleaved poly (ADP-ribose) polymerase and caspase-3, two markers of
apoptosis, has been associated with decreased levels of
phosphorylated-CDC2 (Tyr15) and was a result of MK1775 treatment
(31). Deregulation of CDC2 activity
may trigger the activation of caspase-3 and apoptosis (32). These results together demonstrate a
proof-of-mechanism for this novel inhibitor of WEE1, with evidence
of decreased phosphorylated-CDC2 (Tyr15) levels in ovarian cancer
cells. Furthermore, the cell cycle (Fig.
3B) and phosphorylated-CDC2 (Fig.
3D) in SKOV3 cells with p53 mutations were analyzed in the
present study. Notably, the observed effect of MK1775 treatment on
the cell cycle was increased in SKOV3 cells compared with that
observed in ID8 cells, which was consistent with the results of the
MTT and apoptosis assays, suggesting that a p53 defect may
sensitize tumor cells to MK1775 treatment.
 | Figure 3.MK1775 treatment abrogates the
G2/M checkpoint of ID8 cells, through decreasing the
phosphorylation of cyclin-dependent kinase 1. β-actin was used as a
loading control. (A) Cell cycle analysis, performed using flow
cytometry 48 h after treatment with DMSO, or 0.5 µM or 1 µM MK1775.
(B) Cell cycle analysis, performed using flow cytometry 48 h after
treatment with DMSO, or 0.5 µM or 1 µM MK1775. (C) Phosphorylated
CDC2 (Tyr15) protein levels, determined using western
blot 48 h after treatment with 0.5 µM or 1 µM MK1775. (D)
Phosphorylated CDC2 (Tyr15) protein levels determined
using western blotting, 48 h after treatment with 0.5 µM or 1 µM
MK1775. DMSO, dimethyl sulfoxide; PI, propidium iodide; p-CDC2,
phosphorylated cyclin-dependent kinase 1. |
Antitumor effect of MK1775 in
syngeneic mouse model of ovarian cancer
To determine the in vivo antitumor activity
of MK1775, a syngeneic ID8 ovarian cancer model was used. C57BL/6
mice were challenged with ID8 cells and treated daily with MK1775
or DMSO at day 30 post-challenge. The dose of MK1775 was converted
and defined as 50 mg/kg, on the basis of the data from an initial
phase I clinical trial (33). A
significant inhibition of ovarian tumor progression was identified
from 2 weeks after MK1775 treatment, as indicated by the abdominal
circumferences (Fig. 4A). In
addition, MK1775 treatment alone significantly decreased the
production of malignant ascites from ID8-bearing mice at the
advanced stage (Fig. 4B).
Discussion
Due to the poor prognosis of ovarian cancer
patients, novel therapies are required. DNA damage checkpoints are
critical for maintaining the integrity of the cell genome. As a key
regulator of the G2/M transition in the cell cycle, WEE1
has been considered as a promising target for cancer therapy.
Notably, MK1775 as a selective and potent inhibitor of WEE1, is
currently under clinical evaluation as a single-agent treatment and
potentiates the DNA damage caused by cytotoxic chemotherapies or
radiation therapy. In the present study, the in vitro and
in vivo antitumor effect of MK1775 was evaluated as a
single-agent in ovarian cancer.
Previous studies have identified the function of
MK1775 in enhancing the sensitivity of tumor cells to the
traditional DNA-damaging agents or radiotherapy (11,14–17). In
addition, the synergistic antitumor activity of MK1775 was observed
in combination therapies for ovarian, breast, prostate, pancreatic
and colon cancer, and the effect was identified to be dependent on
the cytotoxicity of other DNA-damaging agents or a p53-defect in
tumor cells (14–16). For ovarian cancer, a phase II clinical
trial is underway to evaluate the antitumor effects exhibited by
the combination of MK1775, paclitaxel and carboplatin, compared
with that exhibited by combined paclitaxel and carboplatin
(18). Furthermore, a previous
clinical trial demonstrated the therapeutic benefit from combining
MK1775 with carboplatin in patients with epithelial ovarian cancer
who are platinum-resistant and exhibit mutated p53 (34). Mutations or lack of functional p53
often cause a defective G1/S checkpoint in cancer cells.
Therefore, the effectiveness of DNA damaging treatment may be
augmented by abrogation of the G2/M checkpoint in these cancer
cells while sparing normal cells with an intact G/S checkpoint
(35–37). Studies have focused on the dual
inhibition of components of the ataxia
telangiectasia-related-checkpoint kinase 1 (Chk1)-WEE1 axis, which
exhibits a synergistic cytotoxic effect and simultaneously inhibits
Chk1 and WEE1 through disrupting the cell cycle checkpoint
regulation (23,38,39).
Previous studies have identified the potential use
of MK1775 as a single antitumor agent (20–22);
however, whether the mono-therapeutic activity of MK1775 is
dependent on mutant p53 remains unknown. The results of the present
study demonstrated that MK1775 serves as a single agent to induce
cell apoptosis in distinct cancer cell lines, independent of p53
status. In addition, a p53 defect may sensitize ovarian cancer
cells to MK1775 treatment. Although no significant inhibition of
tumor viability was determined in MK1775-treated nude mice bearing
OVCAR-5, which is null p53 (25),
additional studies are required to determine the association
between tumor p53 deficient status and the efficacy of MK1775
treatment.
The antitumor effect of MK1775 as a single-agent has
been evaluated in C57BL/6 mice harboring syngeneic ID8 ovarian
tumors with WT BRCA1 (40,41). A significant decrease in the
accumulation of malignant ascites was observed following MK1775
treatment, suggesting that MK1775 treatment alone has a potent
antitumor effect and is tolerable for preclinical ovarian cancer
therapy. These results identified that targeting WEE1 by MK1775
monotherapy is not limited to p53-mutant or BRCA1 mutant cancers.
In addition to DNA damage and the cell cycle, other specific
underlying cellular mechanisms may be implicated when WEE1 is
inhibited, providing insights into the development of novel
combination therapy with WEE1 inhibitors. To the best of our
knowledge, the present study was the first to demonstrate the
efficacy of WEE1 inhibition by MK1775 in ovarian cancer in
immunocompetent mice. As host components, including immune cells,
may express WEE1, the effects of MK1775 on host cells and cancer
cells requires study. The results of the present study indicate
that MK1775, as a single-agent therapy, results in WEE1 inhibition
and the subsequent activation of CDC2, thereby suppressing ovarian
tumor viability. The results of the present study suggest potential
clinical applications for MK1775, which may be a therapeutic
benefit for patients with ovarian cancer, independent of p53
status.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81171985), the
National Institute of Health (grant no. CA149669), Northwestern
Memorial Foundation-Friends of Prentice Grants Initiative, SPORE
Pilot Award (grant no. P50 CA090386) and Northwestern University
RHLCCC Flow Cytometry Facility, a Cancer Center Support Grant (NCI
CA060553).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Parker LL and Piwnica-Worms H:
Inactivation of the p34cdc2-cyclin B complex by the human WEE1
tyrosine kinase. Science. 257:1955–1957. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Watanabe N, Broome M and Hunter T:
Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the
cell cycle. EMBO J. 14:1878–1891. 1995.PubMed/NCBI
|
|
5
|
McGowan CH and Russell P: Cell cycle
regulation of human WEE1. EMBO J. 14:2166–2175. 1995.PubMed/NCBI
|
|
6
|
Nigg EA: Cyclin-dependent protein kinases:
Key regulators of the eukaryotic cell cycle. Bioessays. 17:471–480.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Edgar BA and Lehner CF: Developmental
control of cell cycle regulators: A flys perspective. Science.
274:1646–1652. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Magnussen GI, Holm R, Emilsen E, Rosnes
AK, Slipicevic A and Flørenes VA: High expression of Wee1 is
associated with poor disease-free survival in malignant melanoma:
Potential for targeted therapy. PLoS One. 7:e382542012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Magnussen GI, Hellesylt E, Nesland JM,
Trope CG, Flørenes VA and Holm R: High expression of wee1 is
associated with malignancy in vulvar squamous cell carcinoma
patients. BMC Cancer. 13:2882013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mir SE, De Witt Hamer PC, Krawczyk PM,
Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts
D, Kaspers GJ, et al: In silico analysis of kinase expression
identifies WEE1 as a gatekeeper against mitotic catastrophe in
glioblastoma. Cancer Cell. 18:244–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
PosthumaDeBoer J, Wördinger T, Graat HC,
van Beusechem VW, Helder MN, van Royen BJ and Kaspers GJ: WEE1
inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer.
11:1562011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Iorns E, Lord CJ, Grigoriadis A, McDonald
S, Fenwick K, Mackay A, Mein CA, Natrajan R, Savage K, Tamber N, et
al: Integrated functional, gene expression and genomic analysis for
the identification of cancer targets. PLoS One. 4:e51202009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Slipicevic A, Holth A, Hellesylt E, Tropé
CG, Davidson B and Flørenes VA: Wee1 is a novel independent
prognostic marker of poor survival in post-chemotherapy ovarian
carcinoma effusions. Gynecol Oncol. 135:118–124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirai H, Iwasawa Y, Okada M, Arai T,
Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K,
et al: Small-molecule inhibition of Wee1 kinase by MK-1775
selectively sensitizes p53-deficient tumor cells to DNA-damaging
agents. Mol Cancer Ther. 8:2992–3000. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bridges KA, Hirai H, Buser CA, Brooks C,
Liu H, Buchholz TA, Molkentine JM, Mason KA and Meyn RE: MK-1775, a
novel Wee1 kinase inhibitor, radiosensitizes p53-defective human
tumor cells. Clin Cancer Res. 17:5638–5648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rajeshkumar NV, De Oliveira E, Ottenhof N,
Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H,
Maitra A and Hidalgo M: MK-1775, a potent Wee1 inhibitor,
synergizes with gemcitabine to achieve tumor regressions,
selectively in p53-deficient pancreatic cancer xenografts. Clin
Cancer Res. 17:2799–2806. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirai H, Arai T, Okada M, Nishibata T,
Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et
al: MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor
efficacy of various DNA-damaging agents, including 5-fluorouracil.
Cancer Biol Ther. 9:514–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
http://clinicaltrials.gov/show/NCT01357161Accessed.
11 23–2013.
|
|
19
|
Mueller S, Hashizume R, Yang X, Kolkowitz
I, Olow AK, Phillips J, Smirnov I, Tom MW, Prados MD, James CD, et
al: Targeting Wee1 for the treatment of pediatric high-grade
gliomas. Neuro Oncol. 16:352–360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guertin AD, Li J, Liu Y, Hurd MS, Schuller
AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, et al:
Preclinical evaluation of the WEE1 inhibitor MK-1775 as
single-agent anticancer therapy. Mol Cancer Ther. 12:1442–1452.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Harris PS, Venkataraman S, Alimova I,
Birks DK, Balakrishnan I, Cristiano B, Donson AM, Dubuc AM, Taylor
MD, Foreman NK, et al: Integrated genomic analysis identifies the
mitotic checkpoint kinase WEE1 as a novel therapeutic target in
medulloblastoma. Mol Cancer. 13:722014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kreahling JM, Gemmer JY, Reed D, Letson D,
Bui M and Altiok S: MK1775, a selective Wee1 inhibitor, shows
single-agent antitumor activity against sarcoma cells. Mol Cancer
Ther. 11:174–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carrassa L, Chilà R, Lupi M, Ricci F,
Celenza C, Mazzoletti M, Broggini M and Damia G: Combined
inhibition of Chk1 and Wee1: In vitro synergistic effect translates
to tumor growth inhibition in vivo. Cell Cycle. 11:2507–2517. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin D, Fan J, Wang L, Thompson LF, Liu A,
Daniel BJ, Shin T, Curiel TJ and Zhang B: CD73 on tumor cells
impairs antitumor T-cell responses: A novel mechanism of
tumor-induced immune suppression. Cancer Res. 70:2245–2255. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dominguez D, Ye C, Geng Z, Chen S, Fan J,
Qin L, Long A, Wang L, Zhang Z, Zhang Y, et al: Exogenous IL-33
restores dendritic cell activation and maturation in established
cancer. J Immunol. 198:1365–1375. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dankort D, Curley DP, Cartlidge RA, Nelson
B, Karnezis AN, Damsky WE Jr, You MJ, DePinho RA, McMahon M and
Bosenberg M: Braf(V600E) cooperates with Pten loss to induce
metastatic melanoma. Nat Genet. 41:544–552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hooijkaas AI, Gadiot J, van der Valk M,
Mooi WJ and Blank CU: Targeting BRAFV600E in an inducible murine
model of melanoma. Am J Pathol. 181:785–794. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Van Linden AA, Baturin D, Ford JB, Fosmire
SP, Gardner L, Korch C, Reigan P and Porter CC: Inhibition of Wee1
sensitizes cancer cells to antimetabolite chemotherapeutics in
vitro and in vivo, independent of p53 functionality. Mol Cancer
Ther. 12:2675–2684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim ST, Miller NL, Nam JO, Chen XL, Lim Y
and Schlaepfer DD: Pyk2 inhibition of p53 as an adaptive and
intrinsic mechanism facilitating cell proliferation and survival. J
Biol Chem. 285:1743–1753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Anglesio MS, Wiegand KC, Melnyk N, Chow C,
Salamanca C, Prentice LM, Senz J, Yang W, Spillman MA, Cochrane DR,
et al: Type-specific cell line models for type-specific ovarian
cancer research. PLoS One. 8:e721622013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Do K, Doroshow JH and Kummar S: Wee1
kinase as a target for cancer therapy. Cell Cycle. 12:3159–3164.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu L, Zheng H, Murray SA, Ying H and Jim
Xiao ZX: Deregulation of Cdc2 kinase induces caspase-3 activation
and apoptosis. Biochem Biophys Res Commun. 302:384–391. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Targeting p53 mutant ovarian cancer: Phase
I result of the WEE1 inhibitor MK-1775 with carboplatin plus
paclitaxel in patients (pts) with platinum-sensitive, p53-mutant
ovarian cancer (OC). 45th Annual Meeting of the American Society of
Clinical Oncology, Meeting Library. http://meeting library.asco.org2013.
|
|
34
|
http://clinicaltrials.gov/show/NCT01164995Accessed.
November 23–2013.
|
|
35
|
Bucher N and Britten CD: G2 checkpoint
abrogation and checkpoint kinase-1 targeting in the treatment of
cancer. Br J Cancer. 98:523–528. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawabe T: G2 checkpoint abrogators as
anticancer drugs. Mol Cancer Ther. 3:513–519. 2004.PubMed/NCBI
|
|
37
|
Vance S, Liu E, Zhao L, Parsels JD,
Parsels LA, Brown JL, Maybaum J, Lawrence TS and Morgan MA:
Selective radiosensitization of p53 mutant pancreatic cancer cells
by combined inhibition of Chk1 and PARP1. Cell Cycle. 10:4321–4329.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chilà R, Basana A, Lupi M, Guffanti F,
Gaudio E, Rinaldi A, Cascione L, Restelli V, Tarantelli C, Bertoni
F, et al: Combined inhibition of Chk1 and Wee1 as a new therapeutic
strategy for mantle cell lymphoma. Oncotarget. 6:3394–3408. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Magnussen GI, Emilsen E, Giller Fleten K,
Engesæter B, Nähse-Kumpf V, Fjær R, Slipicevic A and Flørenes VA:
Combined inhibition of the cell cycle related proteins Wee1 and
Chk1/2 induces synergistic anti-cancer effect in melanoma. BMC
Cancer. 15:4622015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang J, Wang L, Cong Z, Amoozgar Z, Kiner
E, Xing D, Orsulic S, Matulonis U and Goldberg MS: The PARP1
inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(−/−)
murine model of ovarian cancer. Biochem Biophys Res Commun.
463:551–556. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Quinn JE, Carser JE, James CR, Kennedy RD
and Harkin DP: BRCA1 and implications for response to chemotherapy
in ovarian cancer. Gynecol Oncol. 113:134–142. 2009. View Article : Google Scholar : PubMed/NCBI
|