Introduction
A total of 12–25% of all cases of breast cancer are
characterized by the lack of expression of the estrogen receptor
and progesterone receptor in addition to the absence of the
overexpression of human epidermal growth factor receptor-2 (HER-2),
and are therefore classified as triple-negative breast cancer
(TNBC) (1). TNBC is amongst the most
aggressive subtypes of breast cancer, which is unresponsive to
hormonal therapy and HER-2-targeted antibody therapy (2). Thus, TNBC is characterized by poor
overall survival time, predominantly due to the unavailability of
strategies for specific therapy. Cytotoxic chemotherapy is
currently the main therapeutic option for TNBC (2), including anthracyclines (for example
doxorubicin) and/or microtubule-binding agents (TBAs; such as
taxans). The molecular mechanism of action for TBAs is associated
with cell cycle arrest, predominantly at G2/M; apoptosis is induced
as a consequence of mitotic exit failure or ‘mitotic catastrophe’
(3–5).
A number of TBAs are in broad use in the clinical treatment of
TNBC. Although certain patients initially respond to these
classical cytotoxic drugs, patients exhibit high rates of systemic
relapse during early stages and decreased overall survival times
following metastasis (6,7). This may be due to drug resistance
development arising from the overexpression of drug-efflux pumps,
including the multidrug resistance (MDR)-associated transporter
P-glycoprotein (MDR-1) or proteins from the MDR-associated protein
(MRP) family, collectively members of the ATP-binding cassette
(ABC) transporter superfamily (8,9).
Furthermore, non-target-specific chemotherapy is
toxic, and the use of anthracyclines and TBAs is limited by
multiple side-effects, including cardiotoxicity, myelo- and
immunosuppression, neutropenia, mucositis and fluid retention; the
drugs also exhibit low bioavailability. Thus, there is an urgent
requirement for the development of novel, safe, and effective
therapeutic options for TNBC. To achieve this goal, appropriate
models for the analysis of the mechanisms to bypass MDR in TNBC are
required.
In the present study, a paclitaxel-resistant (TxR)
HCC1806-derived breast cancer cell subline that harbored a MDR
phenotype, with cross-resistance to anthracyclines and TBAs, was
established and characterized. This HCC1806-TxR TNBC subline may
serve as an appropriate model for analyzing the chemoresistance
mechanisms of TNBC, and to allow the study of potential novel
methods for bypassing MDR.
Materials and methods
Chemical compounds
Doxorubicin, paclitaxel, vinblastine and cisplatin
were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany);
etoposide was obtained from Calbiochem (EMD Millipore, Billerica,
MA, USA).
Antibodies
The primary antibodies used for western blotting and
flow cytometry were as follows: anti-ABC subfamily G member 2
(ABCG-2; cat. no. 42078P; Cell Signaling Technology Inc., Danvers,
MA, USA), Alexa-488-conjugated anti-phosphorylated histone subunit
H3 S10 (pH3 S10; cat. no. 9708S; Cell Signaling Technology Inc.,
Danvers, MA, USA), cleaved caspase-3 (cat. no. 9661S; Cell
Signaling Technology, Inc.), anti-PARP (cat. no. 436400;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
MDR-1 (cat. no. sc-55510) MRP-1 (cat. no. sc-18835) and β-actin
(cat. no. sc-8432; Santa Cruz Biotechnology, Dallas, TX, USA);
HRP-conjugated secondary antibodies [anti-mouse immunoglobulin
(Ig)G, cat. no. sc-2005; anti-rabbit IgG, cat. no. sc-2004] for
western blotting were purchased from Santa Cruz Biotechnology,
Inc.
Cell lines and culture conditions
MES-SA/Dx5 Cell Lysate was obtained from Santa Cruz
Biotechnology (cat. no. sc-2284). The human basal-like TNBC cell
line, HCC1806, was purchased from American Type Culture Collection
(Manassas, VA, USA) and maintained in RPMI-1640 medium supplemented
(Paneco, Moscow, Russia) with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 1% L-glutamine, 50 U/ml
penicillin and 50 µg/ml streptomycin. To establish a TxR TNBC
subline, HCC1806 cells were initially incubated in 0.5 nM
paclitaxel for 4 weeks. When stable growth was achieved, the
paclitaxel concentration was doubled every 4–5 weeks over 8 months
to a maximum dose of 100 µM. Prior to experimental use, TxR-cells
were maintained in paclitaxel-free culture medium as previously
described, and passaged at least 3 times. All cells were cultured
at 37°C in a humidified atmosphere of 5% CO2 in an
incubator (Lamsystems, Miass, Russia) throughout the present
study.
Intracellular drug concentration
НСС1806 and HCC1806-TxR cells were exposed to 100 nM
paclitaxel for 2 h. Cells were rinsed twice with ice-cold PBS,
harvested into centrifuge tubes, centrifuged at 250 × g for 5 min
at room temperature and collected for solid-phase extraction high
performance liquid chromatography (HPLC; LC-20 Prominence; Shimadzu
Corporation, Kyoto, Japan). The detection wavelength was 227 nm and
the limit of detection for paclitaxel was 5.0 ng. Experimental
conditions were as follows: mobile phase; water and acetonitrile in
a ratio of 40:60; Ascentis® C18 column, 4.6×250 mm;
guard cartridge, Ascentis® C18 Supelguard with 5 µm
particle size, 20×4 mm; isocratic mode (flow rate, 1 ml/min;
temperature, 40°C; pressure, 35 bar; Sigma-Aldrich). Paclitaxel
(Sigma-Aldrich) was used as the internal standard. The method of
absolute calibration was used for the quantitation of paclitaxel.
The intracellular concentration of paclitaxel was defined as the
total amount of paclitaxel/106 cultured cells.
Cellular survival assay
Exponentially growing cells were seeded
(3.2×104 cells/well) into the 96-well flat-bottomed
plates (Corning Incorporated, Corning, NY, USA) and incubated for
24 h at 37°C with RPMI-1640 culture medium. The cells were then
cultured for 24 or 48 h at a range of concentrations of
chemotherapeutic agents, including paclitaxel (1 nM-10 µM),
doxorubicin (0, 03125-8 µg/ml), vinblastine (1 nM-10 µM), cisplatin
(0, 03125-80 µM) or etoposide (2, 5-640 µM). All the aforementioned
agents were previously diluted in dimethyl sulfoxide (DMSO). DMSO
was used as a negative control in untreated cells. MTS reagent
(CellTiter 96® Aqueous Non-Radioactive Cell
Proliferation Assay; Promega Corporation, Madison, WI, USA) was
added at 2 mg/ml to assess the relative number of viable cells. The
MTS and cells were incubated for ≥1 h and the production of
formazan, dissolved using DMSO, was assessed by the relative
absorbance at 492 nm on a MultiScan FC plate reader (Thermo Fisher
Scientific, Inc.). The resulting IC50 values were
defined as the compound concentration required to inhibit cellular
growth by 50% after 48 h of post treatment. The data was normalized
to the DMSO control group.
Real-time monitoring of cell
proliferation using an i-CELLigence system
An in vitro growth curve characterization of
HCC1806 parental and TxR cells cultured in the presence of
paclitaxel was performed using an iCELLigence system (ACEA
Biosciences, Inc., San Diego, CA, USA). Cells (5×104)
were seeded in electronic microtiter plates (E-Plate; Roche
Diagnostics, Basel, Switzerland) and incubated for 24 h to obtain
the growth baseline reading. Then cells were treated with 10, 50 or
100 nM paclitaxel in triplicate, and cell index (CI) measurements
were obtained, with a signal detected every 30 min until the end of
the experiment (72 h). Normalized CI values were calculated with
RTCA iCelligence software version 1.1.1 (Roche Diagnostics).
Western blot analysis
For western blot analysis, whole-cell extracts were
prepared by scraping HCC1806 parental and TxR cells, or MES-SA
control cells, into radioimmunoprecipitation buffer (1% NP-40, 50
mM Tris-HCl pH 8.0, supplemented with protease and phosphatase
inhibitors). The cellular lysates were incubated for 1 h at 4°C and
then clarified by centrifugation for 30 min at 14,000 × g at 4°C.
Protein concentrations were measured by the Bradford assay. Samples
containing 30 µg of protein were resolved on 4–12% Bis-Tris or 3–8%
Tris-acetate NuPAGE gels (Invitrogen; Thermo Fisher Scientific,
Inc.), transferred to a nitrocellulose membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA), probed with primary
(1:1,000 and incubated overnight at 4°C), and secondary antibodies
(1:1,000 and incubated for 1 h at room temperature) and visualized
with enhanced chemiluminescence (Western Lightning Plus-ECL
reagent, PerkinElmer, Inc., Waltham, MA, USA). The MES-SA cells
acted as a positive control for ABC protein expression.
Cell cycle analysis
For flow cytometry cell cycle analysis, the cells
were treated with paclitaxel at 10 or 1,000 nM for 24–48 h and
trypsinized. Subsequent to centrifuging at 300 × g for 5 min at
room temperature, the cells were washed in PBS, fixed in 4%
paraformaldehyde and permeabilized with ice-cold 90% methanol. The
washed cells were stained with Alexa-488-conjugated anti-pH3 S10
(1:500 and incubated for 1 h at room temperature in the dark), were
then counterstained with propidium iodide (30 min at room
temperature in the dark) (Sigma-Aldrich) and analyzed by
fluorescence-activated cell sorting on a FC500 flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA). Cells were counted and
analyzed using the Kaluza software version 1.3 (Beckman Coulter,
Inc.).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
At 24 h after plating 1×106 HCC1806 or
HCC1806-TxR cells, total RNA was extracted using TRIzol reagent
(cat. no. BC032; Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol and resuspended in diethyl
pyrocarbonate-treated H2O. RNA was reverse transcribed
to cDNA using the Moloney murine leukemia virus reverse
transcriptase kit (Evrogen JSC, Moscow, Russia) according to the
manufacturer's protocol (cat. no. SK021), and subjected to qPCR. A
total of 1 µl template cDNA was used in the qPCR reaction, with 5X
qPCRmix-HS SYBR (Evrogen JSC) and 10 mM of each forward and reverse
primer (Table I). qPCR was performed
with the CFX96 Real-Time detection system (Bio-Rad Laboratories,
Inc.), according to the manufacturer's protocol. Thermal cycling
conditions were as follows: 3 min at 95°C, 45 cycles (15 sec at
95°C, 10 sec at 56°C, 30 sec at 72°C) and a final extension step of
5 min at 72°C. Each sample was processed in parallel with assays
for GAPDH and the absolute levels of each mRNA were normalized
relative to GAPDH. The 2−∆∆Cq method (10) was then used to calculate relative gene
expression.
 | Table I.Primers for quantitative polymerase
chain reaction. |
Table I.
Primers for quantitative polymerase
chain reaction.
| Gene | Forward | Reverse |
|---|
| GAPDH |
GACCACAGTCCATGCCATCA |
TCCACCACCCTGTTGCTGTA |
| P-glycoprotein |
ATGCTCTGGCCTTCTGGATGGGA |
ATGGCGATCCTCTGCTTCTGCCCAC |
| MRP-1 | GCATGA
TCCCTGAAGACGA |
TAGAGCTGGCCCTTGTACTC |
| MRP-2 |
TAGAGCTGGCCCTTGTACTC |
TCAACTTCCCAGACATCCTC |
| MRP-3 |
CGCCTGTTTTTCTGGTGGTT |
TCCCCCAGTCACAAAGATG |
| MRP-4 |
GCTGAGAATGACGCACAGAA |
TCCCAGCAAGGCACGATATT |
| MRP-5 |
GTCCTGGGTATAGAAGTGTG |
CAGAAGATCCACACAACCCT |
| MRP-6 |
TTGGATTCGCCCTCATAGTC |
TCTTTTGGTCTCAGTGGCCT |
| MRP-7 |
CTCCCACTGGATCTCTCAGC |
TCGCATACACGGTGAGGTAG |
| Bcl-2 |
ATGTCCAGCCAGCTGCACCTGAC |
GCAGAGTCTTCAGAGACAGCCAGG |
| Bcl-2 associated
X |
GCTTCAGGGTTTCATCCAGG |
AAAGTAGGAGAGGAGGCCGT |
| Fas |
CAGGCTAACCCCACTCTATG |
TGGGGGTGCATTAGGCCATT |
| Caspase-8 |
ACTTCAGACACCAGGCAGGGCT |
GCCCCTGCATCCAAGTGTGTTC |
| Y-box binding protein
1 |
GACTGCCATAGAGAATAACCCCAG |
CTCTCTAGGCTGTTTTGGGCGAGGA |
Statistical analysis
All experiments were repeated a minimum of 3 times.
The results are presented as the mean ± standard deviation for each
group. Statistical analyses (Student's t-test, Mann-Whitney U test)
were performed using Statistical software program version 7.0 (S.A.
Glantz, McGraw Hill Education, NY, USA) and GraphPad Prism version
4.0 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Establishment of taxol-resistant
HCC1806 breast cancer cell line
The HCC1806-TxR cell line was established following
a continuous treatment with a gradually increasing concentration of
paclitaxel, from 0.5–100 nM. Approximately 8 months and 55 passages
were required to develop HCC1806 cells with stable drug resistance.
It was then demonstrated that НСС1806-TxR cells were less sensitive
to paclitaxel treatment when compared with parental HCC1806 cells;
the paclitaxel drug resistance index was 16.86 (Table II). In addition to resistance to
paclitaxel, HCC1806-TxR cells exhibited cross-resistance to
vinblastine, doxorubicin and etoposide (drug resistance index,
14.65, 4.36 and 3.18, respectively), whereas there was no
identified cross-resistance to cisplatin. Paclitaxel resistance in
HCC1806-TxR cells remained stable following 1 month of culturing
without paclitaxel and following storage at −80°C for 6 months
(data not shown).
| Table II.IC50 values for HCC1806 and
HCC1806-TxR cells. |
Table II.
IC50 values for HCC1806 and
HCC1806-TxR cells.
| Drug | HCC1806 (%) | HCC1806-TxR (%) | Fold change |
|---|
| Paclitaxel, nM | 4.68±0.44 (9.39) | 78.9±3.8 (4.8) | 16.86 |
| Vinblastine, nM | 0.2±0.04 (20) | 2.93±0.32
(10.92) | 14.65 |
| Doxorubicin,
µg/ml | 0.53±0.03 (5.6) | 2.31±0.23 (9.9) | 4.36 |
| Etoposide, mM | 72.94±9.64
(10.6) | 231.91±15.73
(3.1) | 3.18 |
| Cisplatin, µM | 10.27±1.09
(13.2) | 10.19±0.32 (6.8) | 1.01 |
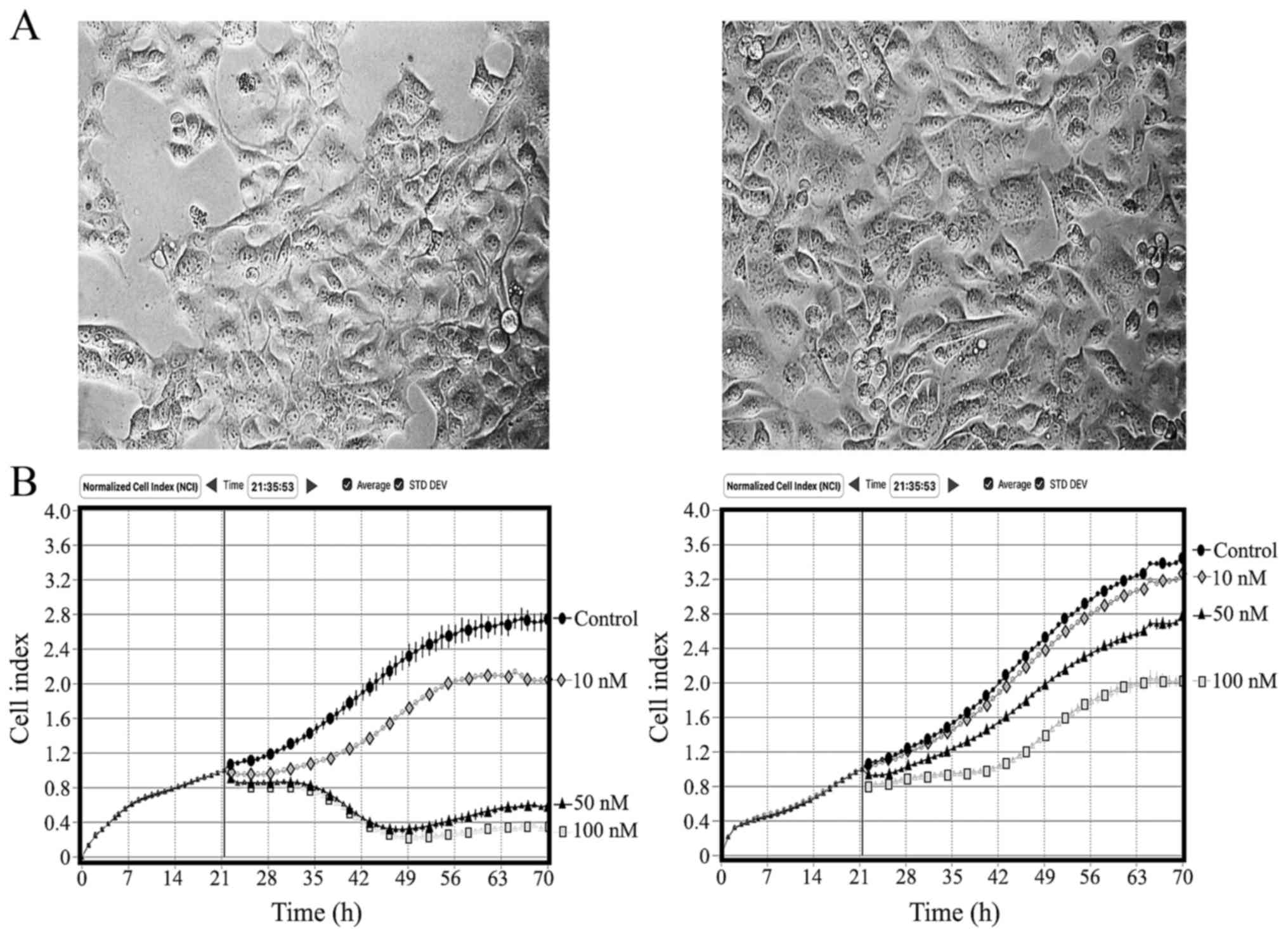
Alterations in cellular morphology and
growth kinetics
The morphology of the TxR cells was distinct from
the parental cells. HCC1806-TxR cells exhibited an enlarged and
oval-shaped morphology, and an increased nucleus/cytoplasm ratio
when compared with the parental cells (Fig. 1A). The resistant cells were consistent
in size and shape in monolayer proliferation, and their growth
kinetics were different from those of the parental cells (Fig. 1B). When cells were cultured with
paclitaxel, a difference in growth kinetics between the TxR and
parental HCC1806 cells was also detected (Fig. 1B), which was consistent with the
MTS-based cytotoxicity data (Table
II).
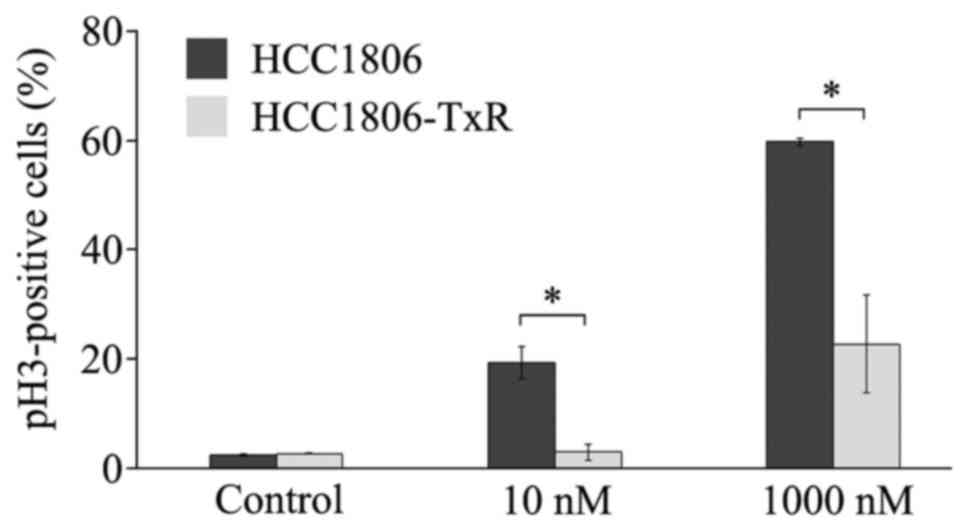
Paclitaxel induced cell accumulation
in M-phase and apoptosis in parental, but not TxR, HCC1806
cells
As the major mechanism for the action of paclitaxel
is the inhibition of tubulin depolymerization to induce apoptosis
due to ‘mitotic catastrophe’, the ability of paclitaxel to induce
the accumulation of cells in M-phase in parental vs.
paclitaxel-resistant HCC1806 cells was compared. pH3 S10 staining
was combined with propidium iodide DNA staining to count the number
of mitotic cells by flow cytometry. An increase of the pH3
S10-positive (i.e., mitotic) HCC1806 cells was observed subsequent
to 10 or 1,000 nM paclitaxel exposure (Fig. 2). In contrast, the number of mitotic
paclitaxel-treated HCC1806-TxR cells was significantly less when
compared with parental cells (P<0.001 for 10 and 1,000 nM;
Fig. 2).
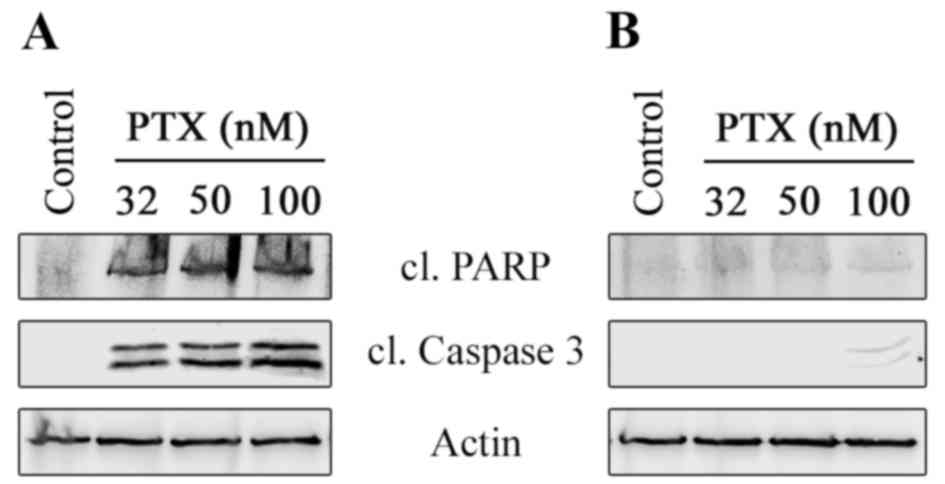
To assess the ability of paclitaxel to induce
apoptosis in parental vs. TxR HCC1806 cells, the expression of
cleaved caspase-3 and PARP, specific markers for apoptosis, was
observed. An increase in the cleavage of PARP and caspase-3 was
detected in paclitaxel-treated HCC1806 cells, reflecting the
ability of paclitaxel to induce apoptosis in HCC1806 parental cells
(Fig. 3A). In contrast, relatively
low cleavage of PARP and caspase-3 were observed in
paclitaxel-treated HCC1806-TxR cells (Fig. 3B), thereby confirming the cytotoxicity
MTS-based data by indicating the resistance of the HCC1806-TxR
subline to paclitaxel.
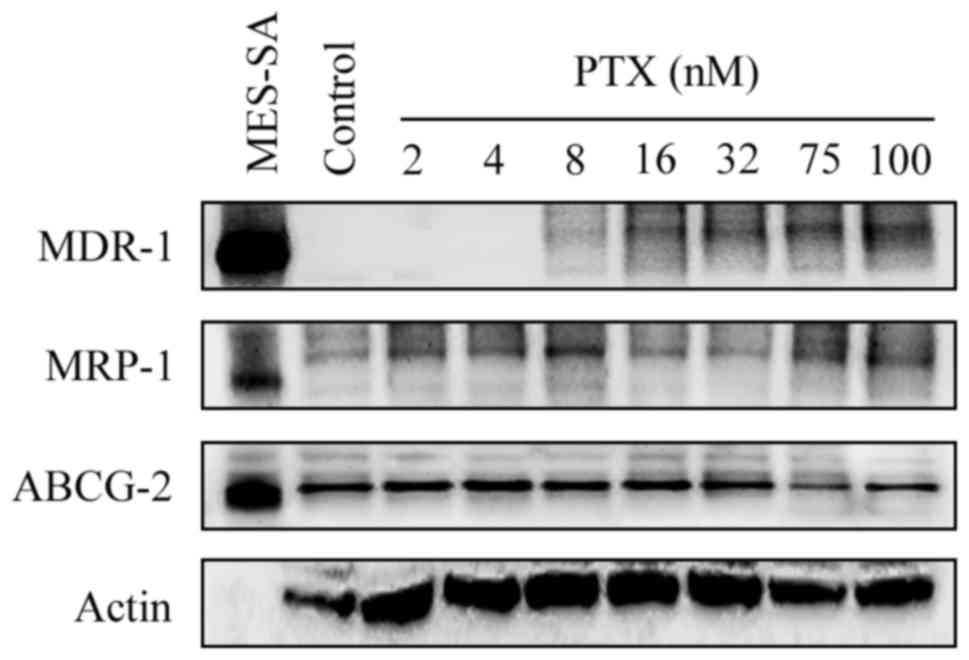
The expression of
chemoresistance-associated genes is altered in HCC1806-TxR
cells
As the resistance of patient tumors to
chemotherapeutic agents is highly dependent on the activities of
the transporters that efflux drugs from tumor cells, the expression
of the MDR-1, MRP-1 and ABCG2 ABC transporter proteins in
HCC1806-TxR cells was assessed. Western blot analysis revealed the
overexpression of the MDR-1 and MRP-1 proteins in HCC1806-TxR cells
when compared with the parental HCC1806 cells (Fig. 4). However, the expression level of
another protein associated with the MDR phenotype, ABCG2, was
unchanged between the parental and TxR lines.
Since a potential mechanism for MDR-1 overexpression
in paclitaxel-resistant cells is the induction by Y-box-binding
protein 1 (YB-1), which binds to a cis-acting element of the MDR-1
promoter to increase MDR-1 mRNA expression (11), YB-1 mRNA levels were compared between
parental HCC1806 cells and the TxR subline. There was an increase
(3.6-fold) of YB-1 mRNA expression in TxR-cells when compared with
the parental HCC1806 cells (Table
III). Similarly, a number of MRP mRNAs were observed to be
overexpressed in HCC1806-TxR cells when compared with the parental
HCC1806 cells (Table III). In TxR
cells, the levels of MRP-1, −5, and −6 were elevated to 2.3, 15.7
and 5.7-fold, respectively. As paclitaxel-induced cell death is
associated with the activation of apoptosis mechanisms, the
expression of the apoptosis-related genes, Bcl-2 associated X
(Bax), Bcl-2, caspase-8 and Fas, were compared in parental vs. TxR
cells. The expression of Bax and caspase-8 mRNA were unchanged in
resistant cells, but a trend towards the downregulation of Fas was
observed when compared with parental cells. In contrast, Bcl-2
expression was upregulated in the TxR subline (Table III).
| Table III.Alterations in the expression level of
chemoresistance and apoptosis associated genes in HCC1806-TxR cells
relative to HCC1806 cells, as determined by reverse
transcription-quantitative polymerase chain reaction. |
Table III.
Alterations in the expression level of
chemoresistance and apoptosis associated genes in HCC1806-TxR cells
relative to HCC1806 cells, as determined by reverse
transcription-quantitative polymerase chain reaction.
| Gene | Fold change |
|---|
| P-glycoprotein | 1.73 |
| MRP-1 | 2.3 |
| MRP-2 | 1.33 |
| MRP-3 | 0.19 |
| MRP-4 | 1.28 |
| MRP-5 | 15.74 |
| MRP-6 | 5.76 |
| MRP-7 | 1.51 |
| Bcl-2 | 1.65 |
| Bcl-2 associated
X | 1.12 |
| Fas | 0.63 |
| Caspase-8 | 0.98 |
| Y-box binding
protein 1 | 3.59 |
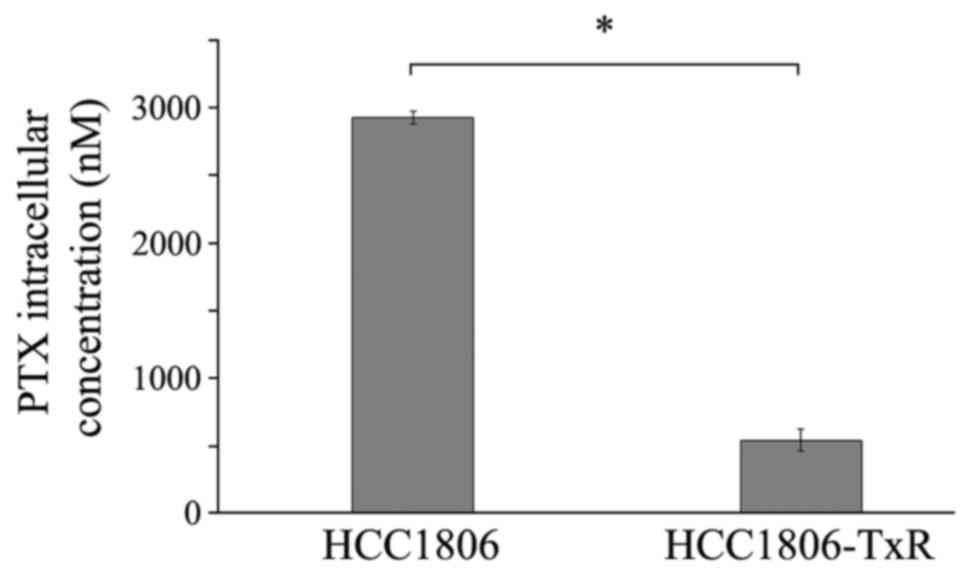
Intracellular drug concentration
As cellular mechanisms for drug resistance include a
decline of intracellular drug concentrations as a result of
increased efflux by overexpressed ABC proteins, and considering the
overexpression of MDR-1 and MRP-related genes in the TxR cells, the
intracellular paclitaxel concentrations in HCC1806 cells were
compared with HCC1806-TxR cells. As presented in Fig. 5, at 2 h of paclitaxel exposure,
HCC1806-TxR-cells contained ~540 ng paclitaxel, which was much
lower than in the parental HCC1806 cells (2,930 ng; P<0.05).
Discussion
In the present study, a paclitaxel-resistant
basal-like TNBC cell line was established from the parental HCC1806
cell line. The paclitaxel IC50 value for HCC1806-TxR
cells was ~17-fold higher when compared with parental HCC1806
cells. Of note, the paclitaxel resistance remained stable after 1
month of culturing without paclitaxel and following storage at
−80°C for 6 months. HCC1806-TxR cells exhibited alterations to
cellular morphology (for example enlarged, oval-shaped) when
compared with parental cells. Additionally, increased growth was
observed in HCC1806-TxR cells exposed to paclitaxel compared with
the parental HCC1806 cells.
Paclitaxel is a commonly used chemotherapeutic drug
for the treatment of patients with a range of types of cancer,
including breast, ovarian and lung cancer (12–14). The
molecular mechanism for the action of paclitaxel is its ability to
bind microtubules and induce their stabilization, thus preventing
their disassembly during mitosis (3,5).
Therefore, in paclitaxel-treated cells, microtubules become locked
in a polymerized state, which leads to G2/M arrest, induces the
accumulation of cells in M-phase and leads to cell death via
apoptosis (15–18). Thus, a substantial decrease of mitotic
and apoptotic cell amount in TxR-subline after paclitaxel exposure
was expected. Indeed, a substantial decrease of pH3 S10-positive
(i.e., mitotic) cells (Fig. 2) and a
low level of PARP and caspase-3 cleavage of in paclitaxel-treated
HCC1806-TxR cells was observed when compared with parental HCC1806
cells (Fig. 3B). This data was
consistent with the cytotoxicity MTS-based data, indicating the
resistance of the HCC1806-TxR subline to paclitaxel.
Additionally, HCC1806-TxR exhibited a
cross-resistance to other types of chemotherapeutic agents,
including vinblastine, and etoposide and doxorubicin topoisomerase
type II inhibitors. The resistance indexes for the drugs indicated
above were 14.65, 4.36 and 3.18, respectively. This data is
consistent with previous study, indicating that cancer cells in
culture becoming resistant to a single drug may become resistant to
a class of drugs with a similar mechanism of action as well
(19). In addition, cancer cells may
acquire a cross-resistance to structurally and mechanistically
unrelated drugs; this phenomenon is known as MDR. For example,
McDonald et al previously demonstrated that
docetaxel-resistant MDA-MB and MCF-7 breast cancer cells developed
a cross-resistance to paclitaxel and vincristine (20), concordant to the findings of the
present study. Multiple reports have indicated that resistant
sublines commonly express MDR-1 and MRP proteins (9,21,22). Clinical studies also report the
development of cross-resistance to multiple anticancer agents
following the initial success of chemotherapy (23,24), which
may also be due to the overexpression of MDR-1 and MRPs.
Thus, the molecular mechanism of cross-resistance of
HCC1806-TxR to vinblastine and topoisomerase type II inhibitors may
be due to the increased expression of ABC transporters, important
mediators of drug efflux. An increased expression of MDR-1 and
MRP-1 proteins in HCC1806-TxR cells was observed (Fig. 4), which may lead to the increased
efflux of paclitaxel and other types of chemotherapeutic drugs (for
example alkylating agents, topoisomerase type II inhibitors),
resulting in MDR (Fig. 5).
In order to investigate the genes involved in
paclitaxel resistance, the gene expression profile between HCC1806
and HCC1806-TxR cells was compared. MDR-1, MRP-1, −5 and −6 were
upregulated in the HCC1806-TxR subline, suggesting that these
factors served a function in the observed MDR phenotype. The
increased activity of ABC transporters in the HCC1806-TxR subline
was additionally confirmed by a marked (5.4-fold) decrease in the
intracellular paclitaxel content in TxR-cells at 2 h of paclitaxel
exposure when compared with parental HCC1806 cells (Fig. 5), thus indicating an increased efflux
of chemotherapeutic drugs in the HCC1806-TxR subline. Furthermore,
differences in expression of genes associated with apoptosis
between the parental and TxR-cells, including Bcl-2 and Fas, were
also detected. This data may also explain the low expression of
apoptotic markers (cleaved PARP and caspase-3) in TxR cells
subsequent to paclitaxel exposure when compared with parental
HCC1806 cells (Fig. 3).
In conclusion, a HCC1806 subline was established
with high resistance to paclitaxel, which also harbored a
cross-resistance to diverse types of chemotherapeutic agents,
including those with similar and different modes of action to
paclitaxel. The MDR phenotype was associated with MDR-1, MRP-1, −5
and −6 overexpression, thus providing an effective efflux of
anticancer drugs from HCC1806-TxR cancer cells. In addition, the
TxR breast cancer subline also resisted apoptosis subsequent to the
exposure to paclitaxel. This may be due to the downregulation of
pro-apoptotic proteins (Fas) and the upregulation of anti-apoptotic
proteins (Bcl-2). Paclitaxel resistance remained stable subsequent
to 1 month of culturing without paclitaxel and following prolonged
storage at −80°C. Taken together, the established MDR in a
HCC1806-derived breast cancer subline may be appropriate for in
vitro screenings of novel anticancer agents for overcoming
MDR-mediated mechanisms in TNBC.
Acknowledgements
The present study was supported by a grant from the
Russian Science Foundation (grant no. 14-15-00342).
References
|
1
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kassam F, Enright K, Dent R, Dranitsaris
G, Myers J, Flynn C, Fralick M, Kumar R and Clemons M: Survival
outcomes for patients with metastatic triple-negative breast
cancer: Implications for clinical practice and trial design. Clin
Breast Cancer. 9:29–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dumontet C and Jordan MA:
Microtubule-binding agents: A dynamic field of cancer therapeutics.
Nat Rev Drug Discov. 9:790–803. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parker A, Kavallaris M and McCarroll JA:
Microtubules and their role in cellular stress in cancer. Front
Oncol. 4:1532014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bosch A, Eroles P, Zaragoza R, Viña JR and
Lluch A: Triple-negative breast cancer: Molecular features,
pathogenesis, treatment and current lines of research. Cancer Treat
Rev. 36:206–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yu KD, Zhu R, Zhan M, Rodriguez AA, Yang
W, Wong S, Makris A, Lehmann BD, Chen X, Mayer I, et al:
Identification of prognosis-relevant subgroups in patients with
chemoresistant triple-negative breast cancer. Clin Cancer Res.
19:2723–2733. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ullah MF: Cancer multidrug resistance
(MDR): A major impediment to effective chemotherapy. Asian Pac J
Cancer Prev. 9:1–6. 2008.PubMed/NCBI
|
|
9
|
Hasanabady MH and Kalalinia F: ABCG2
inhibition as a therapeutic approach for overcoming multidrug
resistance in cancer. J Biosci. 41:313–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–428. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohga T, Uchiumi T, Makino Y, Koike K, Wada
M, Kuwano M and Kohno K: Direct involvement of the Y-box binding
protein YB-1 in genotoxic stress-induced activation of the human
multidrug resistance 1 gene. J Biol Chem. 273:5997–6000. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Holmes FA, Walters RS, Theriault RL,
Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK and Hortobagyi
GN: Phase II trial of taxol, an active drug in the treatment of
metastatic breast cancer. J Natl Cancer Inst. 83:1797–1805. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brown T, Havlin K, Weiss G, Cagnola J,
Koeller J, Kuhn J, Rizzo J, Craig J, Phillips J and Von Hoff D: A
phase I trial of taxol given by a 6-hour intravenous infusion. J
Clin Oncol. 9:1261–1267. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McGuire WP, Rowinsky EK, Rosenshein NB,
Grumbine FC, Ettinger DS, Armstrong DK and Donehower RC: Taxol: A
unique antineoplastic agent with significant activity in advanced
ovarian epithelial neoplasms. Ann Intern Med. 111:273–279. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fuchs DA and Johnson RK: Cytologic
evidence that taxol, an antineoplastic agent from Taxus brevifolia,
acts as a mitotic spindle poison. Cancer Treat Rep. 62:1219–1222.
1978.PubMed/NCBI
|
|
16
|
Schiff PB, Fant J and Horwitz SB:
Promotion of microtubule assembly in vitro by taxol. Nature.
277:665–667. 1979. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haldar S, Jena N and Croce CM:
Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA.
92:4507–4511. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weaver BA: How Taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo B, Villeneuve DJ, Hembruff SL, Kirwan
AF, Blais DE, Bonin M and Parissenti AM: Cross-resistance studies
of isogenic drug-resistant breast tumor cell lines support recent
clinical evidence suggesting that sensitivity to paclitaxel may be
strongly compromised by prior doxorubicin exposure. Breast Cancer
Res Treat. 85:31–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McDonald SL, Stevenson DA, Moir SE,
Hutcheon AW, Haites NE, Heys SD and Schofield AC: Genomic changes
identified by comparative genomic hybridization in
docetaxel-resistant breast cancer cell lines. Eur J Cancer.
41:1086–1094. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharom FJ: ABC multidrug transporters:
Structure, function and role in chemoresistance. Pharmacogenomics.
9:105–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim KY, Kim SH, Yu SN, Park SK, Choi HD,
Yu HS, Ji JH, Seo YK and Ahn SC: Salinomycin enhances
doxorubicin-induced cytotoxicity in multidrug resistant MCF-7/MDR
human breast cancer cells via decreased efflux of doxorubicin. Mol
Med Rep. 12:1898–1904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kröger N, Achterrath W, Hegewisch-Becker
S, Mross K and Zander AR: Current options in treatment of
anthracycline-resistant breast cancer. Cancer Treat Rev.
25:279–291. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yonemori K, Katsumata N, Uno H, Matsumoto
K, Kouno T, Tokunaga S, Yamanaka Y, Shimizu C, Ando M, Takeuchi M
and Fujiwara Y: Efficacy of weekly paclitaxel in patients with
docetaxel-resistant metastatic breast cancer. Breast Cancer Res
Treat. 89:237–241. 2005. View Article : Google Scholar : PubMed/NCBI
|