Introduction
Prostate cancer (PCa) is a common malignant tumor
and the second leading cause of male morbidity and mortality
worldwide. In 2015, PCa alone accounted for ~26% (220,800) of newly
diagnosed cancer cases in men and led to >27,000 deaths
(1). Although numerous methods, such
as biochemical assays (2), biopsies
(3), digital rectal examination (DRE)
(4), and transrectal ultrasonography
(5), have been widely used in PCa
diagnosis, lack of early efficient diagnoses is the major problem
affecting precise therapy of PCa. Therefore, research on the
mechanisms of PCa tumorigenesis and progression will help to
improve prevention strategies and develop effective interventions
to treat PCa.
Bloom syndrome protein (BLM), which is mutated in
patients with Bloom syndrome, is a member of the RecQ helicase
family. The core structure is highly conserved between yeast and
humans (6). Usually, BLM utilizes
energy from ATP-hydrolysis to unwind double-stranded DNA (dsDNA)
with 3′ to 5′ polarity, and to disassemble complex DNA structures,
such as G-quadruplexes (G4s) and Holliday junctions (HJs) that are
formed during the S phase of the cell cycle (7). BLM is a crucial helicase required for
DNA metabolic processes, including DNA recombination, replication
and repair. Such helicases are referred to as ‘the caretakers of
the genome’. BLM is expressed at high levels in tumor cells, and
its level expression is known to be differentially regulated
through the cell cycle stages. Considering that abnormalities in
BLM are linked to genome instability, accumulating evidence has
recently indicated that changes in BLM protein expression are
associated with the development of a variety of tumors (8–10). Using
exome sequencing and gene analysis, Johnson et al (11) identified that BLM missense variants
and PCa status were completely co-segregated. However, the role of
BLM in PCa, and whether it can influence the cell cycle and
apoptosis, still remains unclear. The results of the present study
demonstrate that BLM protein expression is upregulated in PCa
patient tissues and PC3 cells. Furthermore, the downregulation of
BLM can inhibit PC3 cell ischemic proliferation and induce
apoptosis in vitro.
Materials and methods
Tissue collection
A total of 15 cancerous prostate tissues (PCa) and
10 noncancerous tissues (non-PCa) were collected from patients who
underwent surgery at Beijing Chao-Yang Hospital, Capital Medical
University (Beijing, China). The present study was approved by the
ethics committee of Beijing Chao-Yang Hospital, Capital Medical
University, and written informed consent was obtained from each
patient.
Immunohistochemistry
All tissues from PCa patients were paraffin-embedded
and cut to 4 mm thickness. Endogenous peroxidase activity in
deparaffinized sections was blocked by incubation with 3% hydrogen
peroxide. Subsequently, sections were boiled in 1 mM EDTA pH 8.0
for 10 min, followed by cooling at room temperature. Fetal bovine
serum was applied to block non-specific antigen sites. Sections
were incubated with anti-BLM antibody for 3 h at a dilution of
1:200, and then the secondary antibody was applied for 30 min.
Finally, the sections were stained with liquid diaminobenzydine and
counterstained with Mayer's hematoxylin blue.
Cells and culture
The BPH1 benign prostatic hyperplasia cell line and
PC3 PCa cell line were obtained from the American Type Culture
Collection (Manassas, VA, USA). Cells were routinely cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), supplemented with 10% fetal bovine serum (Gibco) in a
standard culture conditions (5% CO2 at 37°C).
BLM short hairpin RNA (shRNA) plasmid
transfection
PC3 cells were transfected with BLM-targeting shRNA
(GeneChem Co. Ltd, Shanghai, China) (target sequence:
AAGGAAGTTGTATGCACTA) or non-targeting shRNA (GeneChem Co. Ltd,
Shanghai, China) using Lipofectamine™ 2000. Transfection with a
negative control (NC) scramble sequence was also conducted.
Transfection efficiency was confirmed by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot.
Cell proliferation assay
PC3 cells (1×103) were seeded in 6-well plates in
RPMI-1640 containing 10% FBS. Cells were harvested and counted
using the EdU assay after 2, 4 and 7 days, as follows. Cell
proliferation was detected by using the Cell-LightTM
5-ethynyl-2′-deoxyuridine (EdU) imaging detection kit according to
the manufacturer's instructions (RiboBio, Guangzhou, China). EdU
was added to cells seeded in 6-well plates and the cells were
cultured for an additional 2 h. The cells were then collected and
washed with PBS twice. Apollo reaction buffer was then incubated
with the cells for 10 min in the dark. Subsequently, cells were
washed once with PBS, stained with Hoechst33342 for 30 min at room
temperature and washed with PBS again. EdU-positive cells were
counted using Image-Pro Plus 6.0 software (Media Cybernetics,
Silver Springs, MD, USA) in five randomly selected fields.
For cell cycle analysis, cells were seeded on 6-well
plates and stained with Vybrant® DyeCycle™ Ruby stain
kit at 37°C for 30 min. The DNA content was measured by flow
cytometry according to the manufacturer's instructions (Thermo
Fisher Scientific, Inc.).
Cell invasion and migration
assays
A cell invasion assay was performed using a 24-well
Transwell (Corning, New York, NY, USA) with a polycarbonate filter
pre-coated with Matrigel at a 1:20 dilution (BD Biosciences,
Franklin Lakes, NJ, USA). Cells (2.5×104) suspended in 0.2 ml
serum-free medium were added to the upper well of the chamber, and
500 ml RPMI-1640 supplemented with 10% FBS was added to the lower
well. After 24 h incubation, cells remaining in the upper well were
removed by scraping. Invaded cells on the bottom of the membrane
were fixed with 4% paraformaldehyde and stained with 0.1% crystal
violet solution. Cells were imaged under a microscope, and the cell
numbers were counted using Image-Pro Plus 6.0 software (Media
Cybernetics, Silver Springs, MD, USA) in five randomly selected
fields. A cell migration assay was conducted in a similar manner
but without the use of Matrigel.
Apoptosis assay
Cell apoptosis was detected using an Annexin V-PE
apoptosis detection kit (BD Pharmingen, San Diego, CA, USA)
according to the manufacturer's protocol. Briefly, cells at density
of 1×106 cells/ml were harvested and washed twice with PBS, then
500 µl of cell suspension in binding buffer was transferred to a
5-ml falcon tube, and 5 µl Annexin V-PE conjugate and 5 µl
propidium iodide were added. The cells were incubated in the dark
for 15 min at room temperature and the fluorescence of the cells
was subsequently determined by flow cytometry.
RNA extraction and RT-qPCR
Total RNA was extracted from tissues or cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). A FastQuant RT Kit (With gDNase) and SuperReal PreMix Plus
(SYBR Green; TIANGEN BIOTECH, Beijing, China) were used. The PCR
primers used were as follows: BLM, forward
5′-GGATCCTGGTTCCGTCCGC-3′, reverse 5′-CCTCAGTCAAATCTATTTGCTCG-3′;
β-actin, forward 5′-TGACGTGGACATCCGCAAAG-3′, reverse
5′-TCTTCATTGTGCTGGGTGCC-3′. PCR cycling conditions were as follows:
Initial denaturation period at 95°C for 15 min, followed by a
two-step PCR program consisting of denaturation at 95°C for 10 sec
and annealing/extension at 60°C for 32 sec for 40 cycles. All
samples were normalized against the internal control (β-actin) and
analyzed using the 2−ΔΔCt method.
Western blot analysis
Total protein was extracted from the cells or
tissues using lysis buffer containing protease inhibitors. Proteins
(30 µg) from each sample were loaded on 10% SDS-polyacrylamide gels
for electrophoresis. Following electrophoresis, the proteins were
transferred onto polyvinylidene difluoride membranes (GE
Healthcare, Chalfont, UK), which were then blocked with 10% non-fat
milk in Tween/Tris-buffered salt solution (TTBS; 20 mm Tris-Cl, pH
7.5, 0.15 M NaCl and 0.05% Tween-20) for 1 h. Following incubation
with the anti-BLM antibody (Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) at 4°C overnight, the membrane was washed and incubated
with IRDye® 800CW donkey anti-goat IgG secondary
antibody at room temperature for 2 h followed by LI-COR Odyssey gel
imaging scanner detection (LI-COR Biosciences, Lincoln, NE, USA).
To verify equal loading of protein, the blots were reprobed with
primary monoclonal antibody against β-actin (ProteinTech Group,
Inc., Chicago, IL, USA).
Statistical analysis
Quantitative analysis of immunoblotting was
performed using Quantity-One software (Gel Doc 2000 imaging system,
Bio-Rad Laboratories, Inc., Hercules, CA, USA). For protein level,
the protein ratio of BLM (band density of protein/band density of
β-actin) was set as 100% in the BPH or NC group. The data from
other groups were expressed as a percentage of the BPH or NC group.
The values are presented as the mean ± standard error. Statistical
analysis was conducted by one-way analysis of variance followed by
all pairwise multiple comparison procedures using the Bonferroni
test and Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Differential expression of BLM in PCa
and non-PCa patients
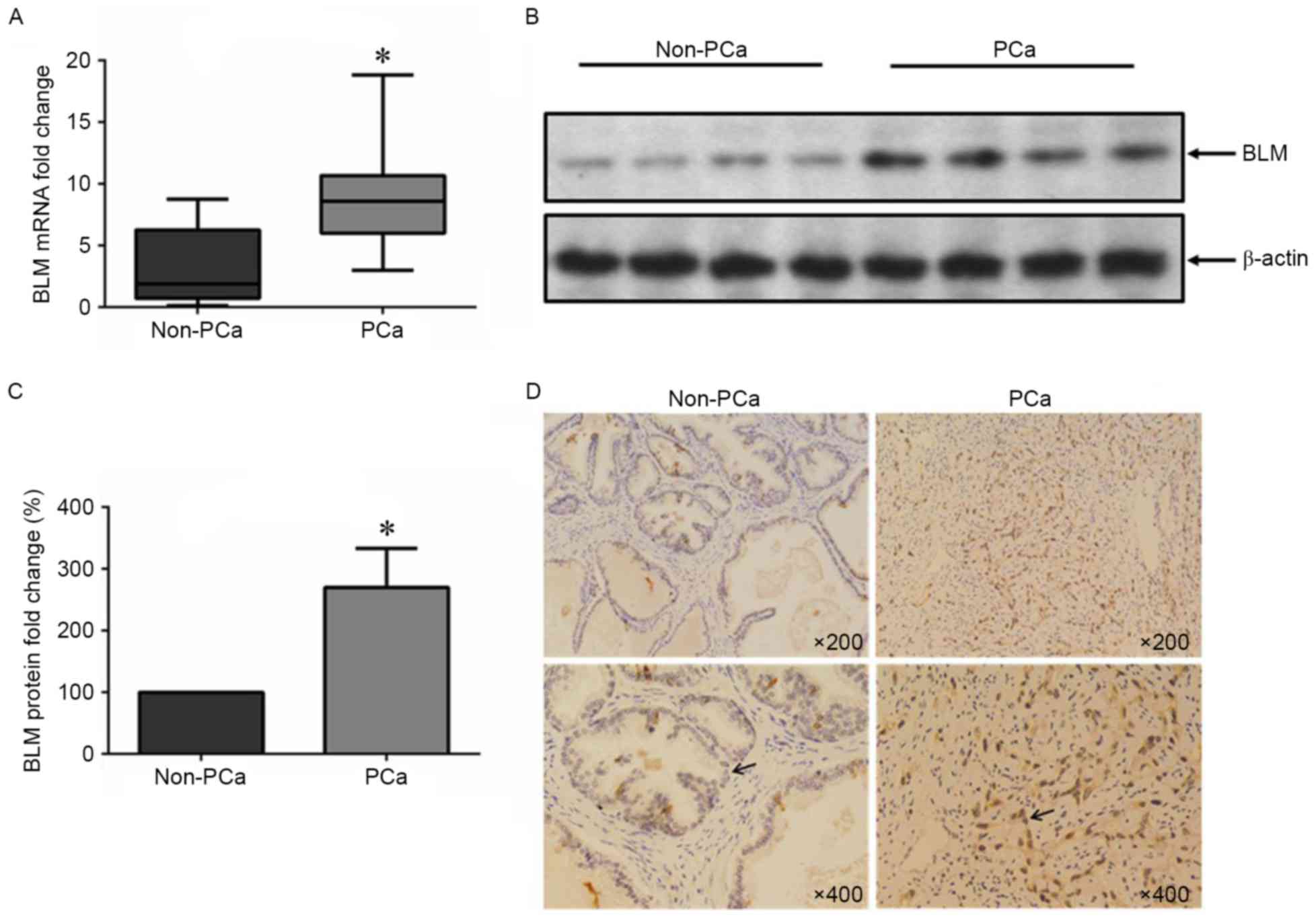
To determine BLM expression status in PCa tissues,
the mRNA and protein levels of BLM in PCa and non-PCa tissue
samples were detected. The patient profiles are summarized in
Table I. We found that the BLM mRNA
and protein expression levels were upregulated in the PCa tissues
compared the non-PCa group (Fig.
1A-C). Similarly, immunohistochemical staining showed that the
expression of BLM protein was increased in the nuclei of tumor
cells, but it almost was undetectable in non-PCa tissue (Fig. 1D).
 | Table I.Clinical and pathological parameters
of PCa and Non-PCa patients. |
Table I.
Clinical and pathological parameters
of PCa and Non-PCa patients.
| Parameter | PCa (n=15) | Non-PCa (n=10) |
|---|
| Age |
|
Range | 58–76 | 49–76 |
| Mean | 68.40 | 64.60 |
| Total PSA |
|
Range | 0.005–75.488 | 1.550–10.383 |
| Mean | 17.400 | 5.126 |
| Clinical stage |
| T1c | 2 | N/A |
| T2c | 5 | N/A |
| T3a | 2 | N/A |
| T3b | 4 | N/A |
| T4b | 2 | N/A |
| Gleason score |
| 3+3 | 1 | N/A |
| 3+4 | 2 | N/A |
| 4+3 | 4 | N/A |
| 4+4 | 5 | N/A |
| 4+5 | 2 | N/A |
| 5+4 | 1 | N/A |
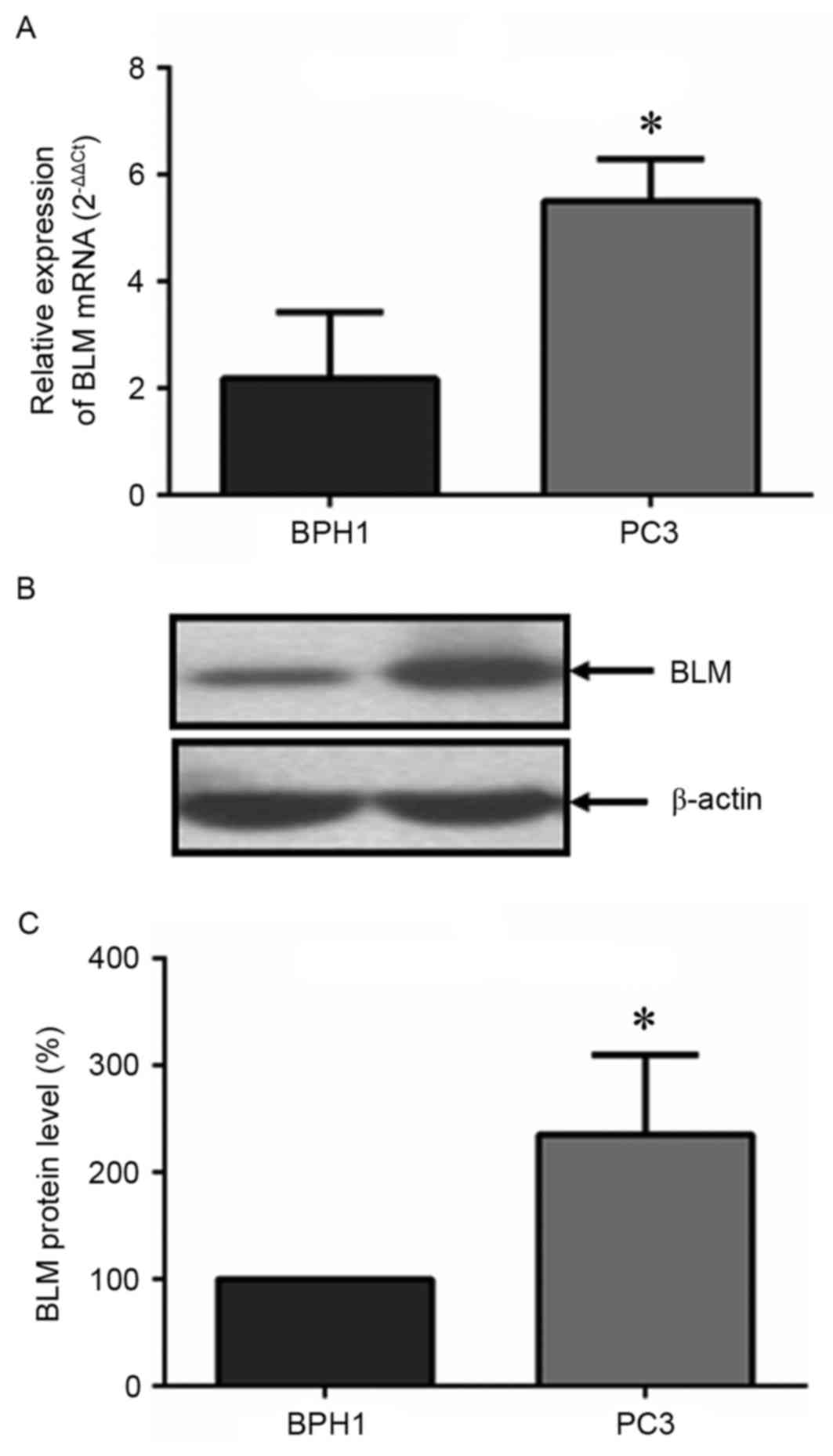
Changes of BLM expression level in PCa
and BPH1 cells
To verify the results from tissue samples, the BLM
expression level was also analyzed in BPH1 and PC3 cells in
vitro. As shown in Fig. 2, the
mRNA and protein levels of BLM in PC3 cells were significantly
higher than that of the BPH1 cells.
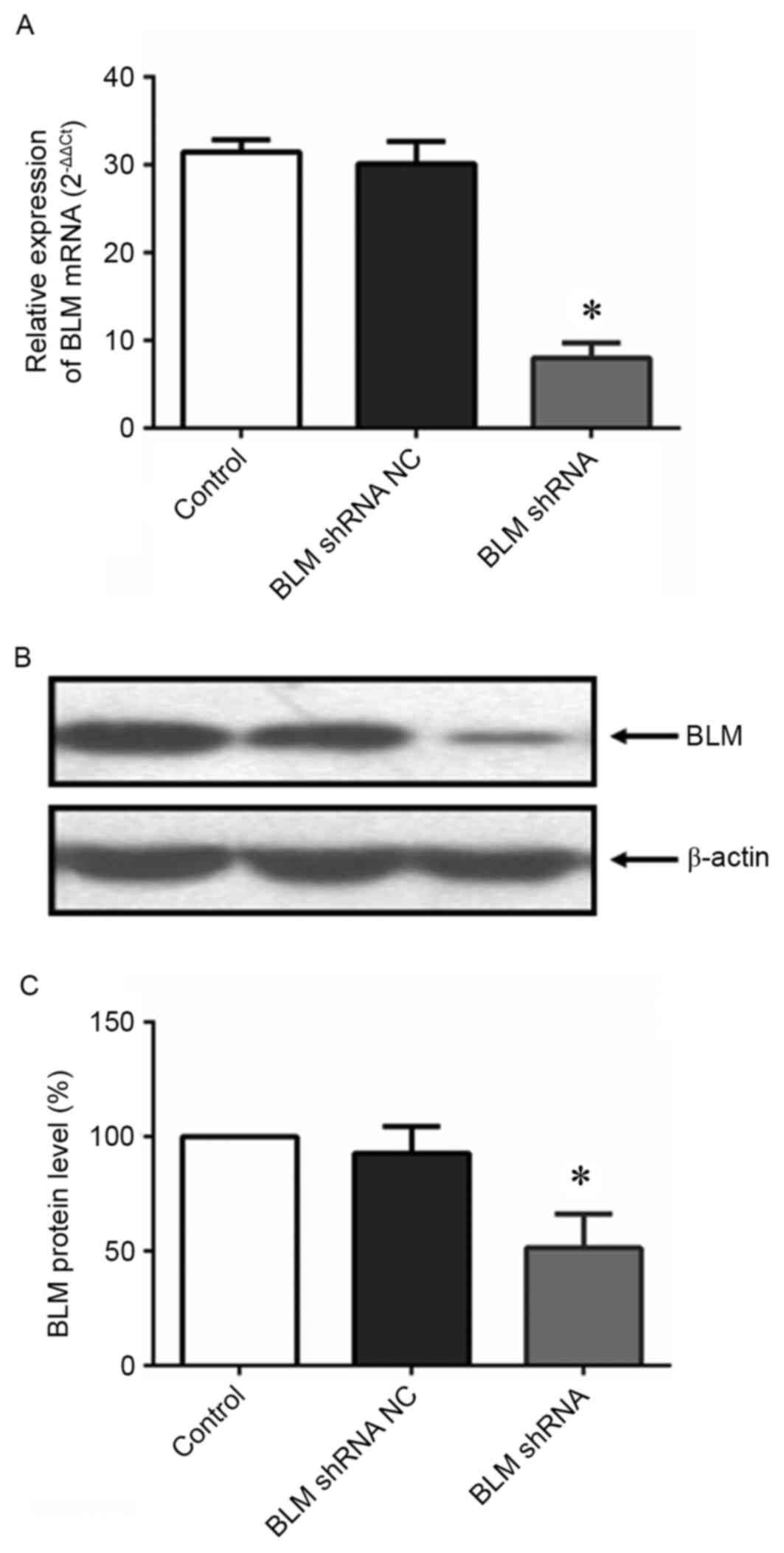
Inhibition of BLM reduced PC3 cell
proliferation in vitro
To explore the role of BLM in PCa cell
proliferation, PC3 cells were transfected with a BLM-targeting
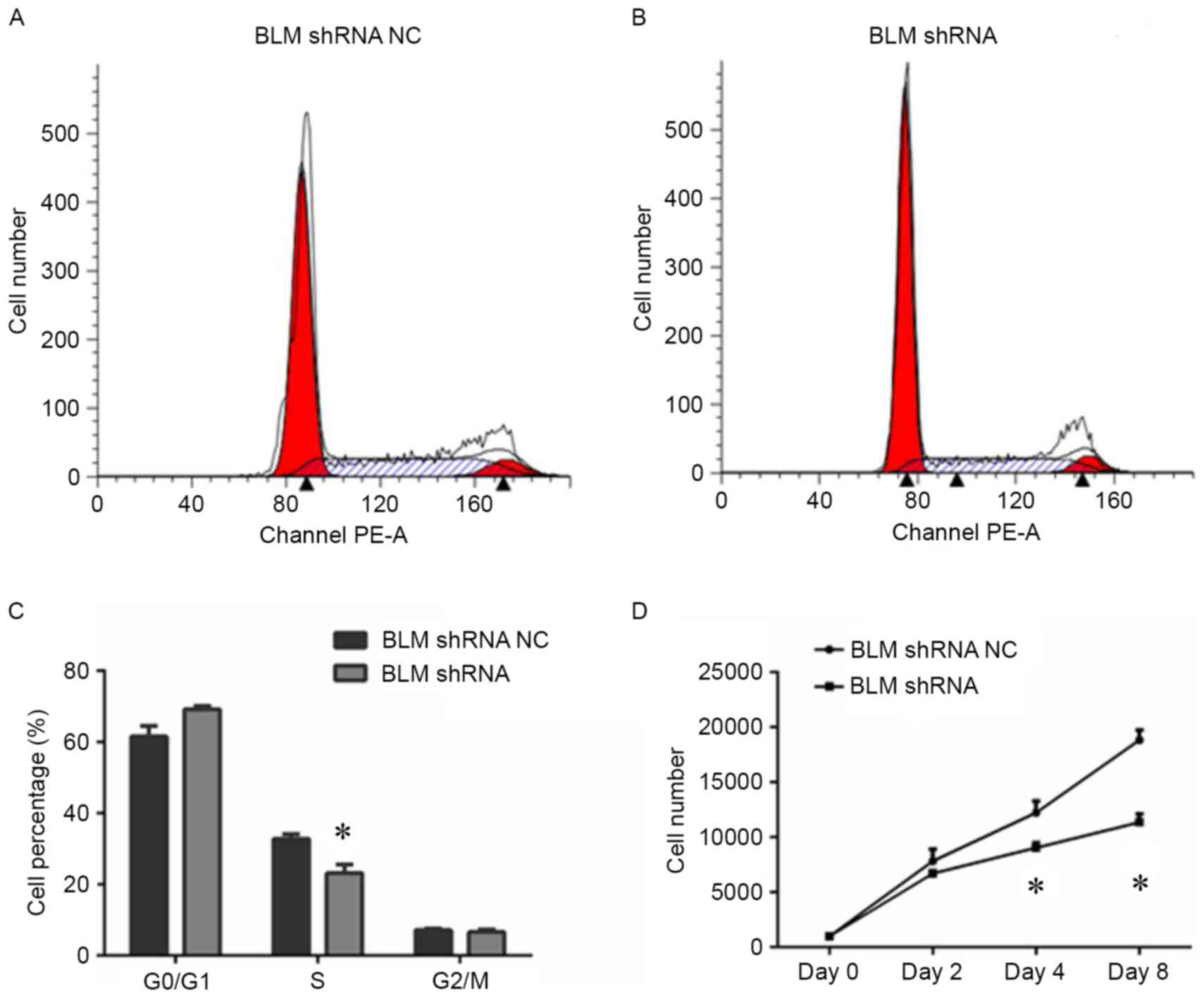
shRNA plasmid. As shown in Fig. 3,
both of the BLM mRNA and protein expression levels were
significantly downregulated in PC3 cells after transfection with
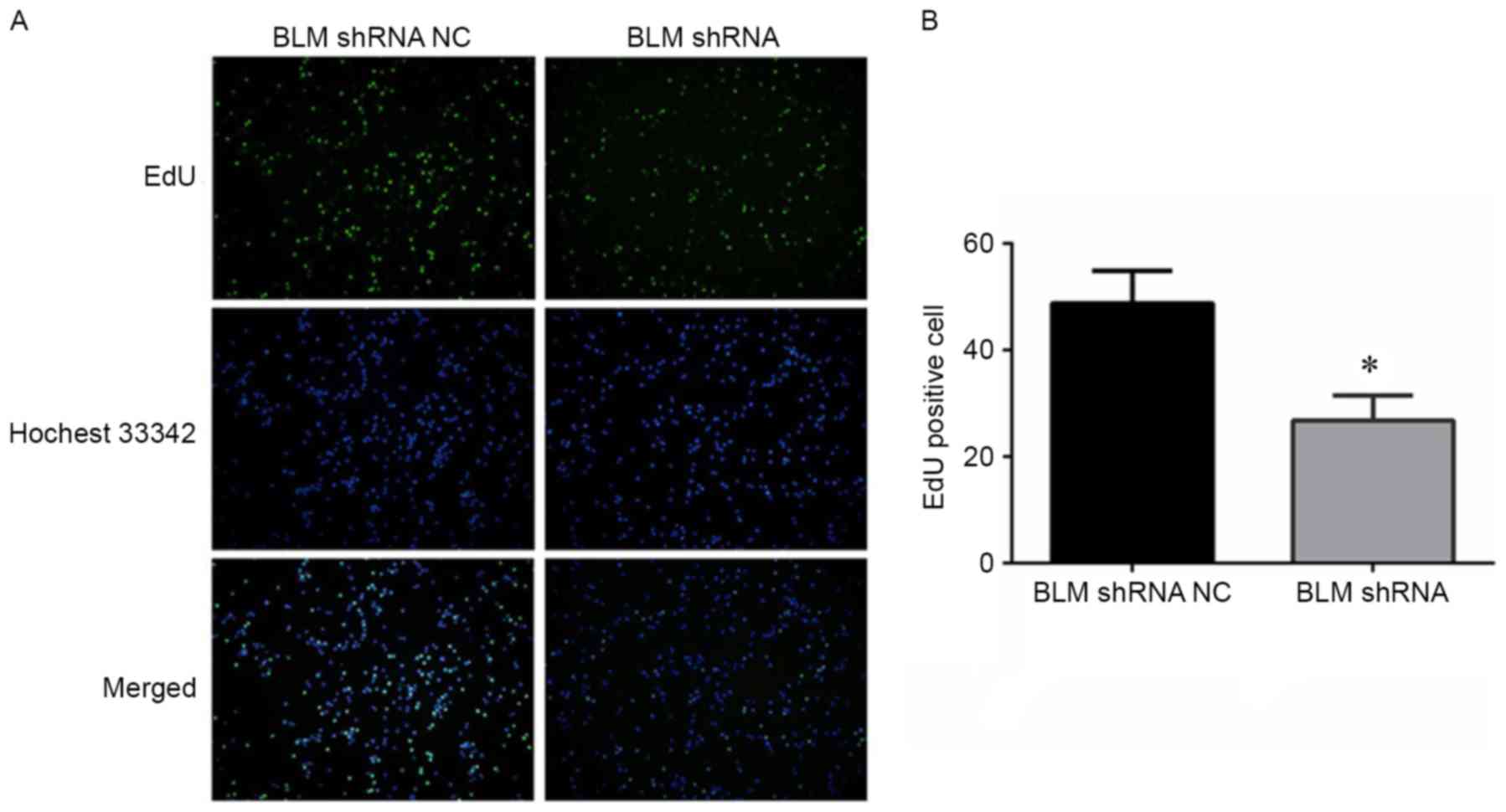
the BLM-targeting shRNA. An EdU proliferation assay was conducted
to determine the effect of BLM inhibition on PC3 cell
proliferation. As shown in Fig. 4A and
B, shRNA-mediated inhibition of BLM significantly reduced PC3
cell proliferation. Additionally, cell cycle analysis was performed
to examine if BLM inhibition influences the cell cycle. The results
in Fig. 5A and B revealed that there
was significant decrease in the number of cells in S-phase after
transfection with BLM shRNA. Similarly, the PC3 cells exhibited a
significantly reduced proliferative capacity when BLM was knocked
down (Fig. 5D).
Downregulation of BLM promoted PCa
cell apoptosis
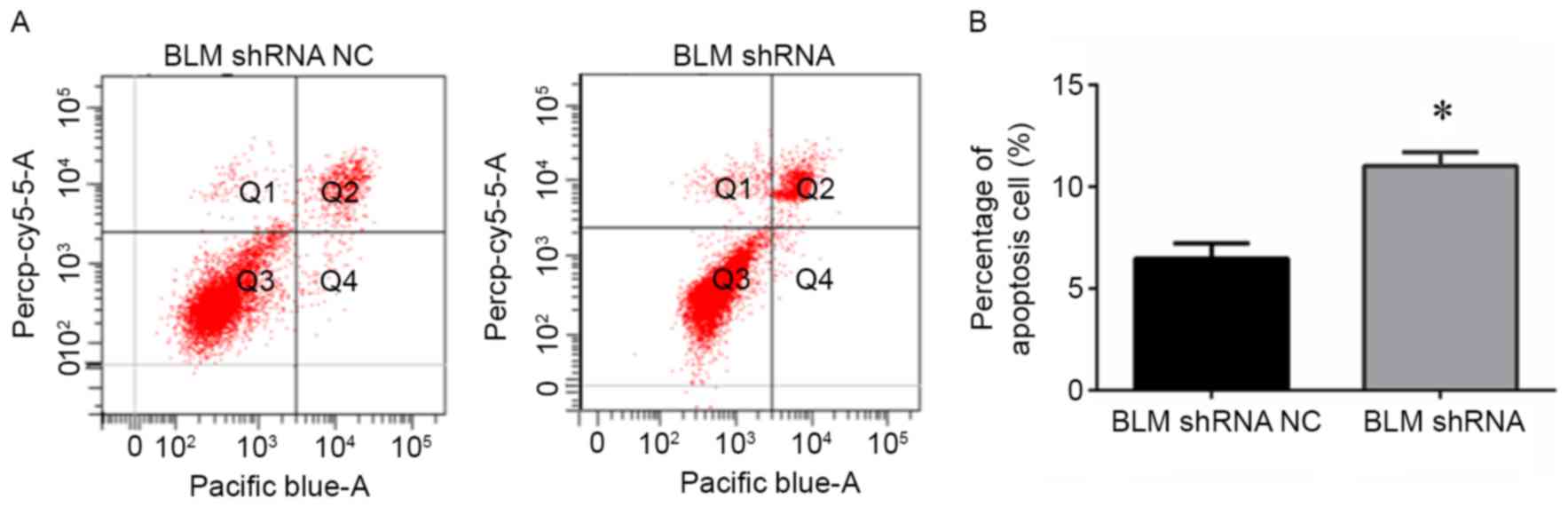
To further elucidate the role of BLM in PCa
apoptosis, PC3 cells transfected with BLM shRNA plasmid were
subjected to Annexin-V and PI staining, and flow cytometry analysis
was performed. The results shown in Fig.
6A and B demonstrated that knockdown of BLM led to an increase
in apoptosis.
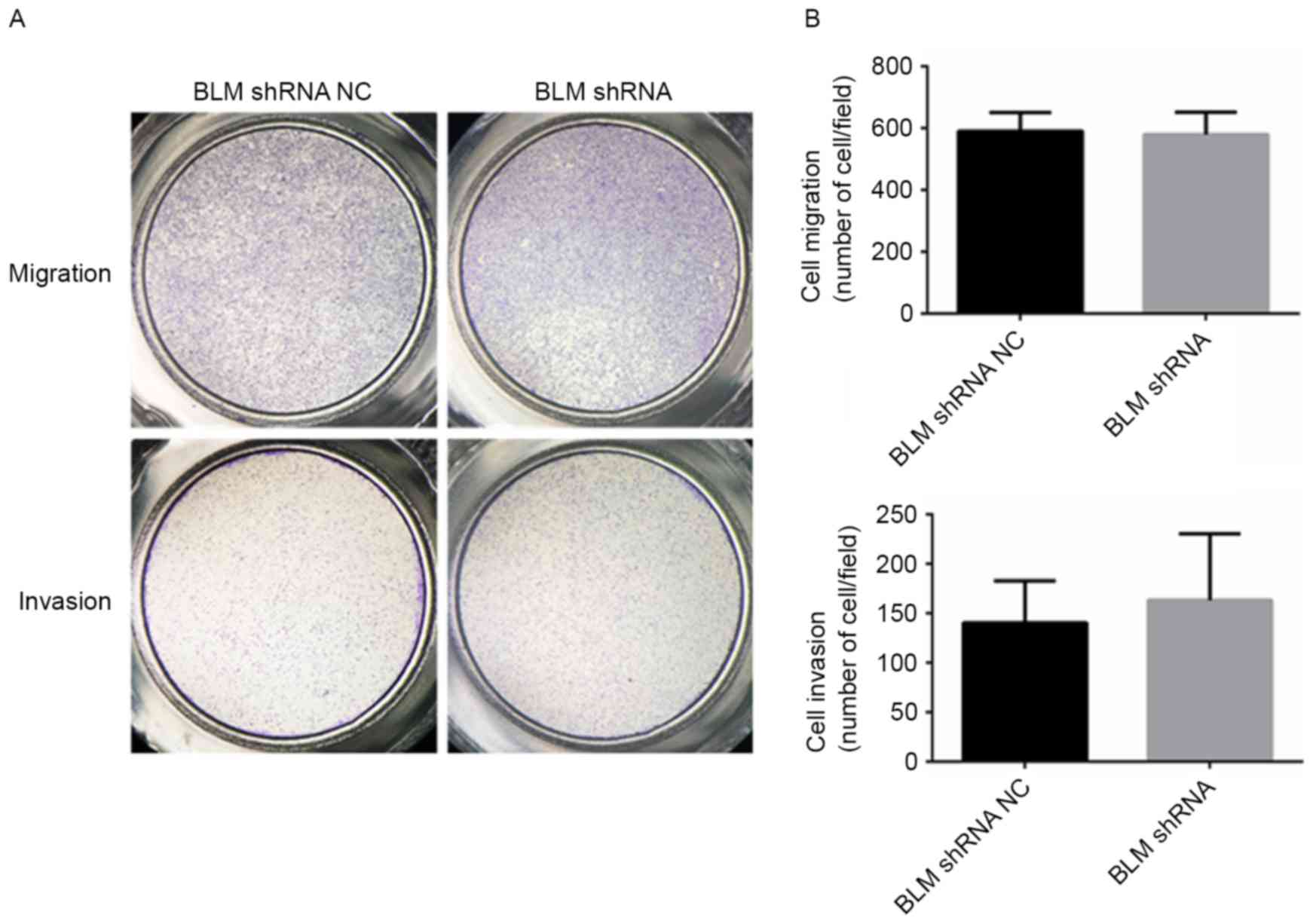
Inhibition of BLM had no effect on PCa
cell migration and invasion
The effect of BLM on PCa cell migration and invasion
was examined using Transwell assays. As presented in Fig. 7A and B, PC3 cells transfected with the
BLM shRNA plasmid did not exhibit any increase or decrease in
migration and invasion compared with the NC cells.
Discussion
It is well-established that PCa development is
affected by genetic inheritance, with numerous genes recognized to
influence PCa susceptibility, including MED12 (12), HOXB13 (13), BRCA1 and BRCA2 (14,15). As a
RecQ helicase, BLM is part of a family of DNA unwinding enzymes,
and has crucial roles at multiple steps in the DNA recombination,
replication and repair processes (16). The human genome contains five RecQ
genes that encode Werner syndrome protein (WRN), BLM, RECQ1, RECQ4,
and RECQ5. It is worth noting that BLM is mutated in Bloom
syndrome, a rare autosomal recessive disorder that is characterized
by proportional dwarfism, sun-sensitive facial erythema, skin
pigmentation abnormalities, immunodeficiency and infertility
(17). In addition, mutations in BLM
are pathogenic due to an increased predisposition for development
of many tumor types, including lymphoid and epithelial-derived
tumors (18,19). Balci et al (20) reported that colon carcinoma was
diagnosed in ~12% of patients with Bloom syndrome, and Thompson
et al (8) reported that
heterozygous truncating mutations in BLM increase the risk of
breast cancer.
Cell proliferation and cell cycle control are the
major regulatory mechanisms of cell growth. A large body of
evidence has established the mechanisms of how BLM mutations affect
tumor development, including changes to the regulation of many
cellular DNA metabolic processes, including DNA replication, DNA
repair, telomere maintenance and RNA transcription. BLM expression
is high in the S-phase of the cell cycle (21), but BLM depletion has been reported to
suppress cell proliferation in human fibroblasts, and BLM-deficient
cells have defective S-phase progression (22). Other studies also report that BLM
plays a role in recovery from S-phase arrest in response to
hydroxyurea. This may due to interaction of BLM and p53-binding
protein 1 in a Chk1-mediated pathway (23). Our study demonstrated that knockdown
of BLM in PC3 cells suppressed cell proliferation and increased
cell S-phase arrest. Apoptosis has an important role in tumor
progression, Chester et al (24) reported that increased apoptosis
occurred in BLM mutant embryos, Kaneko et al (25) suggested that BLM-deficient cells
have abnormal regulation of p53 protein expression and are highly
susceptible to apoptosis. This previous study also suggested that
p53 mediates cell death by inducing mitochondria-related apoptosis
(25). Consistently, the current
study also revealed that BLM deficiency enhanced cell apoptosis.
Taken together, these results provide strong evidence that BLM may
play an important role in the genomic stability and DNA repair of
PCa cells. Therefore, it is tempting to speculate that BLM could be
a useful therapeutic target for treating PCa cases that are
susceptible to specific DNA-damaging chemotherapeutic agents.
However, in the present study, silencing of BLM
expression had no impact on invasion and migration of PC3 cells.
This effect of BLM during tumorigenesis predominantly involves
changes in DNA replication and repair through increased fragility
of hotspots after replication fork stalling. Without functional
BLM, inefficient resolution of DNA linkages at fragile sites gives
rise to increased numbers of anaphase ultra-fine DNA bridges and
fragile site loci that contribute to chromosomal instability and
tumorigenesis (26). However, as
indicated by the results of our study, loss of BLM function may not
influence the invasion and migration potential of PCa cells.
In summary, our results provide a new perspective
for PCa tumorigenesis and development. These findings highlight a
novel phenomenon in the regulation of PCa progression, which is
useful for understanding the effect of BLM on proliferation and
apoptosis of PCa cells. Thus, BLM could be a promising biomarker
and/or a therapeutic target for PCa treatment. However, further
work is necessary to explore the regulatory mechanisms of BLM in
PCa.
Acknowledgements
This work was supported by Beijing Municipal
Administration of Hospitals Clinical Medicine Development of
Special Funding Support (grant no. ZYLX201408), China Postdoctoral
Science Foundation (grant no. 2015M581131).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Melichar B: PSA, PCA3 and the phi losophy
of prostate cancer management. Clin Chem Lab Med. 51:707–712. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elabbady AA and Khedr MM: Extended 12-core
prostate biopsy increases both the detection of prostate cancer and
the accuracy of Gleason score. Eur Urol. 49:49–53. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Green L: Digital rectal examination
screening for prostate cancer. JAMA. 270:13151993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ghai S and Toi A: Role of transrectal
ultrasonography in prostate cancer. Radiol Clin North Am.
50:1061–1073. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mohaghegh P, Karow JK, Brosh RM Jr, Bohr
VA and Hickson ID: The Bloom's and Werner's syndrome proteins are
DNA structure-specific helicases. Nucleic Acids Res. 29:2843–2849.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Böhm S and Bernstein KA: The role of
post-translational modifications in fine-tuning BLM helicase
function during DNA repair. DNA Repair (Amst). 22:123–132. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thompson ER, Doyle MA, Ryland GL, Rowley
SM, Choong DY, Tothill RW, Thorne H kConFab, Barnes DR, Li J, et
al: Exome sequencing identifies rare deleterious mutations in DNA
repair genes FANCC and BLM as potential breast cancer
susceptibility alleles. PLoS Genet. 8:e10028942012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Voer RM, Hahn MM, Mensenkamp AR,
Hoischen A, Gilissen C, Henkes A, Spruijt L, van Zelst-Stams WA,
Kets CM, Verwiel ET, et al: Deleterious germline BLM mutations and
the risk for early-onset colorectal cancer. Sci Rep. 5:140602015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Davari P, Hebert JL, Albertson DG, Huey B,
Roy R, Mancianti ML, Horvai AE, McDaniel LD, Schultz RA and Epstein
EH Jr: Loss of Blm enhances basal cell carcinoma and
rhabdomyosarcoma tumorigenesis in Ptch1+/−mice. Carcinogenesis.
31:968–973. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnson AM, Zuhlke KA, Plotts C, McDonnell
SK, Middha S, Riska SM, Schaid DJ, Thibodeau SN, Douglas JA and
Cooney KA: Mutational landscape of candidate genes in familial
prostate cancer. Prostate. 74:1371–1378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kämpjärvi K, Kim NH, Keskitalo S, Clark
AD, von Nandelstadh P, Turunen M, Heikkinen T, Park MJ, Mäkinen N,
Kivinummi K, et al: Somatic MED12 mutations in prostate cancer and
uterine leiomyomas promote tumorigenesis through distinct
mechanisms. Prostate. 76:22–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ewing CM, Ray AM, Lange EM, Zuhlke KA,
Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, et al:
Germline mutations in HOXB13 and prostate-cancer risk. N Engl J
Med. 366:141–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Douglas JA, Levin AM, Zuhlke KA, Ray AM,
Johnson GR, Lange EM, Wood DP and Cooney KA: Common variation in
the BRCA1 gene and prostate cancer risk. Cancer Epidemiol
Biomarkers Prev. 16:1510–1516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Agalliu I, Kwon EM, Zadory D, McIntosh L,
Thompson J, Stanford JL and Ostrander EA: Germline mutations in the
BRCA2 gene and susceptibility to hereditary prostate cancer. Clin
Cancer Res. 13:839–843. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kitano K: Structural mechanisms of human
RecQ helicases WRN and BLM. Front Genet. 5:3662014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ellis NA and German J: Molecular genetics
of Bloom's syndrome. Hum Mol Genet. 5:1457–1463. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schuetz JM, MaCarthur AC, Leach S, Lai AS,
Gallagher RP, Connors JM, Gascoyne RD, Spinelli JJ and
Brooks-Wilson AR: Genetic variation in the NBS1, MRE11, RAD50 and
BLM genes and susceptibility to non-Hodgkin lymphoma. BMC Med
Genet. 10:1172009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goss KH, Risinger MA, Kordich JJ, Sanz MM,
Straughen JE, Slovek LE, Capobianco AJ, German J, Boivin GP and
Groden J: Enhanced tumor formation in mice heterozygous for Blm
mutation. Science. 297:2051–2053. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balcı S and Aktas D: Mucinous carcinoma of
the colon in a 16-year-old Turkish boy with Bloom syndrome:
Cytogenetic, histopathologic, TP53 gene and protein expression
studies. Cancer Genet Cytogenet. 111:45–48. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singh DK, Popuri V, Kulikowicz T, Shevelev
I, Ghosh AK, Ramamoorthy M, Rossi ML, Janscak P, Croteau DL and
Bohr VA: The human RecQ helicases BLM and RECQL4 cooperate to
preserve genome stability. Nucleic Acids Res. 40:6632–6648. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park SJ, Lee YJ, Beck BD and Lee SH: A
positive involvement of RecQL4 in UV-induced S-phase arrest. DNA
Cell Biol. 25:696–703. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sengupta S, Robles AI, Linke SP, Sinogeeva
NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J,
et al: Functional interaction between BLM helicase and 53BP1 in a
Chk1-mediated pathway during S-phase arrest. J Cell Biol.
166:801–813. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chester N, Kuo F, Kozak C, O'Hara CD and
Leder P: Stage-specific apoptosis, developmental delay, and
embryonic lethality in mice homozygous for a targeted disruption in
the murine Bloom's syndrome gene. Genes Dev. 12:3382–3393. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kaneko H, Fukao T, Kasahara K, Yamada T
and Kondo N: Augmented cell death with Bloom syndrome helicase
deficiency. Mol Med Rep. 4:607–609. 2011.PubMed/NCBI
|
|
26
|
Chan KL, Palmai-Pallag T, Ying S and
Hickson ID: Replication stress induces sister-chromatid bridging at
fragile site loci in mitosis. Nature Cell Biol. 11:753–760. 2009.
View Article : Google Scholar : PubMed/NCBI
|