Introduction
Peroxisome proliferator-activated receptors (PPARs)
are a class of ligand-dependent sequence-specific nuclear
transcription factors that can be divided into three subtypes:
PPARα, PPARβ (PPARδ) and PPARγ. PPARγ has complex biological
functions and is involved in fat and glucose metabolism, monocyte
activation and the inflammatory response; moreover, recent studies
have found that it can also regulate tumor cell differentiation and
inhibit tumor cell proliferation, features that can play roles in
the development of gastrointestinal tumors (1–5). The human
PPARγ gene is located in the P25 region of chromosome 3 and spans
more than 100 kb. Each species of PPARγ protein is highly
conserved, and the amino acid sequences of PPARγ protein in human
and mice share a 95% homology (3).
The post-transcriptional regulatory effect of PPARγ depends on its
binding to ligands, but the exact range of endogenous ligands
remain unidentified (4). There are
two classes of PPARγ ligands known to exist currently, natural and
synthetic ligands. 15-Deoxyprostaglandin J2 (15-d-PGJ2) and
pioglitazone are the representatives of the two classes,
respectively (5). After ligand
binding in the nuclear membrane, heterodimers are formed with
retinoic acid receptors, and those dimers bind to the peroxisome
proliferator or responsive element in target gene promoter regions
to activate their transcription (6).
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is
a recently discovered tumor suppressor gene whose cognate protein
can act as both lipid and protein phosphatase (7). A large number of studies have shown that
PPARγ ligands can induce cell apoptosis, thereby inhibiting the
growth of cancer cells (1–3). The activation of PPARγ can inhibit the
proliferation of pancreatic, breast and colorectal cancers, by
upregulating the expression of PTEN (8–10). We
hypothesized that a PTEN ligand able to stimulate PPARγ could
inhibit proliferation in hepatocellular carcinoma cells. In this
study, the effect of PPARγ ligands on the growth of hepatocellular
carcinoma cells and the changes in PTEN expression during the
process were observed in vitro, in order to help elucidate a
possible mechanism of PPARγ activation regulating the liver cancer
growth.
Materials and methods
Cell culture
SMMC-7721 hepatocellular carcinoma cells (Shanghai
Cell Institute of Chinese Academy of Sciences) were grown adhering
to the walls of flasks, using RPMI-1640 culture solution (Gibco,
Carlsbad, CA, USA) containing 10% newborn calf serum (Lanzhou
Minhai Biology, Gansu, China), 100 U/ml penicillin and 100 µg/ml
streptomycin in an incubator containing 95% O2 and 5%
CO2 at 37°C.
Determination of cell viability via
MTT assay
MTT was prepared by dissolving 50 mg MTT powder
(Amresco LLC, Solon, OH, USA) in 10 ml phosphate-buffered saline
(PBS) and stirring with a magnet for 30 min, followed with
filtration with a 0.22-µm microporous membrane for sterilization.
The sterile solution was stored at 4°C. SMMC-7721 cells in
logarithmic growth phase were inoculated into 96-well plates at
5×103 cells/well and incubated for 24 h. Four groups
were set up for 15-d-PGJ2 and pioglitazone treatments, a control
group with 0 µmol/l of added drug, one with 10 µmol/l, one with 25
µmol/l, and another with 50 µmol/l. 15-d-PGJ2 and pioglitazone were
both purchased from Cayman Chemical, Inc. (Ann Arbor, MI, USA) and
prepared with dimethyl sulfoxide (DMSO) into a storage
concentration of 50 mmol/l. The final concentration of DMSO in cell
culture medium was not more than 0.1%. The drugs were added to the
wells after cells grew adhering to the wall. A blank control group
was set up for zero setting. The cells were incubated in a
CO2 incubator at 37°C for 24, 48 and 72 h after being
treated with either drug, then 100 µg MTT were added to each well
before and cells were incubated for another 4 h. Then the media
were discarded, and the cells were washed with PBS. Finally, 150 µl
DMSO were added. The cells were placed on a vibrating platform for
10 min, and the A490 values were measured with a Model
680 microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). The cell viability was calculated from the equation: Cell
viability = (A490 value in experimental
group/A490 value in control group) × 100%.
Determination of cell cycle via flow
cytometer
SMMC-7721 cells were seeded in 10-cm culture dishes
and the media was changed after culture for 24 h. Fresh medium
containing 50 µmol/l 15-d-PGJ2 or pioglitazone was added to each
dish; cells were incubated for a further 72 h and then washed using
PBS. Propidium iodide (PI) was used for staining the DNA inside the
cells. The cell cycle analysis, giving the proportion of cells on
each phase, was determined by flow cytometry (Bio-Rad Laboratories,
Inc.).
Detection of levels of PTEN mRNA
expression in cells via reverse transcription-polymerase chain
reaction (RT-PCR).RNA extraction
After SMMC-7721 treatment of cells in culture with
0, 25 and 50 µmol/l of either 15-d-PGJ2 or pioglitazone for 72 h,
the total RNA was extracted by Tri-Blue reagent (Shenneng Bocai,
Shanghai, China) using the one-step method. Briefly, the old
culture solution was discarded, and cells were washed twice with
pre-cooled PBS. Cell pellets in 1.5 ml Eppendorf tubes were mixed
with 1 ml Tri-Blue. Next, 200 µl chloroform was added to each tube,
and each tube mixed well in a vortex for 30 sec. After the
centrifugation, the supernatant was retained and isopycnic
isopropanol was added, followed by centrifugal removal of the
supernatant. Cells were finally washed twice using alcohol,
followed by drying at room temperature and addition of DEPC
water.
RT
Total RNA was used for reverse transcription using
MMLV reverse transcriptase. Briefly, 1 µl total RNA, 1 µl Oligo(dT)
18-Primer (0.5 µg/µl) and 10 µl DEPC-treated H2O were
all mixed together uniformly. A low speed centrifugation step was
carried out before incubation at 70°C for 5 min to denature
secondary structures. Then the samples were placed on ice, and the
reactions were prepared by adding 4 µl 5X reaction buffer and 1 µl
R1 bolock ribonuclease inhibitor; then 1 µl 10 nM dNTP mix. The
tubes were briefly centrifuged, and then incubated at 37°C for 5
min. Next, 1 µl RevertAid MuLV Reverse Transcriptase was added,
mixed and incubated for further 60 min. The reactions were stopped
at 70°C; and the tubes were placed on ice. The equivalent cDNA
obtained was amplified by PCR to detect PTEN mRNA levels with
amplified β-actin as the internal reference product. All steps were
repeated at least 3 times for each experimental group.
PCR
PTEN primers were synthesized by Shenneng Bocai.
PTEN: Upstream primer, 5′-AGACCATAACCCACCACAGC-3′ and downstream
primer, 5′-ACCAGTTCGTCCCTTTCCAG-3′; amplified fragment length, 123
bp. β-actin: Upstream primer, 5′-AGCGGGAAATCGTGCGTG-3′ and
downstream primer, 5′-CAGGGTACATGGTGGTGCC-3′; amplified fragment
length, 287 bp. According to the instructions, PTEN primers were
dissolved into 10 µmol/l working solution using DEPC-H2O
to prepare 50 µl PCR reactions. The reactions were placed in a
thermocycler instrument (Bio-Rad Laboratories, Inc.). The protocol
for PTEN amplification was set up with an initial denaturation at
94°C for 4 min, followed by 35 cycles of denaturation at 94°C for
20 sec, annealing at 60°C for 20 sec, and extension at 72°C for 40
sec; and a final extension at 72°C for 10 min. For β-actin
amplification the protocol included an initial denaturation at 94°C
for 3 min, followed by 35 cycles of denaturation at 94°C for 30
sec, annealing at 60°C for 45 sec, and extension at 72°C for 1 min,
with a final extension at 72°C for 10 min. The PCR products were
stored at 4°C. Finally, agarose gel electrophoresis was performed
to analyze the results.
Detection of the expression of PTEN
and pAkt protein via western blot analysis
SMMC-7721 cells were inoculated into 6-well plates
and cultured for 24 h under normal conditions. Once the cells
adhered to the walls, the medium was changed. The cells were next
incubated with 0, 10, 25 and 50 µmol/l of either 15-d-PGJ2 or
pioglitazone for 72 h. After washing cells twice with PBS, 150 µl
cell lysis buffer [20 mmol/l Tris (pH 7.5), 4 mmol/l EDTA (pH 8.0),
2% SDS] were added to each well to crack open the cell membranes
via a cell sonicator (JY96-II; Ningbo Xinzhi Biologic Science and
Technology Co., Ltd., Ningbo, China). After a 10,500 × g
centrifugation step for 10 min, the supernatants containing the
soluble protein fractions were transferred into pre-cooled EP
tubes. Protein concentrations were determined by the BCA method.
Next, 20 µg total protein were added to loading buffer at a ratio
of 4:1, and boiled for 5 min; after the protein denaturation, the
samples were loaded onto a gel for electrophoresis and then
transferred onto a PVDF membrane (Roche Diagnostics, Basel,
Switzerland). Standard western blot protocols were followed
(protein blotting system; Bio-Rad Laboratories, Inc.). Mouse
monoclonal PTEN antibody (dilution, 1:1,000, catalog no. ab79156)
or mouse monoclonal pAkt (dilution, 1:1,000, catalog no. ab105731),
purchased from Abcam (Cambridge, MA, USA) were incubated with the
membranes overnight at 4°C. TBP was used for membrane washing.
Then, HRP-labeled goat anti-mouse secondary antibody (dilution,
1:5,000; catalog no. ab6785; Abcam, Cambridge, MA, USA) was added
and incubated at room temperature for 1 h. TBST was used for
washing the membrane three times for 5 min. Finally, the ECL
chemiluminescence method was used for visualizing the results.
Additionally, an α-tubulin antibody (dilution, 1:5,000; catalog no.
5335) was used as the internal reference. Mouse anti-human PTEN
monoclonal antibody (dilution, 1:1,000; catalog no. 9556) and mouse
anti-human pAkt monoclonal antibody (dilution, 1:1,000; catalog no.
12694) were purchased from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Peroxidase (HRP)-labeled goat anti-mouse
secondary antibody (dilution, 1:5,000; catalog no. 4410) was
purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA).
Statistical analysis
The SPSS 12.0 statistical software (IBM, Armonk, NY,
USA) was used for statistical analyses. All variables were
presented as mean ± standard deviation (mean ± SD); analysis of
variance (ANOVA) and t-test were used for inter-group differences.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of 15-d-PGJ2 and pioglitazone
on proliferation of SMMC-7721 cells
SMMC-7721 cells were treated with different
concentrations of 15-d-PGJ2 or pioglitazone. It was found that both
of the PPARγ ligands had a significant inhibitory effect on the
proliferation of SMMC-7721 cells. The inhibitory effects were more
pronounced with increasing drug concentrations (p<0.05).
Table I obtain details of time- and
dose-dependence on cellular proliferation.
 | Table I.Effects of 15-d-PGJ2 and pioglitazone
on the proliferation of SMMC-7721 cells (mean ± SD). |
Table I.
Effects of 15-d-PGJ2 and pioglitazone
on the proliferation of SMMC-7721 cells (mean ± SD).
| Groups | Drug concentration
(µmol/l) | 24 h | 48 h | 72 h |
|---|
| 15-d-PGJ2 | 10 | 87.61±1.73 | 81.98±1.85 | 77.86±2.21 |
|
| 25 | 65.70±1.81 | 61.83±1.72 | 54.89±1.90 |
|
| 50 | 48.52±1.82 | 47.93±1.94 | 30.79±2.51 |
| Pioglitazone | 10 | 93.56±2.25 | 84.73±2.07 | 82.04±2.19 |
|
| 25 | 70.63±2.22 | 63.67±1.55 | 58.26±2.34 |
|
| 50 | 54.43±1.84 | 40.96±2.29 | 33.79±2.93 |
Cell cycle effects of 15-d-PGJ2 and
pioglitazone on SMMC-7722 cell cycle
15-d-PGJ2 and pioglitazone can inhibit the
proliferation of SMMC-7721 cells, therefore we used PI and flow
cytometry to further investigate the ligand effects on the cell
cycle. The proportion of G0/G1 phase SMMC-7721 cells after
treatment with 50 µmol/l 15-d-PGJ2 or pioglitazone for 72 h was
significantly higher than the same proportion in cells without
treatment (p<0.05). However, the proportion of S phase cells was
significantly decreased (p<0.05), and the proportion of G2/M
phase cells was not increased significantly (p<0.05) in the
high-dose treatment groups. This apparent inhibition at the level
of replication can be seen in Table
II.
| Table II.Effects of 15-d-PGJ2 and pioglitazone
on SMMC-7722 cell cycle (mean ± SD). |
Table II.
Effects of 15-d-PGJ2 and pioglitazone
on SMMC-7722 cell cycle (mean ± SD).
|
| Cell cycle
distribution (%) |
|---|
|
|
|
|---|
| Group | G0/G1 phase | S phase | G2/M phase |
|---|
| Control | 63.45±1.49 | 32.19±1.83 | 4.36±0.64 |
| 15-d-PGJ2 (50
µmol/l) | 76.87±1.09 | 18.28±1.46 | 4.85±0.58 |
| Pioglitazone (50
µmol/l) | 78.31±1.28 | 16.90±1.08 | 4.79±0.73 |
Effects of 15-d-PGJ2 and pioglitazone
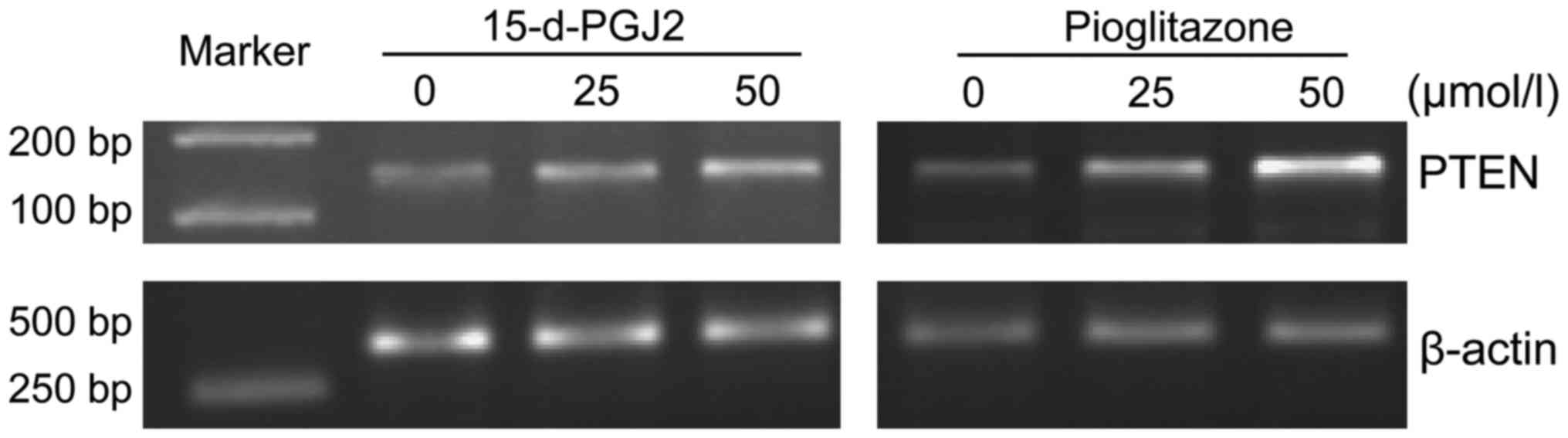
on the expression levels of PTEN mRNA in SMMC-7721 cells
After treating SMMC-7721 cells with 15-d-PGJ2 or
pioglitazone for 72 h, it was found that the expression levels of
PTEN mRNA were significantly increased in a drug concentration
manner for both ligand treatments (Fig.
1).
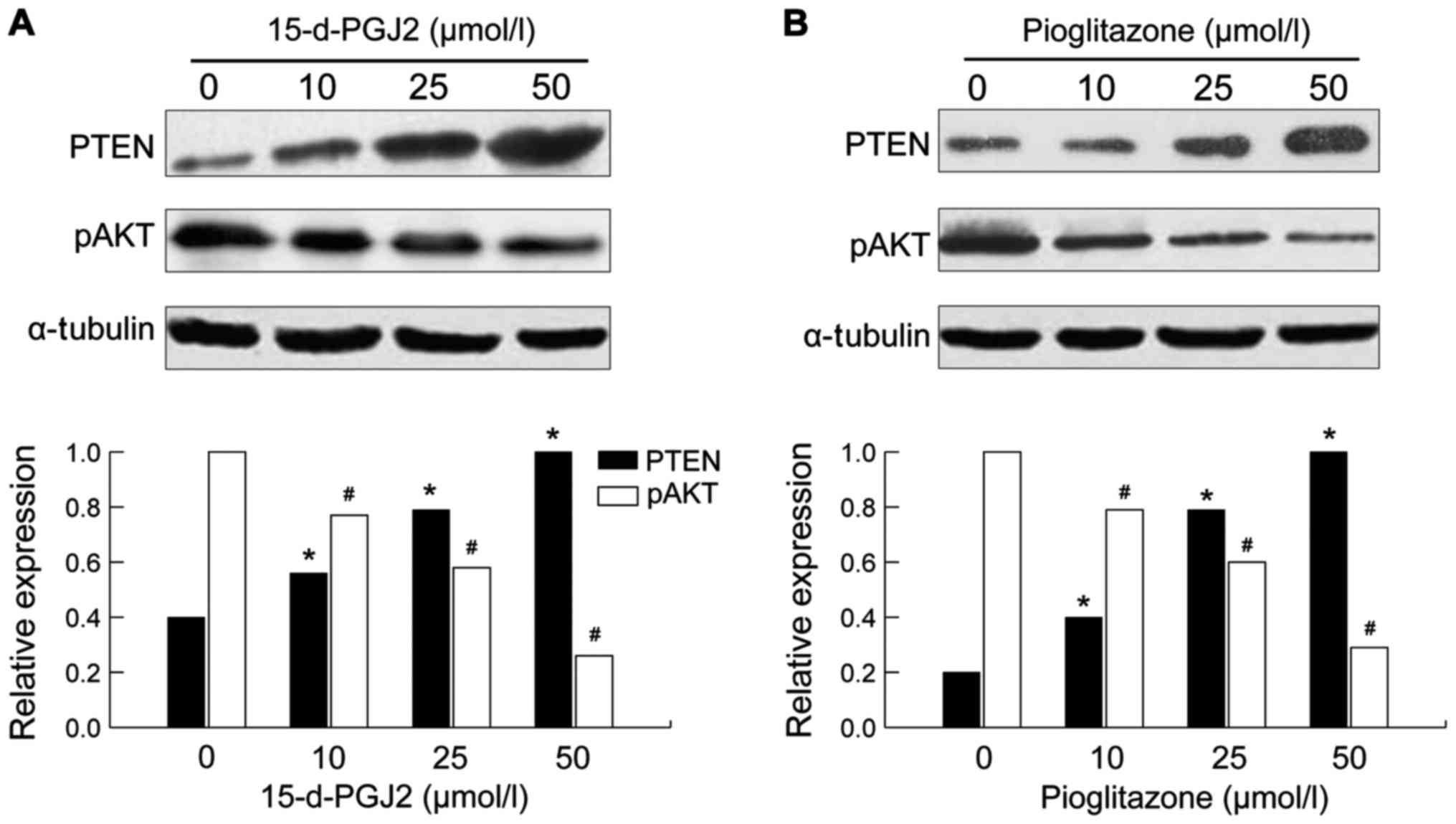
Effects of 15-d-PGJ2 and pioglitazone
on the expression levels of PTEN and pAkt protein in SMMC-7721
cells
After either 15-d-pGJ2 or pioglitazone treatment for
72 h, the expression of PTEN protein in SMMC-7721 cells was
increased in a drug concentration manner (p<0.05). At the same
time, the expression of the protein of the downstream pAkt gene was
decreased and the difference was more significant with higher
amounts of drug concentration used (p<0.05) (Fig. 2).
Discussion
PPARγ is a class of ligand-dependent
sequence-specific nuclear transcription factors, and 15-d-PGJ2 and
pioglitazone are its natural and synthetic ligands, respectively
(11). The effects of 15-d-PGJ2 and
pioglitazone on the growth of SMMC-7721 hepatocellular carcinoma
cells under different concentrations and different treatment
lengths were observed by MTT assay. The results showed that both
drugs had a significant time- and dose-dependent inhibitory effect
on the cell growth. Furthermore, an analysis of the relation
between the inhibitory effect and the cell cycle performed by flow
cytometry, found that 15-d-PGJZ and pioglitazone can both
significantly increase the proportion of G0/Gl phase cells and
decrease the proportion of S phase cells. This suggests that the
inhibitory effect is at least partially related to the inhibition
of cell DNA synthesis and mitosis, which blocks the transition of
cells from G1 to S phase.
The tumor suppressor function of PTEN has been
attributed to its lipid phosphatase activity (12). PTEN can catalyze the dephosphorylation
of phosphorylated phosphatidylphthalide 4,5-diphosphoinositol
(PIP2) and phosphatidic acid 3,4,5-triphosphoinositol (PIP3)
(13). Previous studies have shown
that PTEN can specifically inhibit the activation of
phosphatidylinositol-3-kinase-dependent protein kinase B (PKB or
Akt), and PI3K catalyzes the phosphorylation of phosphoinositide on
cell membranes to produce PIP2 and PIP3, the latter of which
further activates the downstream Akt (14). Therefore, PTEN can antagonize the
activity of PIP3 and PIP2, and reduce their concentration in cells,
thereby inhibiting the phosphorylation of Akt (15,16).
Inactivation of PTEN may promote cell proliferation and tumor
angiogenesis, by inhibiting cell apoptosis (17–19).
It was found in this study that, after treatment
with 15-d-pGJ2 or pioglitazone, SMMC-7721 cells increased the
expression levels of their PTEN mRNA in a ligand concentration
manner. Furthermore, western blot analysis found that the
expression of the PTEN protein was also increased; and
correspondingly, the expression of the downstream pAkt gene product
(belonging to the PI3K pathway) was decreased more significantly
with increasing ligand concentrations. Finally, the results of the
MTT assay showed that the proliferation of SMMC-7721 cells was
inhibited by the PPARγ ligands in a dose-dependent manner. Taken
together, the above results suggest that the inhibitory effect of
the PPARγ ligands, 15-d-pGJ2 or pioglitazone, on the proliferation
of the hepatocellular carcinoma cells may be achieved by
upregulating the expression of PTEN (a cancer suppressor gene),
thus affecting the PI3K pathway and inhibiting the phosphorylation
of Akt.
At the same time, there is evidence that PTEN can
also act as a cell cycle regulator by stimulating P27 expression
(8). P27 is an important cyclin
kinase inhibitor that can inhibit the activity of G1 phase
cyclin-dependent kinase (CDK) and induce the G1 phase cell arrest
(9). The results of flow cyto-metry
in our study showed that the proportion of G0/G1 phase cells was
increased and that of S phase cells was decreased significantly
after addition of PPARγ ligands. This suggests that PPARγ ligands
inhibit the proliferation of SMMC-7721 by blocking the transition
to S phase, and this could be due to upregulation of the expression
of PTEN, leading to an increase in the p27 protein level, which
would induce a G1 phase cell arrest.
In this study, two ligands of PPARγ, 15-d-PGJ2 and
pioglitazone, were used to confirm their inhibitory effect on the
proliferation of SMMC-7721 hepatocellular carcinoma cells, and this
inhibitory effect was related to the expression of PTEN, a tumor
suppressor gene. In conclusion, we believe that PPARγ ligands may
act as tumor suppressors, which categorizes them as potential
clinical treatments against liver cancer.
References
|
1
|
Li M, Lee TW, Mok TS, Warner TD, Yim AP
and Chen GG: Activation of peroxisome proliferator-activated
receptor-gamma by troglitazone (TGZ) inhibits human lung cell
growth. J Cell Biochem. 96:760–774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valentiner U, Carlsson M, Erttmann R,
Hildebrandt H and Schumacher U: Ligands for the peroxisome
proliferator-activated receptor-gamma have inhibitory effects on
growth of human neuroblastoma cells in vitro. Toxicology.
213:157–168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fenner MH and Elstner E: Peroxisome
proliferator-activated receptor-gamma ligands for the treatment of
breast cancer. Expert Opin Investig Drugs. 14:557–568. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu J, Imamura K, Nomura S, Mafune K,
Nakajima A, Kadowaki T, Kubota N, Terauchi Y, Ishii G, Ochiai A, et
al: Chemopreventive effect of peroxisome proliferator-activated
receptor gamma on gastric carcinogenesis in mice. Cancer Res.
65:4769–4774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Toyoda M, Takagi H, Horiguchi N, Kakizaki
S, Sato K, Takayama H and Mori M: A ligand for peroxisome
proliferator activated receptor gamma inhibits cell growth and
induces apoptosis in human liver cancer cells. Gut. 50:563–567.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
IJpenberg A, Jeannin E, Wahli W and
Desvergne B: Polarity and specific sequence requirements of
peroxisome proliferator-activated receptor (PPAR)/retinoid X
receptor heterodimer binding to DNA. A functional analysis of the
malic enzyme gene PPAR response element. J Biol Chem.
272:20108–20117. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Yen C, Liaw D, Podsypanina K, Bose
S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al:
PTEN, a putative protein tyrosine phosphatase gene mutated in human
brain, breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen WC, Lin MS and Bai X: Induction of
apoptosis in colorectal cancer cells by peroxisome
proliferators-activated receptor gamma activation up-regulating
PTEN and inhibiting PI3K activity. Chin Med J (Engl).
118:1477–1481. 2005.PubMed/NCBI
|
|
9
|
Farrow B and Evers BM: Activation of
PPARgamma increases PTEN expression in pancreatic cancer cells.
Biochem Biophys Res Commun. 301:50–53. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Patel L, Pass I, Coxon P, Downes CP, Smith
SA and Macphee CH: Tumor suppressor anti-inflammatory actions of
PPAR gamma agonists are mediated up-regulation of PTEN. Curr Biol.
11:764–768. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okano H, Shiraki K, Inoue H, Yamanaka T,
Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K, et
al: Peroxisome proliferator-activated receptor gamma augments tumor
necrosis factor family-induced apoptosis in hepatocellular
carcinoma. Anticancer Drugs. 13:59–65. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Motomura W, Takahashi N, Nagamine M,
Sawamukai M, Tanno S, Kohgo Y and Okumura T: Growth arrest by
troglitazone is mediated by p27Kip1 accumulation, which
results from dual inhibition of proteasome activity and Skp2
expression in human hepatocellular carcinoma cells. Int J Cancer.
108:41–46. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma H, Sprecher HW and Kolattukudy PE:
Estrogen-induced production of a peroxisome proliferator-activated
receptor (PPAR) ligand in a PPARgamma-expressing tissue. J Biol
Chem. 273:30131–30138. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han B, Dong Z, Liu Y, Chen Q, Hashimoto K
and Zhang JT: Regulation of constitutive expression of mouse PTEN
by the 5′-untranslated region. Oncogene. 22:5325–5337. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cantley LC and Neel BG: New insights into
tumor suppression: PTEN suppresses tumor formation by restraining
the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA.
96:4240–4245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leslie NR and Downes CP: PTEN: The down
side of PI 3-kinase signalling. Cell Signal. 14:285–295. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stambolic V, Suzuki A, de la Pompa JL,
Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM,
Siderovski DP and Mak TW: Negative regulation of PKB/Akt-dependent
cell survival by the tumor suppressor PTEN. Cell. 95:29–39. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: Implications for tumor angiogenesis and
therapeutics. Cancer Res. 60:1541–1545. 2000.PubMed/NCBI
|
|
19
|
Lu Y, Lin YZ, LaPushin R, Cuevas B, Fang
X, Yu SX, Davies MA, Khan H, Furui T, Mao M, et al: The
PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and
induces apoptosis and anoikis in breast cancer cells. Oncogene.
18:7034–7045. 1999. View Article : Google Scholar : PubMed/NCBI
|