Introduction
Maxillary cancer is commonly treated with cisplatin
(CDDP) chemotherapy combined with radiotherapy. Although the tumor
regresses during treatment, in about half of the cases the tumor
histopathologically remains, and ultimately the CDDP-resistant
tumor is surgically resected. CDDP plays a central role in
treatment of maxillary cancer; however, the existence of a
CDDP-resistance mechanism has now been recognized (1). To examine whether known
chemotherapy-resistant genes are involved in CDDP resistance of
maxillary carcinoma, we previously analyzed gene expression in
maxillary squamous cell carcinoma biopsies prior to treatment. The
results showed that expression of a group of genes (multidrug
resistance protein 1; multidrug resistance associated protein 1;
Cu++ transporting, beta polypeptide; xeroderma
pigmentosum; complementation group A; excision repair
cross-complementing rodent repair deficiency, complementation group
1; B-cell CLL/lymphoma 2) associated with treatment resistance was
decreased in these tumors, and that only tumor protein p53
(TP53) mutation was linked to treatment resistance (2). TP53 mutations are associated with
treatment resistance not only in head and neck cancers, but also in
breast cancer, lung cancer, hepatic cancer, and chronic lymphocytic
leukemia (3–8). On the other hand, it has been reported
that there is no such association in small cell lung cancer or
epithelial ovarian cancer (9,10). Thus, the relationship between
TP53 mutation and treatment resistance is not necessarily
clear.
Recently, whole exome sequencing has shown major
driver genes in head and neck squamous cell carcinoma (HNSCC). In
addition to the previously identified TP53, cyclin dependent
kinase inhibitor 2A, Phosphatidylinositol-4,5-bisphosphate 3-kinase
catalytic subunit alpha, and histidyl-tRNA synthetase genes,
mutations in major genes that regulate squamous differentiation,
including notch1, interferon regulatory factor 6, and tumor protein
p63 (TP63), have been newly identified as drivers (11,12). In
particular, TP53 mutations occur at a high frequency in
HNSCC, but many non-TP53 mutated tumors are human
papillomavirus-positive (13). Both
types of tumors may involve a common mechanism mediated by
TP53 dysfunction, but the biological differences between
these cancers are unclear. As a first step in understanding the
biological differences observed between tumors with and without
TP53 mutation, this study aimed to clarify differences at
the gene expression level between maxillary cancers with and
without TP53 mutation.
Materials and methods
Samples
Specimens were used from 14 patients with maxillary
cancer (Table I). Tumor staging and
differentiation was in accordance with the Union for International
Cancer Control TNM classification (14). Maxillary cancer biopsy specimens
before treatment were used in the study. This study was approved by
the Ethics Committee at Nihon University School of Medicine and
conforms to the Declaration of Helsinki (2013). Informed consent
was obtained from all patients.
 | Table I.Clinicopathological features and
TP53 mutation of 14 cases of maxillary squamous cell
carcinoma. |
Table I.
Clinicopathological features and
TP53 mutation of 14 cases of maxillary squamous cell
carcinoma.
| Case | Age | Sex | Stageb | Gradeb | TP53
mutationc |
|---|
| 1T | 46 | F | II | 2 |
|
| M6a | 55 | M | III | 1 |
|
| M2a | 55 | M | III | 2 |
|
| M11a | 58 | M | III | 2 |
|
| 7Ta | 60 | M | III | 2 |
|
| M5a | 64 | M | III | 3 |
|
| M9a | 73 | M | III | 1 | P190T |
| M8 | 64 | M | III | 1 | F285K |
| M1a | 63 | M | III | 2 | R280S |
| M10 | 64 | M | III | 2 | c.782+1G>A |
| M13a | 51 | M | III | 3 | R156AfsX14 |
| M12a | 80 | M | IV A | 2 | c.782+1G>A |
| M14a | 67 | M | IV A | 2 | Y220C |
| 8T | 65 | M | IV A | 2 | H193R |
TP53 mutation analysis
Total RNA was extracted as described previously, and
was used as a template for cDNA synthesis (2). The synthesized cDNA was used to perform
polymerase chain reaction (PCR) analysis of a high mutation region
(aa115-aa342) of the TP53 gene as described previously. The
sequence of the PCR products was analyzed by Sanger sequencing
(2).
Comprehensive gene expression
analysis
Comprehensive gene expression analysis was performed
in 5 patients each with and without TP53 mutations (Table I). Biotin-labeled cRNA was synthesized
from total RNA according to the Affymetrix manual. Hybridization
was performed using a GeneChip Human Genome U133 Plus 2.0 Array
(Affymetrix, Santa Clara, CA). A GeneChip Fluidics Station 400
(Affymetrix, Inc., Santa Clara, CA, USA) and Scanner 3000
(Affymetrix, Inc.) were used for detection. Analysis was performed
using GeneChip Operating Software (Affymetrix, Inc.) and GeneSpring
v7 (Silicon Genetics, Redwood City, CA, USA); the output data were
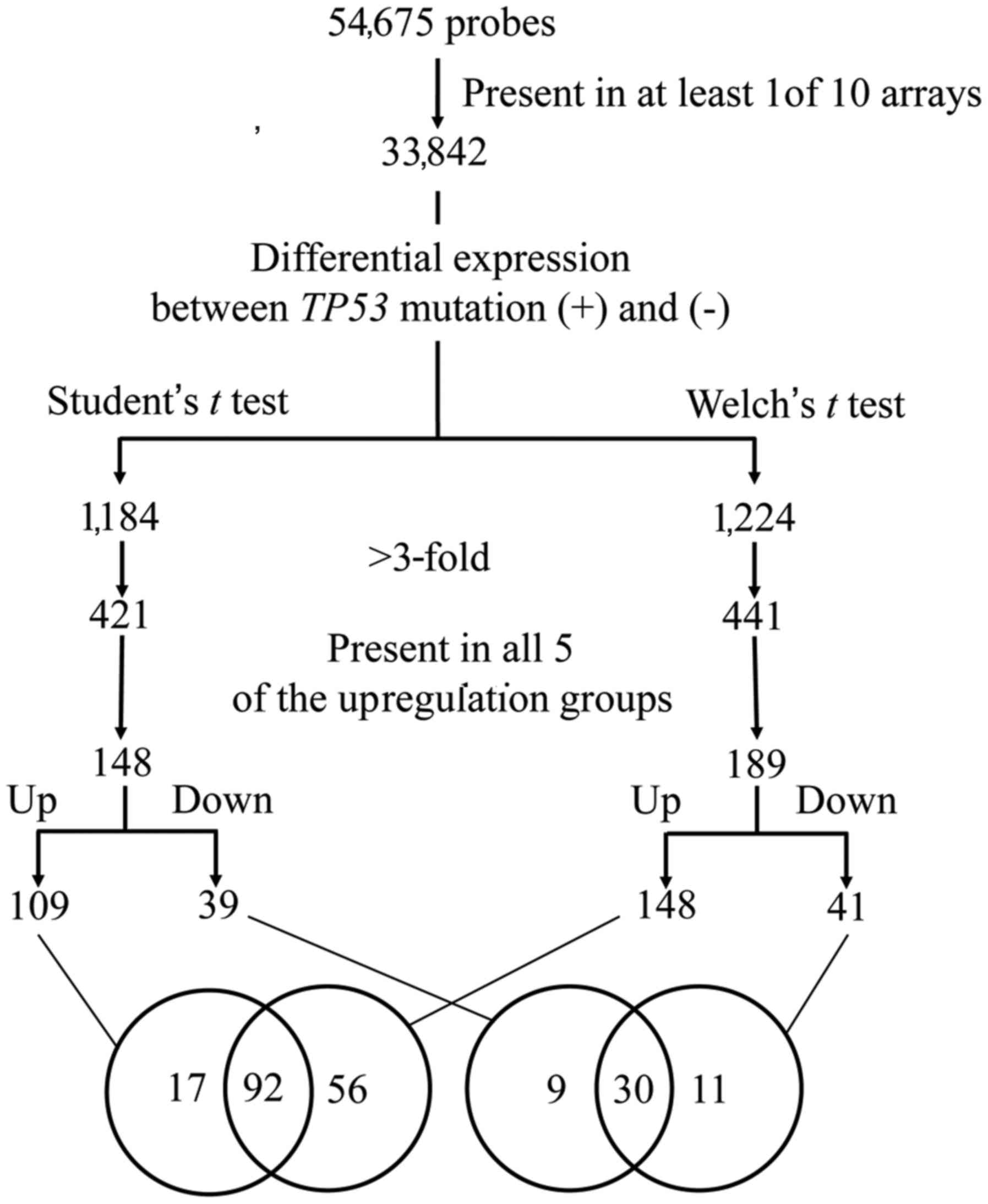
normalized per chip and per gene. Genes with >3-fold
differential expression between TP53 mutation (+) and (−), that
were commonly identified using two parametric tests (Student's
t-test and Welch's t-test), were used as gene candidates with
differential expression (Fig. 1).
Quantification of mRNA
A quantitative PCR (qPCR) assay was carried out
using the SYBR-Green Real-time PCR Master Mix (Life Technologies,
Frederick, MD, USA) as described previously (2). The gene expression level was normalized
against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA.
Table II lists the primer sequences
used.
| Table II.Primer sequences used for
quantitative polymerase chain reaction analysis in this study. |
Table II.
Primer sequences used for
quantitative polymerase chain reaction analysis in this study.
|
| Primer
sequence |
|---|
|
|
|
|---|
| Gene | Forward
(5′-3′) | Reverse
(5′-3′) |
|---|
| CSTA |
ACGGAAAATTGGAAGCTGTG |
TTTGTCCGGGAAGACTTTTG |
| SFN |
CAGGCTACTTCTCCCCTCCT |
TCAATCTCGGTCTTGCACTG |
| DSC3 |
ATTGCAGTCTTGATTCTGCC |
ACGTTTGTAGGGGAGCACAC |
| GRHL1 |
GCTAGTATCAGTCAGATGCA |
GAAGGCTCTGATGCGTGATA |
| EPPK1 |
TCAGCTCAGCCATAATCACG |
ACATGGCCTGGTAGATGCTC |
| PROM2 |
CTGATCCCCAGCATCATCTT |
ACCAGATCACTCCCACAAGG |
| ANXA8 |
AGCTGGTCACAGAGTCTCCT |
GCTGCTGAAGGATGTGTGTT |
| CLCA2 |
TACCTCTTGCTATTTTGTTA |
GCTGCTTTGATGGGAGTAGA |
| SAMD9 |
GACATTATGGGCCTGGAAGT |
TGTGAATTTCCCCTTTCTGG |
| PRRG4 |
AATATTTGTCAGTGCTTAAC |
AAATGACCACACAGGCAGAA |
| DSP |
TAGGAGAAAATTACCCTCCC |
GAAAAGATTGCCGCTGTCAT |
| F2RL1 |
GCCACTTAGAATAGCATTTG |
GATGTGGTCCAAACCCTCTG |
| S100A2 |
ATGAGTGGGAATGGCAAGAG |
GCAGAGACAGACCCAGGAAG |
| MAST4 |
TCTCCTCTCTGTGGGAAGGA |
GCCATCTTTGTGGTTCGTTT |
| JUP |
AACCAGCTGTCGAAGAAGGA |
GTGTCCAGGTCGCTGGTATT |
| SCD |
TGTTCGTTGCCACTTTCTTG |
TAGTTGTGGAAGCCCTCACC |
| TP63 |
GAGGTTGGGCTGTTCATCAT |
GAGGAGAATTCGTGGAGCTG |
| KRT6B |
TGCGAATGTCCTTTTTAGTT |
TAATGGGCAGGATGGTTAGC |
| SFRP4 |
GACTTCCGACTTCCTTACAG |
TCTGTACCAAAGGGCAAACC |
| HMCN1 |
ATCAGCTGAACCACTTATGA |
AAACCAAACCTGTCCCACTG |
| MEST |
GAATCGATCTGGTCGGCTTA |
CATCAGTCGTGTGAGGATGG |
| GAPDH |
GGTCGGAGTCAACGGATTTG |
GGATCTCGCTCCTGGAAGAT |
Immunohistochemistry (IHC)
Biopsy tissue was fixed in formalin and embedded in
paraffin, and then 4-µm thin-sections were prepared. Four cases
with p53 wild-type (1T, M11, 7T, M5) and 4 cases with a p53
mutation (M9, M8, M13, 8T) were used. After deparaffinization and
removal of endogenous peroxidase, antigen activation was performed
using citrate buffer (pH 6) in a 600 W microwave oven for 5 min
[cystain A (CSTA), stratifin (SFN) or an autoclave for 5 min
desmocollin 3 (DSC3)].
The primary antibodies used were rabbit polyclonal
anti-human CSTA IgG (0.1 µg/ml, HPA001031; Atlas Antibodies AB,
Stockholm, Sweden), mouse monoclonal anti-human 14-3-3 sigma IgG1
clone 1.N.6 (1 µg/ml, GTX14123; GeneTex, Irvine, CA, USA), mouse
monoclonal anti-human desmocollin 3 IgG1 clone Dsc3-U114 (0.05
µg/ml, 61093; Progen Biotech, Heidelberg, Germany), and mouse
monoclonal anti-human p53 IgG2b clone DO-7 (0.69 µg/ml, M7001;
Dako, Glostrup, Denmark). The reactions were carried out overnight
at 4°C. A Histofine Simple Stain MAX-PO (R) kit or MAX-PO (M) kit
(Nichirei, Tokyo, Japan) was used for secondary antibodies. The
sections were colored with diaminobenzidine and nuclei were stained
with hematoxylin.
Statistical analysis
Age, stage, grade, and mRNA expression levels were
compared between the two groups with and without TP53 mutation
using the Mann-Whitney U test. P<0.05 was considered to indicate
a statistically significant difference.
Results
Clinicopathological features of
maxillary carcinoma with and without TP53 mutation
Eight of the 14 patients had a TP53 mutation
(Table I). These mutations included 5
point mutations, 2 splicing abnormalities, and one frameshift
mutation. Table III compares the
clinicopathological features of patients with and without
TP53 mutations. Tumor stage and grade were not significantly
related to TP53 mutation status. However, there was a
correlation between TP53 mutations and age; thus,
TP53 mutation-positive patients were significantly older
than those without TP53 mutation (P=0.0273). TP53
mRNA expression levels did not significantly differ between the two
groups (2).
| Table III.Comparison of the clinicopathological
features of maxillary squamous cell carcinoma with and without
TP53 mutation. |
Table III.
Comparison of the clinicopathological
features of maxillary squamous cell carcinoma with and without
TP53 mutation.
|
| TP53
mutation |
|
|---|
|
|
|
|
|---|
| Feature | (−) | (+) |
P-valueb |
|---|
| Agea | 56.3±6.1 | 65.9±8.4 | 0.0273 |
| Stage |
|
| 0.0611 |
| II | 1 | 0 |
|
|
III | 5 | 5 |
|
|
IVA | 0 | 3 |
|
| Grade |
|
| 0.7047 |
| 1 | 1 | 2 |
|
| 2 | 4 | 4 |
|
| 3 | 1 | 1 |
|
Differential gene expression in
maxillary carcinoma with and without TP53 mutation
Comprehensive gene expression analysis was performed
with 10 microarrays using mRNA from the maxillary cancer specimens.
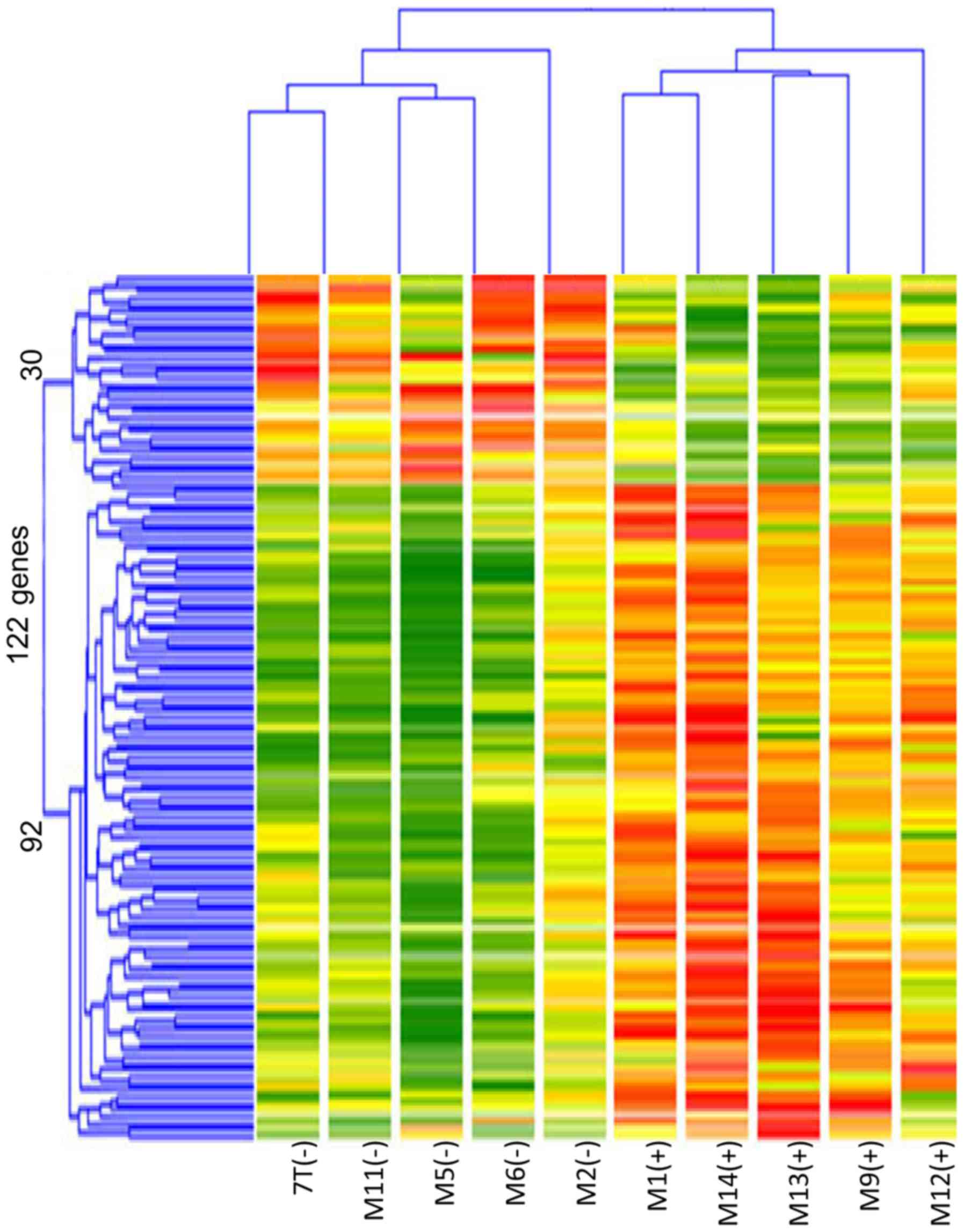
The results showed 92 genes in TP53 mutated tumors with
≥3-fold increased expression and 30 genes whose expression was
decreased to approximately 1/3 compared to non-TP53 mutated
tumors (Fig. 1). Cluster
classification was performed in 10 cases based on the expression
pattern of these 122 genes. As shown in Fig. 2, the 10 cases could be accurately
classified into two clusters based on TP53 mutation status.
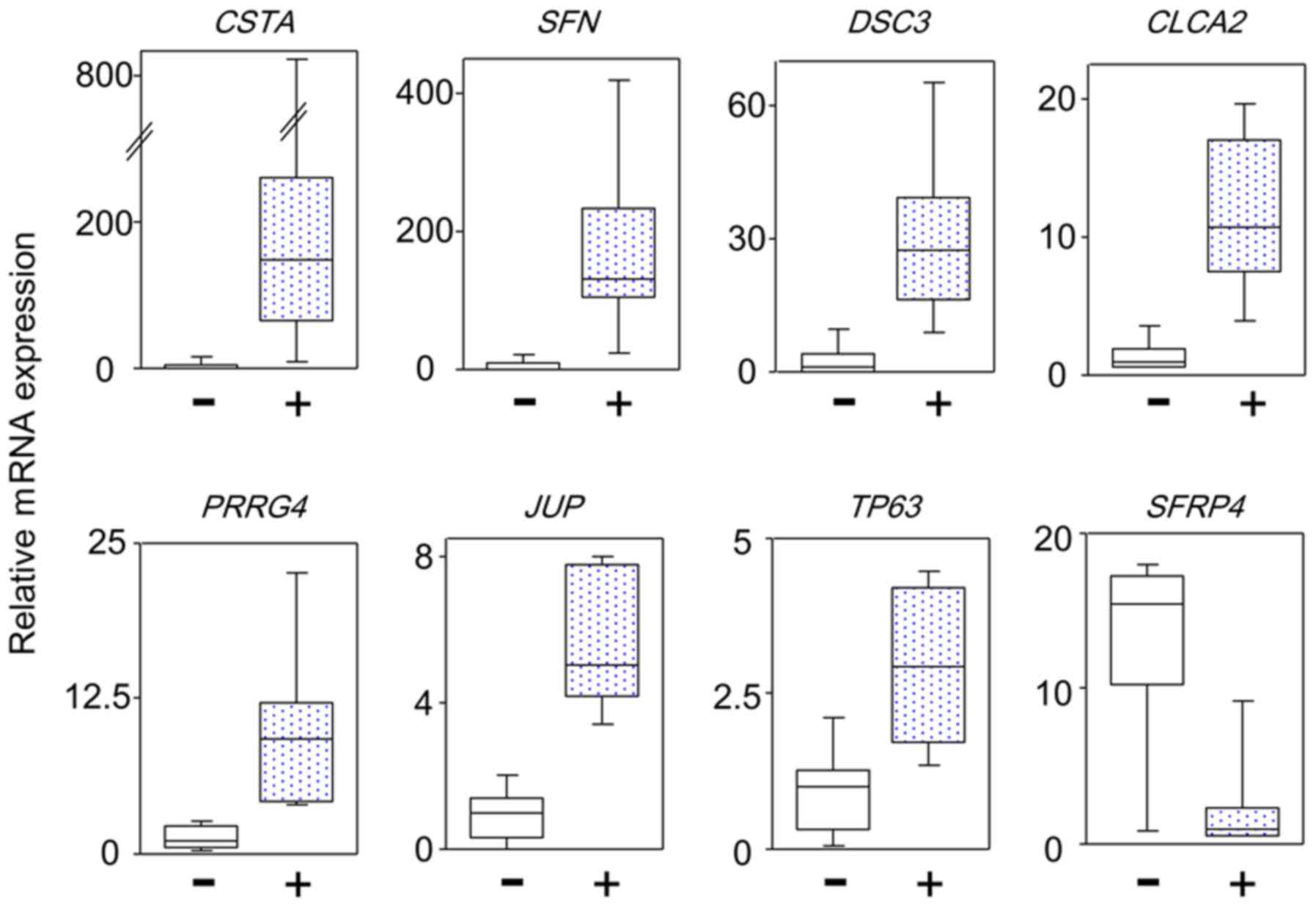
To confirm these gene expression levels, qPCR analysis of the mRNA
expression of 42 genes with ≥4-fold differential expression between
TP53 and non-TP53 mutated tumors based on the
microarray results was performed using patient maxillary squamous
cell carcinoma samples. Twenty-one genes with significant
differences in expression between the two groups were identified.
There were no significant differences in the expression level of
the remaining 21 genes due to the different normalization methods
used. The differential genes commonly identified by different
normalization methods are considered to be reliable. Fig. 3 shows representative results of the
differential gene expression. Table
IV lists 18 genes with high expression and 3 genes with low
expression in TP53 mutated tumors compared to
non-TP53 mutated tumors. The 18 genes included 8 cell
adhesion genes (DSC3; grainyhead like transcription factor
1; epiplakin 1; prominin 2; annexin A8; desmoplakin (DSP);
junction plakoglobin (JUP); and keratin 6B) and 4 cell
growth inhibition genes (SFN, chloride channel accessory 2,
sterile alpha motif domain containing 9, and TP63). Thus, in
TP53 mutated tumors, the expression of genes that inhibited
proliferation, invasion, and metastases was unexpectedly increased,
compared to wild-type tumors.
| Table IV.Twenty-one validated genes with
differential expression in TP53 mutated versus non-mutated
tumors. |
Table IV.
Twenty-one validated genes with
differential expression in TP53 mutated versus non-mutated
tumors.
| A, Genes
upregulated in TP53 mutated cancer |
|---|
|
|---|
| Gene symbol | Gene title | FCa |
P-valueb | Function |
|---|
| CSTA | Cystatin A | 148.8 | 0.0174 | Cysteine protease
inhibitor |
| SFN | Stratifin | 129.9 | 0.0106 | Cell cycle arrest
Tumor progression |
| DSC3 | Desmocollin 3 | 27.3 | 0.0106 | Desmosome |
| GRHL1 | Grainyhead-like
1 | 20.1 | 0.0249 | Transcription
factor Cell adhesion |
| EPPK1 | Epiplakin 1 | 19.9 | 0.0096 | Cell matrix
adhesion |
| PROM2 | Prominin 2 | 17.2 | 0.0176 | Membrane
glycoprotein |
| ANXA8 | Annexin A8 | 11.7 | 0.0106 | Adherens
junction |
| CLCA2 | Chloride channel
accessory 2 | 10.7 | 0.0106 | p53-inducible
senescence |
| SAMD9 | Sterile alpha motif
domain containing 9 | 9.5 | 0.0176 | Regulation of cell
proliferation |
| PRRG4 | Proline rich Gla
4 | 9.3 | 0.0062 | Unknown |
| DSP | Desmoplakin | 9.3 | 0.0285 | Desmosome |
| F2RL1 | Coagulation factor
II receptor-like 1 | 7.5 | 0.0062 |
Pro-inflammation |
| S100A2 | S100 calcium
binding protein A2 | 7.2 | 0.0176 | Tumor suppressor or
promoter |
| MAST4 | Microtubule
associated serine/threonine kinase family member 4 | 7.1 | 0.0456 | Unknown |
| JUP | Junction
plakoglobin | 5.1 | 0.0007 | Desmosome |
| SCD | Stearoyl-CoA
desaturase | 3.2 | 0.0446 | Fatty acid
biosynthesis |
| TP63 | Tumor protein
p63 | 2.9 | 0.0200 | ΔNp63: Cell growth
Tap63: Apoptosis |
| KRT6B | Keratin 6B | 2.7 | 0.0285 | Intermediate
filament cytoskeleton |
|
| B, Genes
downregulated in TP53 mutated cancer |
|
| Gene symbol | Gene title | FCa |
P-valueb | Function |
|
| SFRP4 | Secreted
frizzled-related protein 4 | −15.4 | 0.0112 | Regulation of Wnt
signal |
| HMCN1 | Hemicentin 1 | −8.5 | 0.0285 | Extracellular
matrix |
| MEST | Mesoderm specific
transcript homolog | −6.7 | 0.0104 | α/β-fold hydrolase
(imprinting gene) |
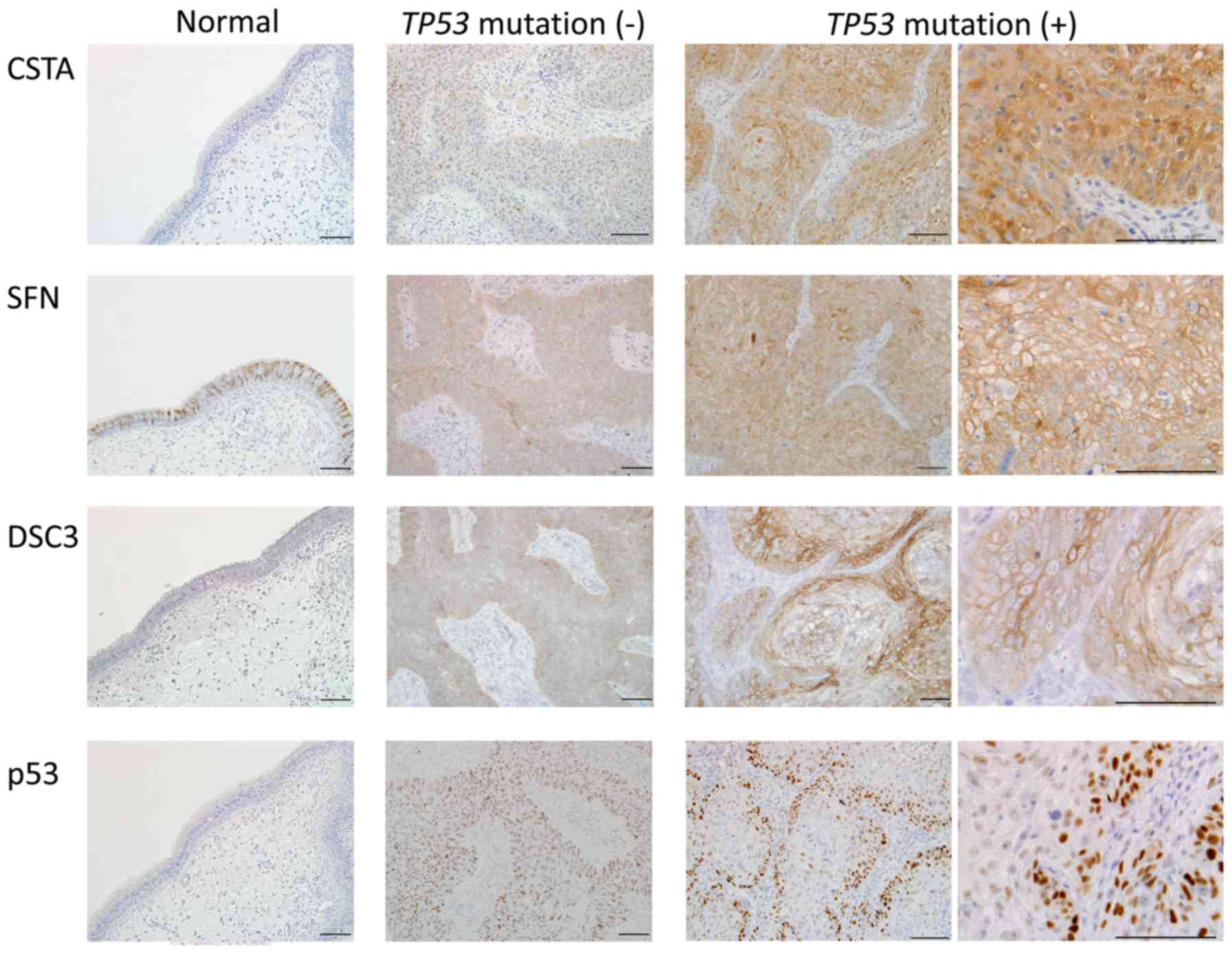
IHC analysis of DSC3, CSTA, SFN and
TP53
IHC analysis of the 3 genes with the highest
differential expression between TP53 mutated and non-mutated
tumors was performed. Fig. 4 shows
representative staining images. CSTA was negative in normal
paranasal sinus mucosa, and staining was also weak in the
TP53 wild-type tumors. In TP53 mutated tumors, CSTA
was strongly expressed throughout the entire tumor. SFN was
strongly stained in cell membranes in normal paranasal sinus
mucosa. Staining of SFN was stronger in mutated tumors than in the
wild-type tumors, and was localized more in the cell membrane than
in the cytoplasm. DSC3 expression was weakly positive in cell
membranes in normal paranasal sinus mucosa. Staining in tumors was
stronger in cell membranes, with stronger staining in mutated
tumors compared to wild-type tumors. For all these genes, the genes
were strongly expressed at the protein as well as at the mRNA level
in TP53 mutated tumors.
Localization of the p53 protein was also examined
using IHC. p53 expression was negative in normal paranasal sinus
mucosa, whereas in TP53 wild-type tumors, positive p53
nuclear expression was observed in all cancer cells. Immunostaining
of TP53 mutated tumors did not show p53 staining throughout
the tumor; instead, strong nuclear p53 staining was only observed
in tumor cells in the margins adjacent to the stroma. The stroma
and tumor interior were p53 negative. Even in biopsy specimens
after treatment, p53 staining in residual tumor was observed in the
tumor margins (data not shown). p53 IHC staining was negative in
case M13 (R156AfsX14) because of the frameshift mutation.
Discussion
This study found clear differences in gene
expression between TP53 mutated and TP53 wild-type
maxillary squamous cell carcinoma tumors. A characteristic finding
was increased expression of cell growth inhibition genes and
increased expression of cell adhesion genes such as DSC3 in the
TP53 mutated tumors. Takahashi et al compared gene
expression using microarrays in breast cancer with and without
TP53 mutations (15). They
found that the expression of genes that stimulate the cell cycle
and cell division was increased in TP53 mutated tumors, thus
suggesting that TP53 mutation was a poor prognostic factor
in breast cancer. However, our results were the opposite; i.e., we
found increased expression of 8 tumor suppressor genes including
SFN in TP53 mutated tumors compared to wild type tumors. In
particular, there was ≥100-fold differential expression of CSTA and
SFN based on TP53 mutation status (Table IV).
CSTA is a cysteine protease inhibitor that
specifically inhibits cathepsin B (16). Cathepsin B, because it localizes on
tumor cell surfaces and degrades the extracellular matrix (ECM), is
involved in cancer progression (17).
Our study showed markedly increased expression of CSTA in
TP53 mutated tumors, with strong expression in the
cytoplasm. This result suggested that the overexpressed CSTA might
more efficiently inhibit ECM degradation, resulting in a decrease
in cancer progression of TP53 mutated tumors. Increased CSTA
has been shown to inhibit the migration, invasion, and
proliferation of laryngeal cancer (18). CSTA expression has been reported to
reduce distant metastases in breast cancer, and this may be due to
inhibition of cysteine cathepsins (19). On the other hand, increased CSTA was
found to be associated with a poorer prognosis in nasopharyngeal
cancer (16). Thus, the effects of
CSTA differ depending on the type of tumor, and in our study, CSTA
may have similar effects as those reported for nasopharyngeal
cancer.
SFN is a gene that is induced by TP53 and is also
called the 14-3-3 σ protein. SFN obstructs G2 cell cycle entry by
sequestering Cdc2-cyclin B and Cdc/Cdk complexes in the cytoplasm
(20,21). Based on these functions, it was
surmised that SFN is a tumor suppressor protein. In addition,
14-3-3 σ opposes tumor-promoting metabolic programs by enhancing
c-Myc poly-ubiquitination and subsequent degradation. Thus, cancer
metabolic reprogramming occurs in tumors with low 14-3-3 σ
expression (22). However, tumors
with high SFN expression have also been reported (23–26).
Cancers with high SFN expression have increased proliferation and
anti-cancer drug resistance and thus SFN can be regarded as a tumor
progressive protein (23,24). Roberts et al reported that SFN
binds with plakophilin-3 in desmosomes and decreases incorporation
of plakophilin-3 into desmosomes, thereby decreasing desmosomal
adhesion and increasing cell migration (27). However, in our study, TP53
mutated tumors were associated with increased expression of DSC3,
DSP, and JUP, which are three genes that encode desmosomal
structural proteins, suggesting increased cell adhesion. Genes that
were overexpressed in the TP53-mutated tumors in our study
are often associated with increased adhesion and cell growth
inhibition. This means that TP53-mutated tumors may have
increased mesenchymal-epithelial transition compared to TP53
wild-type tumors. A difference in the level of
mesenchymal-epithelial transition also occurs depending on HPV
infection. HPV-positive oropharyngeal squamous cell carcinomas have
been reported to lose their epithelial cell phenotype compared with
HPV-negative tumors (28). Other
studies also have shown that HPV-positive tumors have increased
epithelial mesenchymal transition (29,30).
HPV-related carcinogenesis is associated with TP53 inactivation,
and thus many non-TP53 mutated tumors are HPV positive (13). Therefore, our study findings regarding
the expression of genes involved in mesenchymal-epithelial
transition in TP53-mutated maxillary carcinoma are consistent with
the results of previous studies that showed loss of the epithelial
cell phenotype in HPV-positive patients.
TP53 immunostaining in our study showed strong
expression in tumor cells regardless of mutation status. Staining
was unevenly distributed in TP53 mutated tumors, with
negative staining in the tumor center, but strongly positive
staining in the tumor margins. This type of uneven distribution has
not previously been reported. This staining differed from that of
TP53 wild-type tumors in which the entire tumor was
uniformly stained. In mutated tumors, TP53 degradation may be more
likely, or synthesis may be inhibited, in the tumor center. It is
also possible that the interaction of tumor cells with the stroma
may affect TP53 expression/localization in mutated tumors. This
phenomenon whereby tumor cells with different phenotypes are
produced may be linked to chemotherapy resistance. Indeed,
regression of mutated tumor also occurs with treatment. However,
unlike complete regression of wild-type tumors, mutated tumors have
some residual treatment-resistant tumor. This may be due to the
heterogeneity of TP53 mutated tumors. Gain-of-function
TP53 mutants have recently been shown to have upregulated
chromatin regulatory genes that result in genome-wide increases in
histone methylation and acetylation. Knockdown or pharmacological
inhibition of these chromatin regulatory genes can markedly lower
cancer cell proliferation (31).
Mutated TP53 can become a new transcription
factor leading to transcription activation that does not occur with
wild-type TP53. Our group has shown that this new
transcription activity does actually occur. However, the genes with
increased expression in the present study mostly played a role in
adhesion and cell growth inhibition. Thus this result suggested
that TP53 mutation in tumors results in a tumor phenotype
that is opposite to that of cancer progression and malignant
transformation. As represented by SFN, expression of tumor
suppressor genes has in fact been observed in
chemotherapy-resistant cancer. The significance of this paradoxical
phenomenon will require further investigation.
Acknowledgements
We thank Miyoko Maeda and Mariko Ishibashi for their
assistance with IHC and microarray analysis, respectively.
Glossary
Abbreviations
Abbreviations:
|
PCR
|
polymerase chain reaction
|
|
CDDP
|
cisplatin
|
|
TP53
|
tumor protein p53
|
|
HNSCC
|
head and neck squamous cell
carcinoma
|
|
TP63
|
tumor protein p63
|
|
IHC
|
immunohistochemistry
|
|
CSTA
|
cystain A
|
|
SFN
|
stratifin
|
|
DSC3
|
desmocollin 3
|
|
DSP
|
desmoplakin
|
|
JUP
|
junction plakoglobin
|
|
ECM
|
extracellular matrix
|
References
|
1
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kudo I, Esumi M, Kida A and Ikeda M: p53
mutation, but not in vitro predictor genes of therapeutic efficacy
of cisplatin, is clinically relevant in comparing partial and
complete responder cases of maxillary squamous cell carcinoma.
Oncol Rep. 24:851–856. 2010.PubMed/NCBI
|
|
3
|
Bergamaschi D, Gasco M, Hiller L, Sullivan
A, Syed N, Trigiante G, Yulug I, Merlano M, Numico G, Comino A, et
al: p53 polymorphism influences response in cancer chemotherapy via
modulation of p73-dependent apoptosis. Cancer Cell. 3:387–402.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu J, Ma Q, Zhang M, Wang X, Zhang D, Li
W, Wang F and Wu E: Alterations of TP53 are associated with a poor
outcome for patients with hepatocellular carcinoma: Evidence from a
systematic review and meta-analysis. Eur J cancer. 48:2328–2338.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohnishi K, Ota I, Takahashi A, Yane K,
Matsumoto H and Ohnishi T: Transfection of mutant p53 gene
depresses X-ray- or CDDP-induced apoptosis in a human squamous cell
carcinoma of the head and neck. Apoptosis. 7:367–372. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Varna M, Bousquet G, Plassa LF, Bertheau P
and Janin A: TP53 status and response to treatment in breast
cancers. J Biomed Biotechnol. 2011:2845842011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Viktorsson K, De Petris L and Lewensohn R:
The role of p53 in treatment responses of lung cancer. Biochem
Biophys Res Commun. 331:868–880. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zenz T, Krober A, Scherer K, Habe S,
Buhler A, Benner A, Denzel T, Winkler D, Edelmann J, Schwanen C, et
al: Monoallelic TP53 inactivation is associated with poor prognosis
in chronic lymphocytic leukemia: results from a detailed genetic
characterization with long-term follow-up. Blood. 112:3322–3329.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salani R, Kurman RJ, Giuntoli R II,
Gardner G, Bristow R, Wang TL and Shih IM: Assessment of TP53
mutation using purified tissue samples of ovarian serous carcinomas
reveals a higher mutation rate than previously reported and does
not correlate with drug resistance. Int J Gynecol Cancer.
18:487–491. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steels E, Paesmans M, Berghmans T, Branle
F, Lemaitre F, Mascaux C, Meert AP, Vallot F, Lafitte JJ and
Sculier JP: Role of p53 as a prognostic factor for survival in lung
cancer: A systematic review of the literature with a meta-analysis.
Eur Respir J. 18:705–719. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stransky N, Egloff AM, Tward AD, Kostic
AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C,
McKenna A, et al: The mutational landscape of head and neck
squamous cell carcinoma. Science. 333:1157–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Westra WH, Taube JM, Poeta ML, Begum S,
Sidransky D and Koch WM: Inverse relationship between human
papillomavirus-16 infection and disruptive p53 gene mutations in
squamous cell carcinoma of the head and neck. Clin Cancer Res.
14:366–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
International Union against Cancer: TNM
classification of malignant tumours. Sobin LH, Gospodarowicz MK and
Wittekind C: 7th. Wiley-Blackwell; New York, NY: 2009
|
|
15
|
Takahashi S, Moriya T, Ishida T, Shibata
H, Sasano H, Ohuchi N and Ishioka C: Prediction of breast cancer
prognosis by gene expression profile of TP53 status. Cancer Sci.
99:324–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang KP, Wu CC, Chen HC, Chen SJ, Peng
PH, Tsang NM, Lee LY, Liu SC, Liang Y, Lee YS, et al:
Identification of candidate nasopharyngeal carcinoma serum
biomarkers by cancer cell secretome and tissue transcriptome
analysis: Potential usage of cystatin A for predicting nodal stage
and poor prognosis. Proteomics. 10:2644–2660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dickinson DP: Cysteine peptidases of
mammals: Their biological roles and potential effects in the oral
cavity and other tissues in health and disease. Crit Rev Oral Biol
Med. 13:238–275. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li C, Chen L, Wang J, Zhang L, Tang P,
Zhai S, Guo W, Yu N, Zhao L, Liu M and Yang S: Expression and
clinical significance of cathepsin B and stefin A in laryngeal
cancer. Oncol Rep. 26:869–875. 2011.PubMed/NCBI
|
|
19
|
Parker BS, Ciocca DR, Bidwell BN, Gago FE,
Fanelli MA, George J, Slavin JL, Moller A, Steel R, Pouliot N, et
al: Primary tumour expression of the cysteine cathepsin inhibitor
Stefin A inhibits distant metastasis in breast cancer. J Pathol.
214:337–346. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chan TA, Hermeking H, Lengauer C, Kinzler
KW and Vogelstein B: 14-3-3Sigma is required to prevent mitotic
catastrophe after DNA damage. Nature. 401:616–620. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Laronga C, Yang HY, Neal C and Lee MH:
Association of the cyclin-dependent kinases and 14-3-3 sigma
negatively regulates cell cycle progression. J Biol Chem.
275:23106–23112. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Phan L, Chou PC, Velazquez-Torres G,
Samudio I, Parreno K, Huang Y, Tseng C, Vu T, Gully C, Su CH, et
al: The cell cycle regulator 14-3-3 σ opposes and reverses cancer
metabolic reprogramming. Nat Commun. 6:75302015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perathoner A, Pirkebner D, Brandacher G,
Spizzo G, Stadlmann S, Obrist P, Margreiter R and Amberger A:
14-3-3sigma expression is an independent prognostic parameter for
poor survival in colorectal carcinoma patients. Clin Cancer Res.
11:3274–3279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Neupane D and Korc M: 14-3-3sigma
modulates pancreatic cancer cell survival and invasiveness. Clin
Cancer Res. 14:7614–7623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakamura Y, Oshima K, Naoi Y, Nakayama T,
Kim SJ, Shimazu K, Shimomura A, Maruyama N, Tamaki Y and Noguchi S:
14-3-3 σ expression is associated with poor pathological complete
response to neoadjuvant chemotherapy in human breast cancers.
Breast Cancer Res Treat. 134:229–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mikami T, Maruyama S, Abé T, Kobayashi T,
Yamazaki M, Funayama A, Shingaki S, Kobayashi T, Jun C and Saku T:
Keratin 17 is co-expressed with 14-3-3 sigma in oral carcinoma in
situ and squamous cell carcinoma and modulates cell proliferation
and size but not cell migration. Virchows Arch. 466:559–569. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Roberts BJ, Reddy R and Wahl JK III:
Stratifin (14-3-3 σ) limits plakophilin-3 exchange with the
desmosomal plaque. PloS One. 8:e770122013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hatakeyama H, Mizumachi T, Sakashita T,
Kano S, Homma A and Fukuda S: Epithelial-mesenchymal transition in
human papillomavirus-positive and -negative oropharyngeal squamous
cell carcinoma. Oncol Rep. 32:2673–2679. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wakisaka N, Yoshida S, Kondo S, Kita M,
Sawada-Kitamura S, Endo K, Tsuji A, Nakanish Y, Murono S and
Yoshizaki T: Induction of epithelial-mesenchymal transition and
loss of podoplanin expression are associated with progression of
lymph node metastases in human papillomavirus-related oropharyngeal
carcinoma. Histopathology. 66:771–780. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kranjec C and Banks L: A systematic
analysis of human papillomavirus (HPV) E6 PDZ substrates identifies
MAGI-1 as a major target of HPV type 16 (HPV-16) and HPV-18 whose
loss accompanies disruption of tight junctions. J Virol.
85:1757–1764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhu JJ, Sammons MA, Donahue G, Dou ZX,
Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-Awar R, Katona BW,
Shilatifard A, et al: Gain-of-function p53 mutants co-opt chromatin
pathways to drive cancer growth. Nature. 525:206–211. 2015.
View Article : Google Scholar : PubMed/NCBI
|