Introduction
Glioblastoma multiforme (GBM), or grade IV
astrocytoma as classified by the World Health Organization
(1), is the most common and malignant
primary brain tumor in humans, with an incidence rate of 3.19 cases
per 100,000 person/year in the United States 2005–2009 (1). The important characteristics of GBM are
the high cellular proliferation rate, genetic instability, diffuse
infiltration and high angiogenesis, conferring high levels of
aggression and drug resistance in GBM (2). In spite of previous significant
improvements in treatment, including surgery, radiotherapy and
chemotherapy, the overall median survival rate remains only 12–16
months at present (3). This poor
prognosis suggests that therapeutic resistance is a significant
problem of GBM. Thus, it is urgent to identify novel candidate
factors involved in glioma proliferation and angiogenesis that may
assist to develop effective targeted therapies for GBM.
A growing number of studies have demonstrated that
exposure to stress may promote tumor progression in many types of
cancers (4–6) and the adrenergic system has been widely
recognized to serve a significant role in stress signaling
(7). Exposure to stressful events
induces the activation of the hypothalamic-pituitary-adrenal (HPA)
axis, which in turn results in the release of glucocorticoids and
catecholamines, including norepinephrine (NE) and epinephrine (E),
from the adrenal gland and from the brain and sympathetic nerve
terminals (8). Concurrently, the
secretion of dopamine, which assists to control the reward and
pleasure centers of the brain, is reduced. This alteration in
homeostasis leads to a microenvironment that is beneficial to tumor
growth and progression in experimental models of disease (5). The functions of NE and E are primarily
mediated by the activation of adrenergic receptors (ARs) including
α-ARs and β-ARs (9). There are 3
subtypes of β-ARs, specifically β1-AR, β2-AR
and β3-AR, which have been identified to date (10). β1-ARs and β2-ARs
are widely expressed in the majority of mammalian cell types, while
β3-ARs are almost exclusively expressed in adipocytes
that have seldom been studied (11).
β-ARs are members of the superfamily of G
protein-coupled receptors (GPCR), which activate the Gαs protein to
initiate multiple signaling cascades and therefore lead to numerous
pathological conditions such as cardiac, psychiatric, immunological
and endocrine disorders (12). In
previous decades, numerous studies have demonstrated that β-ARs may
also regulate different processes of cancer initiation and
progression, through activating the classical cyclic adenosine
monophosphate (cAMP)/protein kinase A (PKA) pathway and
mitogen-activated protein kinases (MAPK) pathway (13,14). For
example, in pancreatic ductal adenocarcinomas cells, it has been
suggested that β-AR agonists promote cell growth by activating
signaling via adenylyl cyclase (AC) and its downstream effectors
cAMP, PKA and nuclear transcription factor cAMP-responsive
element-binding protein (15). In
addition, the extracellular signal-related kinase 1/2 (ERK1/2)
signaling pathway, activated by transactivating epidermal growth
factor receptor (EGFR), is also associated with this process
(16,17). Previously, the potential involvement
of β-ARs in the modulation of astrocytoma cancer cell proliferation
has been suggested (18). However,
the detailed underlying mechanisms by which these events occur
remain unknown, but are becoming characterized.
The present study was designed to investigate the
function and mechanism of β-ARs in human glioblastoma U251 cells.
The results demonstrated that the β1-AR and
β2-AR subtypes were expressed in U251 cells but not
U87-MG cells. In addition, the activation of the β-ARs by
isoproterenol (ISO) significantly enhanced the rate of cell
proliferation in U251 cells via activation of the ERK1/2 pathway
and matrix metalloproteinase (MMP)-2 and MMP-9 mRNA expression.
These results may provide additional insight into the specific
roles of β-ARs in glioblastoma.
Materials and methods
Antibodies and reagents
Primary antibodies including phospho-ERK1/2
(Thr202/Tyr204) (cat no. 9101; 1:2,000), ERK1/2 (cat no. 9102;
1:2,000) and β-actin (cat no. 4970; 1:4,000), the secondary
antibody horseradish peroxidase (HRP)-linked goat anti-rabbit IgG
antibody (cat no. 7074; 1:20,000) and U0126 (a specific ERK1/2
inhibitor) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). ISO and propranolol (PRO) were purchased from
Tocris Bioscience (Bristol, UK). The culture medium and other
solutions used for cell culture were purchased from Invitrogen,
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). MTT was
purchased from Carl Roth GmbH & Co., KG (Karlsruhe,
Germany).
Cell culture
Human glioblastoma cell lines U87-MG and U251 were
grown in Dulbecco's modified Eagle's medium (DMEM) containing 4.5
g/l glucose supplemented with 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.), 2 mM glutamine, 50 µg/ml
streptomycin and 50 IU/ml penicillin. Cultures were maintained at
37°C in a humidified atmosphere containing 5% CO2.
Reverse transcription polymerase chain
reaction (RT-PCR)
The total cellular RNA from the U251 and U87-MG
cells was extracted using TRIzol reagent according to the
manufacturer's protocol (Thermo Fisher Scientific, Inc.). The RNA
concentration was measured using NanoDrop 2000 (Thermo Fisher
Scientific, Inc.). Then the RNA was reverse transcribed into cDNA
using SuperScript III reverse transcriptase according to the
manufacturer's protocol (Thermo Fisher Scientific, Inc.). PCR
amplification was performed using 5 µl cDNA, 12.5 µl 2X Taq PCR
Master Mix (Promega Corporation, Madison, WI, USA), 0.5 µl 10 µM
forward primer, 0.5 µl 10 µM reverse primer and 6.5 µl
ddH2O. The specific primers used are summarized in
Table I. The thermocycling protocol
was as follows: Amplification step, 94°C for 5 min; 30 cycles of
denaturation for 1 min at 95°C; 1 min of annealing at 55°C;
elongation at 72°C for 1 min; and extension at 72°C for 1 min. The
PCR products were electrophoresed on a 1.5% agarose gel and the
bands in the gels that were stained with ethidium bromide were
visualized under ultraviolet light transilluminators (ChemiDoc XRS+
system; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The gene
β-actin was used as a control.
 | Table I.Primers used for reverse
transcription PCR and quantitative PCR. |
Table I.
Primers used for reverse
transcription PCR and quantitative PCR.
| Gene | Forward | Reverse | Size, bp |
|---|
| β1-AR |
5′-GGGAGAAGCATTAGGAGGG-3′ |
5′-CAAGGAAAGCAAGGTGGG-3′ | 270 |
| β2-AR |
5′-CAGCAAAGGGACGAGGTG-3′ |
5′-AAGTAATGGCAAAGTAGCG-3′ | 334 |
| β-actin |
5′-ATCGTGCGTGACATTAAGGAGAAG-3′ |
5′-AGGAAGGAAGGCTGGAAGAGT-3′ | 179 |
| MMP-2 |
5′-CCGTCGCCCATCATCAAGTTC-3′ |
5′-GCAGCCATAGAAGGTGTTCAGG-3′ | 90 |
| MMP-9 |
5′-TGGTCCTGGTGCTCCTGGTG-3′ |
5′-GCTGCCTGTCGGTGAGATTGG-3′ | 111 |
RT-quantitative PCR (qPCR)
The total RNA and cDNA were obtained as
aforementioned. The PCR primers used for the analysis are
summarized in Table I. The 20 µl PCR
system was composed of 2 µl cDNA, 10 µl SYBR® Premix Ex
Taq II, 0.4 µl ROX Reference Dye II (Takara Bio, Inc., Otsu,
Japan), 0.8 µl 10 µM forward primer, 0.8 µl 10 µM reverse primer
and 6 µl double-distilled H2O. The RT-qPCR amplification
was performed using the ViiA7 system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) under the following thermocycler
conditions: 95°C for 30 sec; 40 cycles of denaturation for 5 sec at
95°C; 34 sec of annealing at 60°C; elongation at 95°C for 15 sec;
and extension at 60°C for 1 min. The quantification method for qPCR
data was the 2−ΔΔCq method (19). At least three independent experiments
were conducted and samples were assessed in triplicate for each
experiment. The same PCR system as above was used without template
cDNA was used as a negative control.
Western blot analysis
Cells were cultured in serum-free DMEM overnight at
37°C prior to drug treatment. To determine the time course effect
of ISO on ERK1/2 activation, cells were treated with ISO (10 µΜ)
for 1, 2, 5, 10, 20 and 30 min at 37°C. To determine the effects of
propranolol (PRO) and U0126 on ISO-mediated ERK1/2 activation,
cells were pretreated with PRO (10 µΜ) or U0126 (20 µΜ) for 30 min
and then with ISO (10 µΜ) for 5 min at 37°C. Following treatment,
cells were washed with ice-cold PBS twice, and lysed in ice-cold
lysis buffer. The lysate was sonicated and centrifuged at 12,000 ×
g at 4°C for 5 min. The protein concentration of extracts was
determined by using the Bradford reagent from Bio-Rad Laboratories,
Inc.. Equal amounts of protein (20 µg/lane) were loaded and
separated by 12% SDS-PAGE. Proteins were transferred to
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA), and
blocked in 5% non-fat dry milk for 1 h at room temperature.
Subsequently, the membrane was incubated with different primary
antibodies including phosphor-ERK1/2 (Thr202/Tyr204), ERK1/2 and
β-actin overnight at 4°C and then incubated with HRP-conjugated
goat anti-rabbit IgG secondary antibody (1:20,000) for 2 h at room
temperature. Immunoreactive bands were visualized by enhanced
chemiluminescence (Pierce; Thermo Fisher Scientific, Inc.) and
semi-quantified using ImageJ software (version 1.47t; National
Institutes of Health, Bethesda, MD, USA).
MTT assay
Cellular viability was measured using the MTT assay
as described previously (20).
Briefly, cells were seeded in flat-bottomed 24-well plates
(5×104 cells/well) and grown for 24 h in DMEM
supplemented with 10% FBS. Following 24 h, cells were starved
overnight at 37°C and incubated with different drugs. To determine
the dose course effect of ISO on the proliferation of U251 and
U87-MG cells, cells were treated with 0.1, 1, 5, 10, 30 or 50 µM
ISO for 24 h at 37°C. To determine the time course effect of ISO on
the proliferation of U251 cells, cells were treated with ISO (10
µM) for 24, 48, 72 and 96 h at 37°C. To determine the effects of
PRO and U0126 on ISO-mediated proliferation, cells were pretreated
with PRO (10 µΜ) or U0126 (20 µΜ) for 30 min and then with ISO (10
µΜ) for 48 h at 37°C. Following drug treatment, 20 µl MTT (5 mg/ml)
was added to each well, and the cells were allowed to grow in
complete media at 37°C for 3 h. The supernatant was removed, then
500 µl dimethyl sulfoxide was added to each well and mixed for 10
min to dissolve the crystal. Subsequently, the absorbance was
determined using a microplate spectrophotometer assay reader at 570
nm.
Statistical analysis
Data are indicated as the mean ± standard error of
the mean from at least three independent experiments. Statistical
analysis was performed using the unpaired two-tailed Student's
t-test (comparison of two groups) or one-way analysis of variance
followed by the Tukey's test (comparison of >2 groups).
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were conducted using Prism
version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Expression of β-ARs in human
glioblastoma cell lines
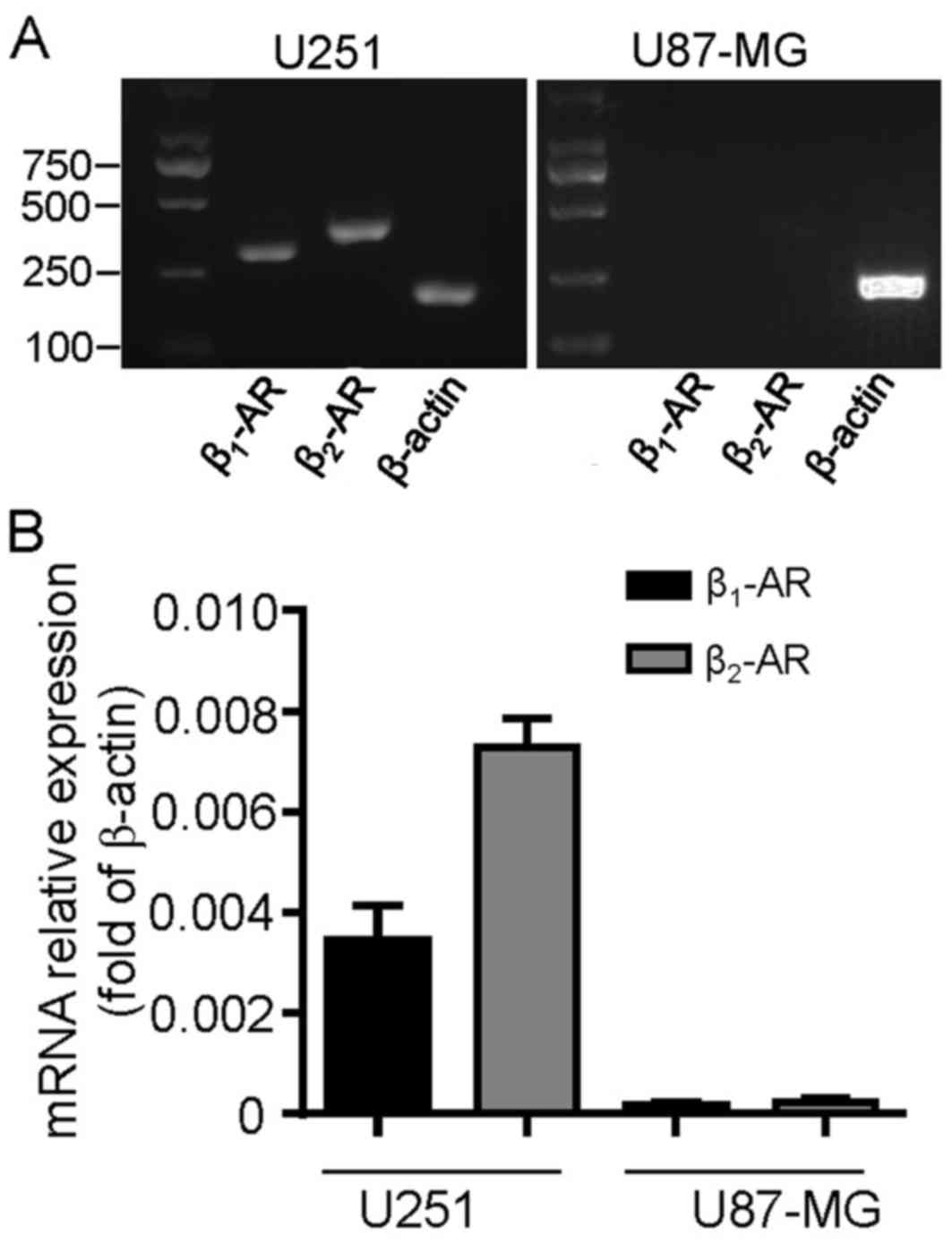
To detect whether β-ARs were expressed in cultured
glioblastoma cells, the two cell lines U251 and U87-MG were
profiled for β1-AR and β2-AR mRNA expression.
The RT-qPCR analysis revealed that β1-AR and
β2-AR transcripts were expressed in U251 cells; however,
they were undetectable in U87-MG cells (Fig. 1A). These results were confirmed by
RT-qPCR analysis (Fig. 1B). The
β2-AR transcript levels were increased compared with
β1-AR in U251 cells (Fig.
1B; P=0.0135). These data suggest that β-ARs may be functional
and serve a role in the development process of U251 cells.
Isoproterenol promotes the
proliferation of U251 cells
Based on the expression profile of β-ARs in U251,
but not U87-MG cells, whether β-ARs activation was involved in U251
cell proliferation was then investigated. Firstly, the
dose-dependent effect of ISO, an agonist of β-ARs, on the
proliferation of U251 cells was analyzed. The cells were treated
with 0.1, 1, 5, 10, 30 or 50 µM ISO for 24 h and then proliferation
was measured by MTT assay. These results revealed that ISO
significantly increased the proliferation of U251 cells, which
peaked at 10 µM ISO and decreased at higher concentrations of ISO
(Fig. 2A). Notably, no effect of
ISO-mediated cell proliferation was detected in U87-MG cells
(Fig. 2B). This is in accordance with
the results demonstrating no expression of β-ARs in U87-MG cells
(Fig. 1A). Secondly, the effect of
different time treatment of ISO on U251 cell proliferation was
detected. The cells were incubated with 10 µM ISO for 24, 48, 72 or
96 h. As indicated in Fig. 2C, ISO
significantly increased the proliferation of U251 cells, with the
most marked effect at 48 h. Thus, 10 µM ISO was chosen as the
reference concentration, and 48 h as the treatment time for the
following studies unless specifically indicated.
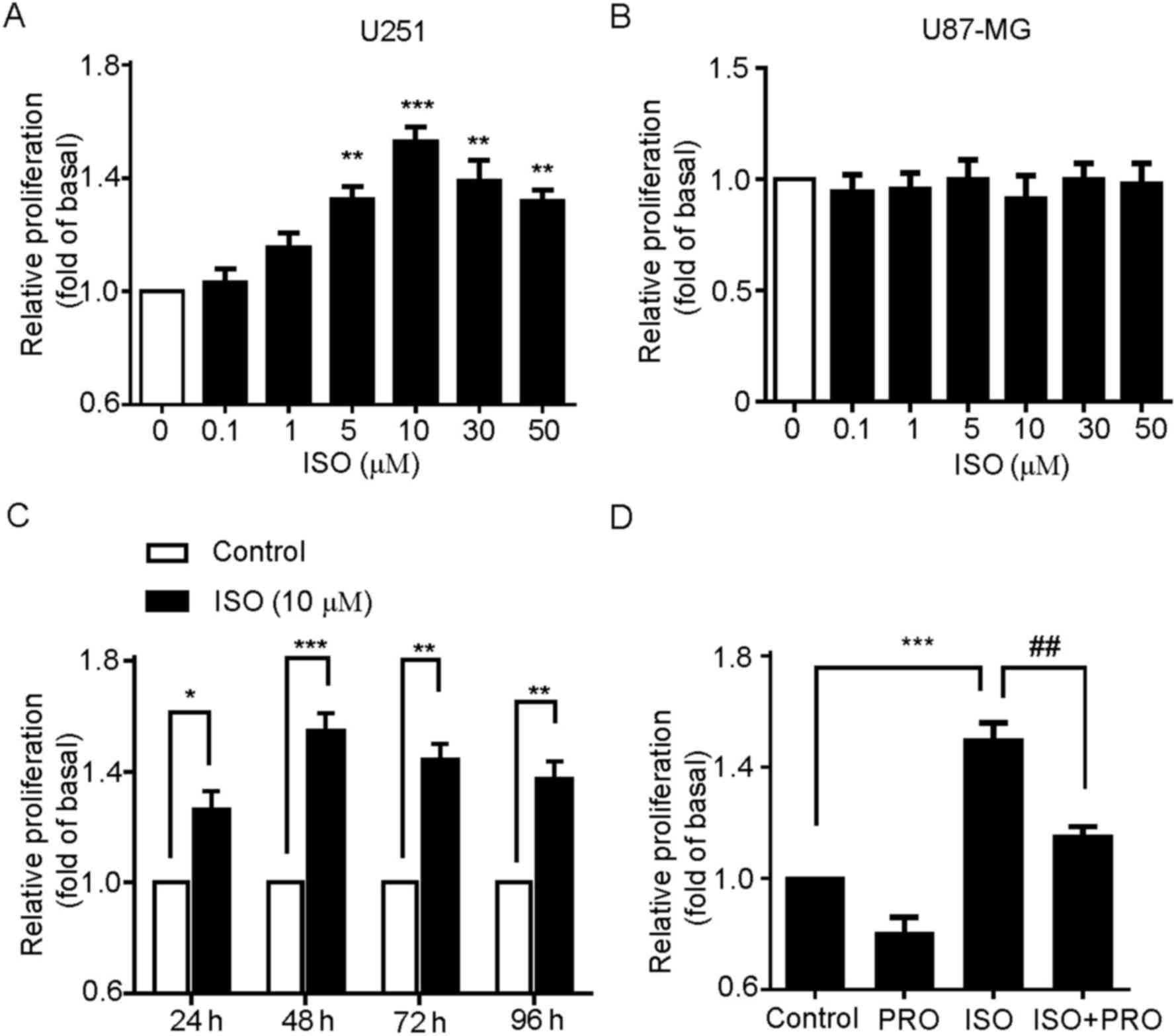 | Figure 2.β-AR activation promotes U251 cell
proliferation. (A) Effects of ISO on the cell proliferation of U251
cells. Cells were treated with various concentrations of ISO as
indicated for 48 h, and cell proliferation was assessed by MTT
assay. **P<0.01, ***P<0.001 vs. control group. (B) Effects of
ISO on the cell proliferation of U87-MG cells. (C) Time-course
stimulation of U251 cells with 10 µM ISO for 24, 48, 72 and 96 h.
*P<0.05, **P<0.01, ***P<0.001 vs. control groups. (D)
Effects of the β-AR antagonist PRO on ISO-induced cell
proliferation. Cells were incubated with PRO for 30 min prior to
and during treatment with ISO for 48 h. Values represent the mean ±
standard error of the mean from at least three triplicate
experiments. ***P<0.001 vs. control group,
##P<0.01 vs. ISO-treated group. β-AR, β-adrenergic
receptors; ISO, isoproterenol; PRO, propranolol. |
To additionally confirm that ISO-induced
proliferation was specifically mediated by β-ARs, the U251 cells
were pretreated with β-AR antagonist PRO and then stimulated by
ISO. As expected, PRO pre-treatment to block endogenous β-ARs
activity significantly suppressed ISO-induced U251 cell
proliferation (Fig. 2D). Taken
together, these results indicate that specific activation of β-ARs
is able to promote the cell proliferation in U251 cells.
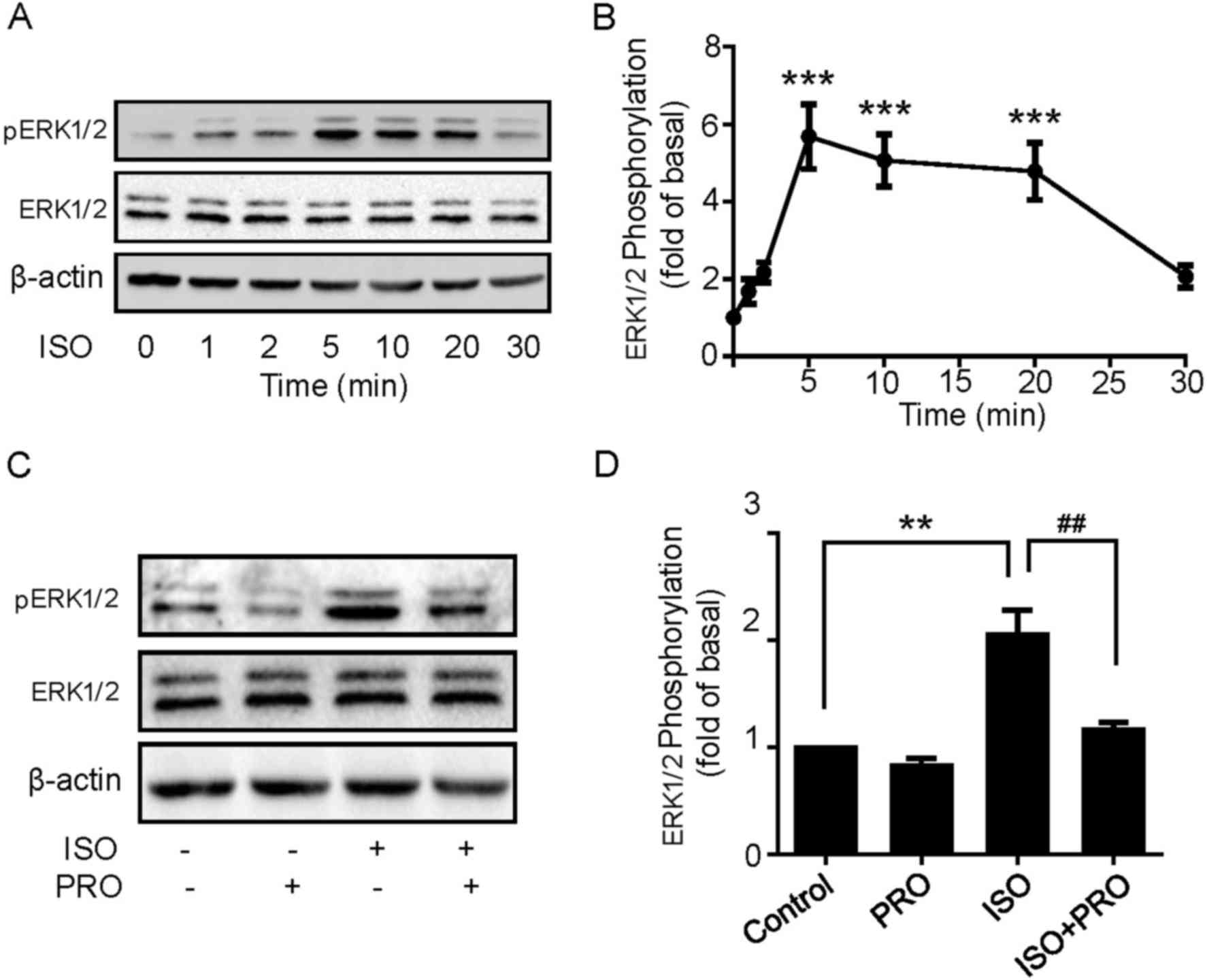
β-ARs activation induces
ERK1/2 phosphorylation in U251 cells
The aforementioned results indicated that ISO
treatment may significantly promote U251 cell proliferation. The
underlying molecular mechanisms of ISO-induced cell proliferation
in U251 cells were then explored. MAPK pathways have been widely
recognized to regulate a variety of physiological processes
including cell proliferation, differentiation and apoptosis in
cancer cells (21). Thus, to define
the role of MAPK cascades in the effect elicited by ISO, cells were
treated with 10 µM ISO for 1, 2, 5, 10, 20 and 30 min, and then
ERK1/2 phosphorylation levels were detected using western blotting.
ISO induced ERK1/2 phosphorylation in a rapid and transient manner,
which peaked at 5 min and decreased following 20 min, while ERK1/2
expression levels remained unchanged (Fig. 3A and B). Additionally, ERK1/2
phosphorylation was significantly blocked by PRO pretreatment
(Fig. 3C and D), suggesting β-ARs
activation is able to induce ERK1/2 phosphorylation in U251
cells.
ISO-induced proliferation is mediated
by ERK1/2 pathway
Whether ISO-mediated ERK1/2 activation was involved
in cell proliferation of U251 cells was investigated. The effects
of U0126, a specific MAPK/ERK (MEK)1/2 inhibitor, on ISO-induced
ERK1/2 phosphorylation were first explored. As demonstrated in
Fig. 4A and B, pretreating U251 cells
with U0126 effectively abolished ISO-induced ERK1/2
phosphorylation. Next, the MTT assay showed that the proliferative
effects of ISO were also significantly suppressed by U0126
(Fig. 4C). These results indicate
that ERK1/2 pathway is essential for ISO-mediated proliferation in
U251 cells.
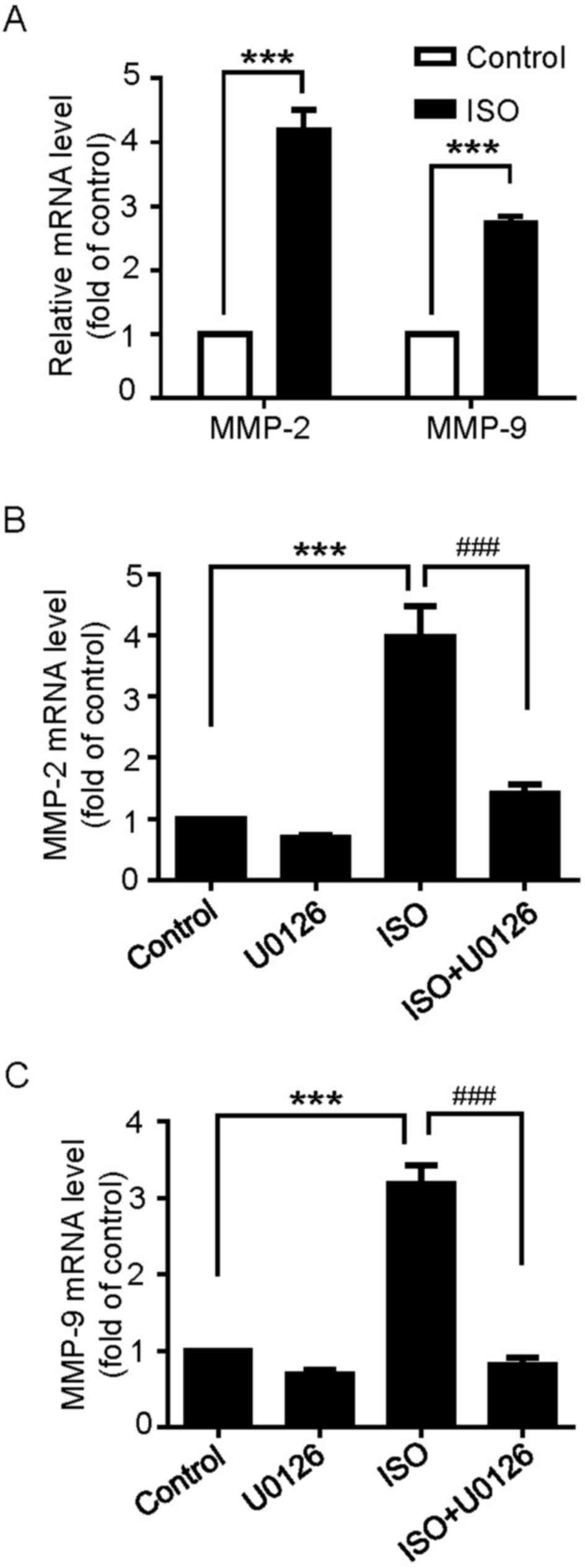
β-ARs activation increases MMP-2 and
MMP-9 mRNA expression through ERK1/2 activation
MMPs, particularly the gelatinases MMP-2 and MMP-9,
have been documented as key factors to the proliferation and
invasion of cancer cells (22,23).
Whether β-ARs activation regulates the expression of MMP-2
and MMP-9 mRNA in U251 cells was then examined. As
demonstrated in Fig. 5A, the results
indicated that MMP-2 and MMP-9 mRNA expression was
significantly upregulated following ISO treatment. In addition, the
pretreatment of U251 cells with U0126 inhibited ISO-induced
MMP-2 and MMP-9 mRNA expression (Fig. 5B and C). These results suggest that
β-ARs-mediated MMP-2 and MMP-9 mRNA expression occurs
through the ERK1/2 pathway.
Discussion
The main results of the present study concern the
mechanism of β-ARs-mediated cell proliferation of glioblastoma
cells. It was demonstrated that: i) β1-AR and
β2-AR are expressed in U251 cells but not in U87-MG
cells; ii) activation of β-ARs by ISO promoted the proliferation of
U251 cells through the ERK1/2 pathway; and iii) β-ARs activation
increased MMP-2 and MMP-9 mRNA expression, which may be important
for U251 cell proliferation.
Previous studies have demonstrated that β-ARs are
expressed in human clinical glioblastoma specimens obtained from
operated patients (24,25). Additionally, studies have indicated
that β-ARs are also expressed in glioblastoma cell lines and
primary cultures derived from human biopsies (26) and in the human-derived 1321N1
astrocytoma cell line (27). In
addition, studies with the human-derived U118 glioma cell line
indicate that there is a low but significant expression of β-ARs in
these cells, but to a lesser extent compared with 1321N1 cells
(18). However, the function of β-ARs
in U87-MG cells remains debated. Previous studies demonstrate that
in vitro U87-MG cells do not express functional β-ARs
(18,24). However, all three subtypes of β-ARs
appear to be detectable in U87-MG tumors in vivo, indicating
that β-ARs expression processes may take place and may be
functional during tumor development (24). Additionally,
(R,R')-4-methoxy-1-naphthylfenoterol, a selective β2-AR
agonist, was identified to inhibit cellular proliferation of U87-MG
cells (28). All these data suggest
that β-ARs may also be functional, by an unknown mechanism.
Nevertheless, the exact function of β-ARs in U251 glioma cells and
the underlying mechanism are not fully understood. In the present
study, the expression of β-ARs in U251 and U87-MG cells was first
measured by RT-PCR and RT-qPCR. The results demonstrated that
β1-ARs and β2-ARs were expressed in U251
cells, but not in U87-MG cells, which was consistent with a
previous study (24).
In previous years, evidence has suggested that β-AR
agonists and antagonists affect cell growth and function, which may
lead to the inhibition or induction of malignant diseases (29). The effect of β-AR activation is
cell-specific as β2-AR antagonists and agonists have
been demonstrated to attenuate cell growth. For example, β-ARs
activation may promote cell proliferation of certain cancer cells
including lung cancer cells (20),
ovarian carcinoma (5), human
hepatocellular carcinoma cells (30),
pancreatic (31), prostate (32), gastric (33) and colorectal cancer cells (34). By contrast, several studies have
demonstrated that the β-AR agonist ISO suppresses the proliferation
of MDA-MB-231 human breast cancer cells (35,36). In
addition, in U118 glioma cells and 132N1 astrocytoma cells,
treatment with β2-AR agonists (R,R')-fenoterol or
isoproterenol inhibited cell proliferation (18). However, the results of the present
study indicated that the activation of β-ARs by ISO promoted U251
cell proliferation. How this difference occurs, and the underlying
mechanism, requires additional investigation. As aforementioned,
obtaining β-AR expression in U87-MG tumors in vivo would
imply that β-AR may facilitate tumor formation by promoting
cellular proliferation. Another potential probability is that this
cell type-specific divergence on cellular proliferation may be due
to cross-talk between β-ARs and other GPCR-linked signaling
cascades including gamma-aminobutyric acid B receptors
(GABABR) (37),
α2-AR (38), bradykinin
B2 receptor (39),
oxytocin (40), and cannabinoid
receptors (28). For example, the
ISO-induced signaling cascade, cellular proliferation and cellular
migration in pancreatic ductal adenocarcinoma cells may be potently
inhibited following stimulation of GABABR signaling
(37).
The MMPs are a family of proteolytic enzymes that
regulate various cell behaviors that are relevant in cancer biology
through the degradation of the extracellular matrix surrounding
tumors (41). Although MMPs,
particularly MMP-2 and MMP-9, have been considered to be important
factors in facilitating invasion and metastases through the
degradation of type IV collagen, a major component of the basement
membrane, additional evidence also demonstrates that MMPs may
promote cancer cell proliferation (23). Previous, studies have suggested that
NE may affect the progression of ovarian cancer by modulating the
expression of MMPs and the angiogenic cytokine, vascular
endothelial growth factor, in ovarian cancer cells (42). Additionally, β-ARs inhibition
suppressed the expression of MMP-2 and MMP-9 in human brain
microvascular endothelial cells (24)
thus increasing the proliferative, invasive and metastatic
potential of these cells. In the present study, it was revealed
that MMP-2 and MMP-9 mRNA expression was significantly increased
upon β-ARs activation. Consistently, the proliferation of U251
cells was also enhanced through the activation of β-ARs. In
addition, pretreatment with U0126 to block ERK1/2 activity
significantly reduced the upregulation of MMP-2 and MMP-9. Previous
studies have demonstrated that the activation of the EGFR-MEK-ERK
signaling pathway causes the overexpression of MMP-2 and MMP-9 in
prostate cancer cells (43). However,
whether EGFR transactivation pathway or/and other signaling
cascades are involved in β-ARs-induced cell proliferation in
glioblastoma cells requires additional study.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China (grant no. 81601179), Natural Science
Foundation of Jiangxi Province of China (grant nos. 20171BAB215039,
20122BAB215020, 20161BAB204166 20161BAB205212 and
20171BAB214022).
References
|
1
|
Dolecek TA, Propp JM, Stroup NE and
Kruchko C: CBTRUS statistical report: Primary brain and central
nervous system tumors diagnosed in the United States in 2005–2009.
Neuro Oncol. 14:(Suppl 5). v1–v49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R and Roila F: ESMO Guidelines
Working Group: Malignant glioma: ESMO clinical recommendations for
diagnosis, treatment and follow-up. Ann Oncol. 20:(Suppl 4).
S126–S128. 2009. View Article : Google Scholar
|
|
4
|
Nagaraja AS, Armaiz-Pena GN, Lutgendorf SK
and Sood AK: Why stress is BAD for cancer patients. J Clin Invest.
123:558–560. 2013.PubMed/NCBI
|
|
5
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hassan S, Karpova Y, Baiz D, Yancey D,
Pullikuth A, Flores A, Register T, Cline JM, D'Agostino R Jr,
Danial N, et al: Behavioral stress accelerates prostate cancer
development in mice. J Clin Invest. 123:874–886. 2013.PubMed/NCBI
|
|
7
|
Timmermans W, Xiong H, Hoogenraad CC and
Krugers HJ: Stress and excitatory synapses: From health to disease.
Neuroscience. 248:626–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Kloet ER, Joëls M and Holsboer F:
Stress and the brain: From adaptation to disease. Nat Rev Neurosci.
6:463–475. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fitzgerald PJ: Is norepinephrine an
etiological factor in some types of cancer? Int J Cancer.
124:257–263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Woo AY and Xiao RP: β-adrenergic receptor
subtype signaling in heart: From bench to bedside. Acta Pharmacol
Sin. 33:335–341. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schuller HM and Al-Wadei HA:
Neurotransmitter receptors as central regulators of pancreatic
cancer. Future Oncol. 6:221–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cohen S, Janicki-Deverts D and Miller GE:
Psychological stress and disease. JAMA. 298:1685–1687. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schuller HM: Beta-adrenergic signaling, a
novel target for cancer therapy? Oncotarget. 1:466–469. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Braadland PR, Ramberg H, Grytli HH and
Taskén KA: β-adrenergic receptor signaling in prostate cancer.
Front Oncol. 4:3752015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Al-Wadei HA, Al-Wadei MH and Schuller HM:
Prevention of pancreatic cancer by the beta-blocker propranolol.
Anticancer Drugs. 20:477–482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Askari MD, Tsao MS and Schuller HM: The
tobacco-specific carcinogen,
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates
proliferation of immortalized human pancreatic duct epithelia
through beta-adrenergic transactivation of EGF receptors. J Cancer
Res Clin Oncol. 131:639–648. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weddle DL, Tithoff P, Williams M and
Schuller HM: Beta-adrenergic growth regulation of human cancer cell
lines derived from pancreatic ductal carcinomas. Carcinogenesis.
22:473–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Toll L, Jimenez L, Waleh N, Jozwiak K, Woo
AY, Xiao RP, Bernier M and Wainer IW: {Beta}2-adrenergic receptor
agonists inhibit the proliferation of 1321N1 astrocytoma cells. J
Pharmacol Exp Ther. 336:524–532. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu P, He J, Liu S, Wang M, Pan B and Zhang
W: β2-adrenergic receptor activation promotes the proliferation of
A549 lung cancer cells via the ERK1/2/CREB pathway. Oncol Rep.
36:1757–1763. 2016.PubMed/NCBI
|
|
21
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dong W, Li H, Zhang Y, Yang H, Guo M, Li L
and Liu T: Matrix metalloproteinase 2 promotes cell growth and
invasion in colorectal cancer. Acta Biochim Biophys Sin (Shanghai).
43:840–848. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Annabi B, Lachambre MP, Plouffe K,
Moumdjian R and Béliveau R: Propranolol adrenergic blockade
inhibits human brain endothelial cells tubulogenesis and matrix
metalloproteinase-9 secretion. Pharmacol Res. 60:438–445. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sardi I, Giunti L, Bresci C, Buccoliero
AM, Degl'innocenti D, Cardellicchio S, Baroni G, Castiglione F, Ros
MD, Fiorini P, et al: Expression of β-adrenergic receptors in
pediatric malignant brain tumors. Oncol Lett. 5:221–225.
2013.PubMed/NCBI
|
|
26
|
Prenner L, Sieben A, Zeller K, Weiser D
and Häberlein H: Reduction of high-affinity beta2-adrenergic
receptor binding by hyperforin and hyperoside on rat C6
glioblastoma cells measured by fluorescence correlation
spectroscopy. Biochemistry. 46:5106–5113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wakshull E, Hertel C, O'Keefe EJ and
Perkins JP: Cellular redistribution of beta-adrenergic receptors in
a human astrocytoma cell line: A comparison with the epidermal
growth factor receptor in murine fibroblasts. J Cell Biochem.
29:127–141. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paul RK, Ramamoorthy A, Scheers J, Wersto
RP, Toll L, Jimenez L, Bernier M and Wainer IW: Cannabinoid
receptor activation correlates with the proapoptotic action of the
β2-adrenergic agonist (R,R')-4-methoxy-1-naphthylfenoterol in HepG2
hepatocarcinoma cells. J Pharmacol Exp Ther. 343:157–166. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Evans BA, Sato M, Sarwar M, Hutchinson DS
and Summers RJ: Ligand-directed signalling at beta-adrenoceptors.
Br J Pharmacol. 159:1022–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yuan A, Li Z, Li X, Yi S, Wang S, Cai Y
and Cao H: The mitogenic effectors of isoproterenol in human
hepatocellular carcinoma cells. Oncol Rep. 23:151–157.
2010.PubMed/NCBI
|
|
31
|
Pham H, Chen M, Takahashi H, King J, Reber
HA, Hines OJ, Pandol S and Eibl G: Apigenin inhibits NNK-induced
focal adhesion kinase activation in pancreatic cancer cells.
Pancreas. 41:1306–1315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang P, He X, Tan J, Zhou X and Zou L:
β-arrestin2 mediates β-2 adrenergic receptor signaling inducing
prostate cancer cell progression. Oncol Rep. 26:1471–1477.
2011.PubMed/NCBI
|
|
33
|
Shin VY, Wu WK, Chu KM, Koo MW, Wong HP,
Lam EK, Tai EK and Cho CH: Functional role of beta-adrenergic
receptors in the mitogenic action of nicotine on gastric cancer
cells. Toxicol Sci. 96:21–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Coelho M, Moz M, Correia G, Teixeira A,
Medeiros R and Ribeiro L: Antiproliferative effects of β-blockers
on human colorectal cancer cells. Oncol Rep. 33:2513–2520.
2015.PubMed/NCBI
|
|
35
|
Slotkin TA, Zhang J, Dancel R, Garcia SJ,
Willis C and Seidler FJ: Beta-adrenoceptor signaling and its
control of cell replication in MDA-MB-231 human breast cancer
cells. Breast Cancer Res Treat. 60:153–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Carie AE and Sebti SM: A chemical biology
approach identifies a beta-2 adrenergic receptor agonist that
causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2
pathway. Oncogene. 26:3777–3788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schuller HM, Al-Wadei HA and Majidi M:
GABA B receptor is a novel drug target for pancreatic cancer.
Cancer. 112:767–778. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cottingham C, Lu R, Jiao K and Wang Q:
Cross-talk from β-adrenergic receptors modulates α2A-adrenergic
receptor endocytosis in sympathetic neurons via protein kinase A
and spinophilin. J Biol Chem. 288:29193–29205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hanke S, Nürnberg B, Groll DH and Liebmann
C: Cross talk between beta-adrenergic and bradykinin B(2) receptors
results in cooperative regulation of cyclic AMP accumulation and
mitogen-activated protein kinase activity. Mol Cell Biol.
21:8452–8460. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wrzal PK, Goupil E, Laporte SA, Hébert TE
and Zingg HH: Functional interactions between the oxytocin receptor
and the β2-adrenergic receptor: Implications for ERK1/2 activation
in human myometrial cells. Cell Signal. 24:333–341. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deryugina EI and Quigley JP: Matrix
metalloproteinases and tumor metastasis. Cancer Metastasis Rev.
25:9–34. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moreno-Smith M, Lutgendorf SK and Sood AK:
Impact of stress on cancer metastasis. Future Oncol. 6:1863–1881.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiao LJ, Lin P, Lin F, Liu X, Qin W, Zou
HF, Guo L, Liu W, Wang SJ and Yu XG: ADAM17 targets MMP-2 and MMP-9
via EGFR-MEK-ERK pathway activation to promote prostate cancer cell
invasion. Int J Oncol. 40:1714–1724. 2012.PubMed/NCBI
|