Introduction
Although the incidence of gastric cancer (GC) has
declined, it remains the third-leading cause of cancer-associated
mortality (1). Despite the use of
numerous treatment modalities, including surgery, chemotherapy,
radiotherapy and immunotherapy, disease prognosis and treatment
efficacy remain poor (2,3). Evidence indicates that vascular
endothelial growth factor receptor 2 (VEGFR2) serves a critical
role in GC oncogenesis and angiogenesis, suggesting that this
molecule may represent a potential therapeutic target (4). Stimulation of VEGER2 by VEGF can
simultaneously activate several molecular pathways, including the
Raf/Mitogen-activated protein kinase kinase/extracellular-related
signal kinase, p38-mitogen activated protein kinase and
phosphoinositide-3 kinase (PI3K)/protein kinase B (AKT)/mechanistic
target of rapamycin signaling pathways, which mediate cell
proliferation, migration, and survival, respectively (5,6). Apatinib
targets VEGFR2 in chemoresistant GC, improving the survival of GC
patients (7,8).
MicroRNAs (miRNAs/miRs) are ~22 nucleotide in
length, non-coding RNA molecules that regulate gene expression at
the post-transcriptional level by binding to the 3′ untranslated
region (3′UTR) of their target mRNAs (9). Numerous studies have demonstrated roles
for miRNAs in human cancer (10). In
particular, miR-21 was found to be aberrantly overexpressed in
numerous cancer types, including those of the prostate, breast and
lung (11). miR-21 also mediates
tumor cell growth and metastasis by activating AKT signaling
(12). Moreover, VEGF upregulates
miR-21 expression in human umbilical vein endothelial cells
(13) by an unknown mechanism.
However, miR-21 was also found to regulate peroxisome
proliferator-activated receptor-α (PPARα) in the process of
endothelial inflammation (14).
PPARα is a pleiotropic molecule that
transcriptionally regulates genes involved in lipid and glucose
homeostasis (15). PPARα also
exhibits anti-inflammatory properties, with previous studies
implicating it in the progression of several types of cancer,
including hepatic, kidney, breast and lung cancer (16–19).
However, the role of PPARα in GC has not been studied. The
anti-inflammatory effects of aspirin are reported to be mediated by
PPARα activation (20). Moreover,
aspirin has also been demonstrated to reduce the risk of developing
GC (21), although the mechanisms
underlying this effect remain unknown.
In the present study, aspirin was used to activate
PPARα as a step towards elucidating the effects of miR-21 and PPARα
in GC. The present study analyzed the association between PPARα,
miR-21 and VEGF in patients with GC. It further identified the
effect of apatinib and aspirin on GC cell proliferation. Treatment
with a combination of apatinib and aspirin may represent a novel
strategy to treat gastric cancer.
Materials and methods
Oligonucleotides, antibodies, reagents
and kits
Oligonucleotides encoding miR-21 mimics (hsa-miR-21
mimics; cat no. HMI0371; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), a non-coding (NC) miRNA (miR-control:
AGUACUGCUUACGAUACGGTT), miR-21 inhibitor (anti-miR-21; cat no.
HSTUD0371; Sigma-Aldrich; Merck KGaA), and NC inhibitor (cat no.
B04003; anti-miR-control; Shanghai GenePharma Co., Ltd., Shanghai,
China). The pcDNA3.1-PPARα plasmid, a PPARα-specific siRNA (cat no.
AM16708; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and the
pGL3-PPARα-3′UTR plasmid (containing miR-21-binding sequences) were
gifts from Dr Han-Yang Hu (Wuhan University, Hubei, China). Mouse
monoclonal antibodies (mAbs) against β-actin (cat no. sc70319,
1:4,000), PPARα (cat no. sc130640, 1:2,000) were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Phosphorylated
(p)-VEGFR2 antibody (cat no. ab38473, 1:1,000) was purchased from
Abcam (Cambridge, MA, USA). Mouse mAbs against p-AKT (cat no. 4051,
1:2,000), AKT (cat no. 2920, 1:2,000), rabbit mAb against p-PI3K
(cat no. 4228, 1:2,000) and PI3K (cat no. 4249, 1:2,000) were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Horseradish peroxidase (HRP)-conjugated goat anti-mouse
immunoglobulin (Ig)G (cat no. sc2005, 1:3,000) and HRP-conjugated
goat anti-rabbit IgG (cat no. sc2004, 1:3,000) secondary antibodies
were obtained from Santa Cruz Biotechnology, Inc. VEGF, aspirin and
apatinib were purchased from Sigma-Aldrich; Merck KGaA. For cell
transfection, Lipofectamine 2000 was purchased from Invitrogen;
Thermo Fisher Scientific, Inc. Finally, the human peroxisome
proliferators activator receptors α ELISA kit (CSB-E09754h) and,
human vascular endothelial cell growth factor ELISA kit
(CSB-E11718h) were purchased from Cusabio (College Park, MD,
USA).
Human brain tissue, cell culture, and
transfection
A total of 30 patients (19 male, 11 female; age
range, 45–60 years; mean age, 55.7) undergoing GC surgery at Inner
Mongolia Autonomous Region People's Hospital (Hohhot, China) were
enrolled in the present study. Tumor tissues and cancer-adjacent
normal tissues, as well as a blood sample, were obtained for use in
the study. Written informed consent was obtained from all
participants. In addition, 30 blood samples from individuals
without cancer were used as controls in ELISA detection. All tissue
samples divided into two parts. One section was used for RNA
isolation, while the other was used for protein extraction. The
human GCMKN1, MKN45, MKN74, and IM95 cell lines were purchased from
the American Type Culture Collection (Manassas, VA, USA) and
cultured in RPMI-1640 medium (Hyclone; GE Healthcare Life Sciences,
Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (EMD Millipore, Billerica, MA, USA) at 37°C
in a humidified chamber with 5% CO2.
Transfection procedures were performed using
Lipofectamine 2000 reagent according to the manufacturer's
protocol. Briefly, MKN45 cells were cultured in 6-well plates in
RPMI-1640 medium at 37°C in a humidified chamber with 5%
CO2. When the cells were 80% confluent, the culture
medium was changed for OPTI-MEM (cat no. 31985088; Gibco; Thermo
Fisher Scientific, Inc.), and MKN45 cells transfected with miR-21
mimics, miR-21 inhibitors, or NC controls at a concentration of 150
pmol/ml. To analyze the effect of PPARα on AKT expression, PPARα
plasmid (7 µg/ml) and PPARα specific siRNA (150 pmol/ml) were
transfected into MKN45 cells. After 48 h, the cells were harvested
for the luciferase reporter assay and western blot analysis. All
experiments were approved by the Inner Mongolia Autonomous Region
People's Hospital Ethics Committee.
ELISA
The serum was obtained from patients with GC and
healthy individuals, and the levels of VEGF and PPARα in the
peripheral blood were detected using ELISA kits according to the
manufacturer's instructions. Absorbance was detected at 450 nm
using a microplate reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Each assay was performed in triplicate, and the results
were averaged over three independent experiments.
Reverse transcription-quantitative PCR
(RT-qPCR) and western blotting
Tumor tissues and cancer-adjacent normal tissues
were washed twice with ice-cold TBS, and RNA was extracted with
TRIzol reagent (Takara Bio, Inc., Otsu, Japan) according to the
manufacturer's instructions. VEGF (8 ng/ml)-treated MKN45 cells or
transfected MKN45 cells were used in PCR analysis. cDNA was
synthesized using a Mir-X™ miRNA FirstStrand Synthesis kit (cat no.
638315; Takara Bio, Inc.) according to the manufacturer's protocol.
A Mir-X™ miRNA RT-qPCR SYBR® kit (cat no. 638314; Takara
Bio, Inc.) was used to amplify mature miR-21. The sequences of the
PCR primers used are as follows: miR-21 forward,
5′-ACGTTGTGTAGCTTATCAGTG-3′ (the reverse primer was supplied in the
Mir-X™ miRNA RT-qPCR SYBR® kit); U6 forward,
5′-CTCGCTTCGGCAGCACA-3′ and reverse, 5′-AACGCTTCACGAATTTGCGT-3′.
The expression levels of miR-21 were normalized to the U6 RNA.
Thermocycling conditions were as follows and according to the
manufacturer's protocol: Denaturation at 95°C for 10 sec followed
by 40 cycles at 95°C for 5 sec and at 65°C for 20 sec. Dissociation
curve conditions were as follows; 95°C for 60 sec, 55°C for 30 sec
and 95°C for 30 sec. Data were analyzed using the 2−ΔΔCq
method (22).
For protein detection, the cells were lysed using
ice-cold RIPA buffer (cat no. 89900; Thermo Fisher Scientific,
Inc.) and centrifuged at 4°C, 12,000 × g, for 5 min. Protein
concentrations were determined using a BCA protein assay kit
(Beyotime Biotechnology, Shanghai, China). Samples were heated at
95°C for 5 min and then supersonicated. Total cellular proteins (30
µg/lane) were subjected to electrophoresis in 12% polyacrylamide
gel. The proteins in the gel were then transferred to a
polyvinylidene difluoride membrane. Following the transfer, the
membrane was blocked at 37°C for 1 h with 5% non-fat milk in
tris-buffered saline (pH 7.6), with 0.05% Tween-20. The blots were
then separately incubated with primary mouse anti-human β-actin
(1:4,000), PPARα (1:2,000), VEGFR (1:1,000), AKT (1:2,000) and PI3K
(1:2,000) antibodies, and then with HRP-conjugated goat anti-mouse
IgG secondary antibody (1:3,000). Chemiluminescence signals were
detected using an enhanced chemiluminescence western blotting kit
(cat no. 32106; Thermo Fisher Scientific, Inc.). Scanned western
blot images were analyzed semi-quantitatively using QuantityOne
software (cat no. 1709600; Bio-Rad Laboratories, Inc.). The
relative intensity values of bands were calculated using FluorChem
2.0 software (ProteinSimple, San Jose, CA, USA) and normalized to
β-actin. Each assay was performed in triplicate, and the results
were averaged over three independent experiments.
Luciferase activity assay
To elucidate the regulatory effects of miR-21 on
PPARα, bioinformatic methods were used to identify the targets of
miR-21 in GC cells. The binding target of miR-21 was predicted
using the online software Target Scan Human 7.0 (http://www.targetscan.org/vert_71/).
For the luciferase reporter assay, 2×105
MKN45 cells were seeded in 24-well plates, and PGL3-PPARα-3′UTR
plasmid and Renilla luciferase vector (cat no. E1751; Promega
Corporation, Madison, WI, USA) were used to co-transfect MKN45
cells for 48 h. Renilla luciferase was used as an internal
control for normalization. Luciferase activity was detected using
the Dual-Luciferase® Reporter Assay system (cat no.
E1910; Promega Corporation) according to the manufacturer's
protocol. In brief, the cells were co-transfected with 14 µg
pGL3-PPARα-3′UTR plasmid and 150 pmol either miR-21mimic, inhibitor
or negative control using Lipofectamine 2000. The cells were lysed
12 h after transfection, and luciferase activity was measured using
the Dual-Luciferase Reporter Assay system (Promega Corporation)
according to the manufacturer's protocol. Additionally, MKN45 cells
were transfected with miRNA mimics or inhibitor (150 pmol/ml) and
collected 48 h later for analysis of PPARα by western blotting.
Each assay was performed in triplicate, and the results were
averaged over three independent experiments.
Immunofluorescence and confocal
microscopy
MKN45 cells (1×106) were cultured in
culture dishes in RPMI-1640, and the cells were treated with
aspirin (1 mM) or apatinib (0.1 mM) for 24 h. The control cells
were treated with DMSO at 37°C. Immunofluorescence was performed as
reported by Zhang et al (23).
Briefly, the cells were fixed in 4% paraformaldehyde at room
temperature for 15 min and then washed with PBS, permeabilized with
0.1% Triton X-100 and subsequently blocked with 10% normal goat
serum for 1 h at 37°C. The cells were subsequently incubated for 1
h at 37°C with primary antibodies against PPARα (1:200), p-AKT
(1:200), p-VEGFR2 (1:100), p-PI3K (1:500) followed by 3 washes with
PBS. Subsequently, the cells were incubated with fluorescein
isothiocyanate-labeled goat anti-mouse IgG (1:300) and
phycoerythrin-labeled goat anti-rabbit IgG (1:200) at 37°C for 1 h.
The cells were then washed with PBS, and Hoechst (cat no.
23491-52-3; Sigma-Aldrich; Merck KGaA) staining was performed to
visualize the nuclei at room temperature for 10 min. The stained
cells were analyzed using confocal microscopy (magnification, ×600;
Leica Microsystems, Inc., Buffalo Grove, IL, USA).
Cell proliferation, migration,
invasion and colony formation assays
Briefly, cell proliferation was detected using the
Cell Proliferation Assay kit (Promega Corporation). The 50%
inhibitory concentration (IC50) values of aspirin and
apatinib were calculated using GraphPad Prism 5 (GraphPad Software,
Inc., La Jolla, CA, USA). MKN45 cells (1×105) were
cultured in Biocoat™ 24-well chambers (BD Biosciences,
San Jose, CA, USA) in RPMI-1640 medium and treated with aspirin (1
mM) or apatinib (0.1 mM) for 72 h. Migration assays were performed,
and cell invasion assays were performed using Biocoat Matrigel
invasion chambers with 8-µm pores and polycarbonate membranes (BD
Biosciences). The cells in the lower chamber were counted under a
light microscope (magnification, ×200) For the colony formation
assay, the MKN45 cells were cultured in 12-well plates (200
cells/well) and treated with aspirin (1 mM), apatinib (0.1 mM), or
aspirin (1 mM) together with apatinib (0.1 mM). Colonies >75 µm
in diameter or containing >50 cells were counted as 1 positive
colony. The cells were grown for 10 days (37°C with 5%
CO2), and colony formation was visualized with crystal
violet staining in less than 30 min at room temperature and counted
using an inverted light microscope. Plate clone formation
efficiency was calculated as (number of colonies/number of cells
inoculated) ×100. All experiments were performed according to the
manufacturer's protocol. Each assay was performed in triplicate,
and the results were averaged over three independent
experiments.
Statistical analysis
Statistical analysis was performed using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA) or GraphPad Prism 5 (GraphPad
Software, Inc.). Spearman's rank correlation coefficient analyses
were performed to test for a statistically significant positive or
negative correlation between VEGF, PPARα and miR-21 using GraphPad
Prism. Data are expressed as the mean ± standard deviation.
Statistical analysis of all examined variables was performed using
analysis of variance. Post-hoc t-tests were performed using an
unpaired Student's t-test or one-way analysis of variance.
P<0.05 was considered to indicate a statistically significant
result.
Results
VEGF and PPARα expression are
associated with miR-21 levels in patients with GC
The levels of miR-21 expression were determined in
GC and normal-adjacent tissue specimens by RT-qPCR. It was observed
that miR-21 expression levels were higher in GC samples compared
with cancer-adjacent normal tissues (Fig.
1A). Subsequently, ELISA was performed to quantify the
concentrations of VEGF and PPARα in the peripheral blood of gastric
cancer patients and healthy controls (Fig. 1B and C). The data show that VEGF
expression was higher in GC patients compared with individuals
without cancer. By contrast, the levels of PPARα in the peripheral
blood of GC patients were lower compared with healthy controls.
Next, the association between VEGF, PPARα and miR-21 expression was
assessed and R-values were evaluated using linear regression with
GraphPad Prism 5 software. It was identified thatmiR-21 levels were
positively correlated with levels of VEGF expression (Fig. 1D) but negatively correlated with
levels of PPARα expression (Fig.
1E).
VEGF inhibits PPARα by inducing miR-21
expression in GC cells
To investigate the effects of VEGF expression on the
levels of miR-21 and PPARα, the levels of miR-21 and PPARα in GC
cell lines (MKN1, MKN45, MKN74 and IM95) were quantified using
RT-qPCR and immunoblot analysis. The basal levels of miR-21 were
not significantly different across the four GC cell lines (Fig. 2A). However, three out of the four cell
lines (MKN1, MKN45 and MKN74) tested expressed high levels of PPARα
protein, with only IM95 cells expressing negligible levels of PPARα
(Fig. 2B). Furthermore, treatment of
MKN45 cells with VEGF (8 ng/ml) significantly increased miR-21
expression (Fig. 2C) but reduced the
levels of PPARα protein (Fig. 2D).
Subsequently, MKN45 cells were transfected with a miR-21 mimic to
determine whether miR-21 is able to regulate the expression of
PPARα. The expression of PPARα protein was significantly suppressed
by the miR-21 mimic, and PPARα expression increased when miR-21 was
inhibited (Fig. 2E). To confirm
whether VEGF is able to inhibit PPARα via induction of miR-21,
MKN45 cells were treated with VEGF alone, or a combination of VEGF
and amiR-21 inhibitor. Levels of PPARα protein were lower in cells
treated with VEGF alone but this inhibitory effect was attenuated
when treatment of VEGF was combined with that of the miR-21
inhibitor (Fig. 2F). Taken together,
these findings indicate that VEGF inhibits PPARα via induction of
miR-21.
 | Figure 2.VEGF inhibits PPARα expression by
inducing miR-21 in GC cells. (A) RT-qPCR was performed to detect
the baseline levels of miR-21 expression in four GC cell lines. (B)
Western blotting was performed to detect the baseline levels of
PPARα protein in four GC cell lines. (C) RT-qPCR was performed to
detect the effect of 8 ng/ml VEGF on the expression of miR-21 in
MKN45 cells. The control cells were treated with PBS.
***P<0.001, VEGF treated group vs. control group. (D) Western
blotting was used to assess the levels of PPARα protein in MKN45
cells treated with VEGF. The control cells were treated with PBS.
**P<0.01, VEGF treated group vs. control group. (E) Western
blotting was used to evaluate the levels of PPARα protein in MKN45
cells treated with a miR-21 mimic, a miR-21 inhibitor or negative
control. **P<0.01, miR-21 mimic group vs. negative control
group; miR-21 inhibitor group vs. miR-21 inhibitor inhibitor group.
(F) Western blotting was used to analyze the levels of PPARα
protein in MKN45 cells treated either with VEGF, or with VEGF
together with a miR-21 inhibitor. The control cells were treated
with PBS. *P<0.05, miR-21 inhibitor with VEGF group;
**P<0.01, VEGF group vs. control group. The results shown are
from three representative independent experiments. VEGF, vascular
endothelial growth factor; PPARα, peroxisome proliferator-activated
receptor-α; miR-21, microRNA-21; GC, gastric cancer; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction
analysis; SD, standard deviation. |
Aspirin and apatinib induced PPARα and
attenuated PI3K/AKT signaling in GC cells
To elucidate the regulatory effects of miR-21 on
PPARα, bioinformatic methods were used to identify the targets of
miR-21 in GC cells. Using this approach, the 3′UTR of PPARα was
identified to contain two miR-21 binding sites (Fig. 3A). A luciferase reporter assay was
subsequently performed to verify the functional interaction of
miR-21 with the PPARα 3′UTR, and it was demonstrated that miR-21
significantly inhibited the luciferase activity of the PPARα 3′UTR
luciferase reporter in MKN45 cells (Fig.
3B). Next, MKN45 cells were transfected with a PPARα expression
plasmid and found that PPARα overexpression was able to suppress
the levels of miR-21 expression (Fig.
3C).
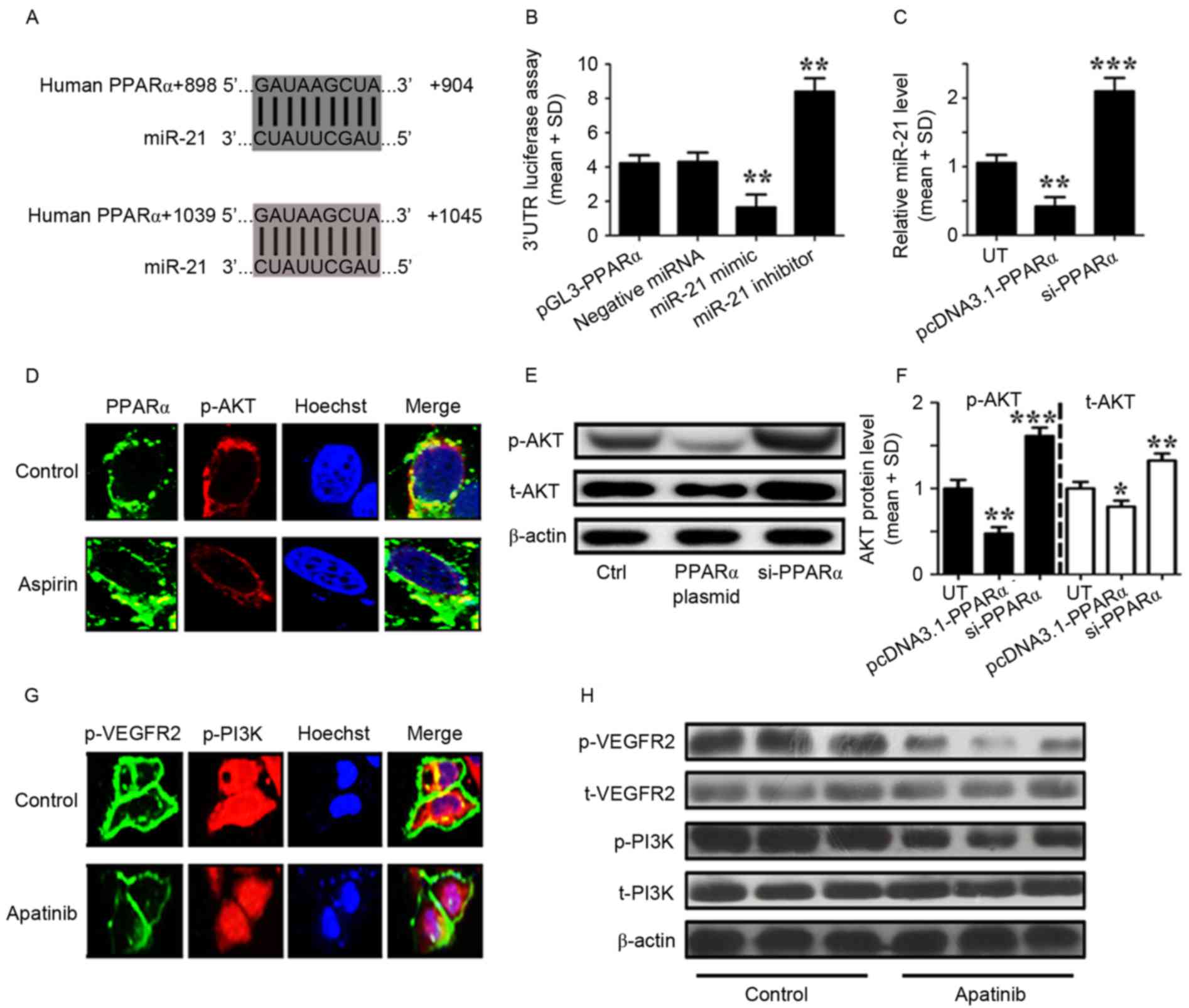 | Figure 3.Aspirin and apatinib induced PPARα
expression and attenuated PI3K/AKT signaling in GC cells,
respectively. (A) The PPARα 3′UTR contains binding sites for
miR-21. (B) The luciferase activity of MKN45 cells was measured
following co-transfection with the indicated PPARα 3′UTR reporter
constructs and a miR-21 mimic, miR-21 inhibitor or negative control
for 12 h. **P<0.01, miR-21 mimic group vs. negative miRNA group;
miR-21 inhibitor group vs. negative miRNA group. (C) RT-qPCR was
performed to detect the expression of miR-21 in MKN45 cells
transfected with PPARα plasmid or siPPARα for 24 h. **P<0.01,
pcDNA3.1-PPARα transfected group vs. UT group; ***P<0.001,
siPPARα transfected group vs. UT group. (D) Immunofluorescence
demonstrates the effects of aspirin on PPARα (green) and p-AKT
(red) expression in MKN45 cells. (E) Western blotting was used to
analyze the levels of p-AKT in MKN45 cells transfected with a PPARα
plasmid or siPPARα. (F) The levels of p-AKT relative to the
internal control (β-actin), according to western blotting results.
*P<0.05, pcDNA3.1-PPARα transfected group vs. UT. (G)
Immunofluorescence images showingthe effects of apatinib on
p-VEGFR2 (green) and p-PI3K (red) expression in MKN45 cells. (H)
Western blotting was used to analyze the levels of phosphorylated
and total VEGFR2 and PI3K in MKN45 cells treated with apatinib.
Control cells were treated with DMSO. The results depicted are from
three representative independent experiments. VEGF, vascular
endothelial growth factor; PPARα, peroxisome proliferator-activated
receptor-α; miR-21, microRNA-21; PI3K, phosphoinositide-3 kinase;
AKT, protein kinase B; 3′UTR, 3′ untranslated region; GC, gastric
cancer; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction analysis; SD, standard deviation; si, small
interfering; t, total; p, phosphorylated; UT, untreated cells. |
As aforementioned, aspirin and apatinib induce PPARα
expression and inhibit the phosphorylation of VEGFR2, respectively.
In addition, it was reported that apatinib may inhibit VEGFR2 as a
tyrosine kinase inhibitor (24).
However, the mechanisms by which aspirin and apatinib induce PPARα
expression remain to be fully understood. MKN45 cells were treated
with aspirin and immunofluorescence microscopy was performed to
visualize the effects of aspirin on the levels of PPARα and p-AKT.
Following aspirin treatment (Fig.
3D), PPARα expression was induced (green) and the levels of
p-AKT (red) were suppressed, compared with the control cells
(Fig. 3D). Subsequently, western blot
analysis was performed to quantify the levels of p-AKT protein in
MKN45 cells following PPARα overexpression or knockdown. PPARα
expression was able to reduce the levels of phosphorylated and
total AKT in MKN45 cells (Fig. 3E and
F). The effects of apatinib treatment on p-VEGFR2 and p-PI3K
were further assessed in MKN45 cells. Following apatinib treatment
(Fig. 3G), the levels of p-VEGFR2
expression in the cell membrane (green), as well as the levels of
p-PI3K (red) were suppressed compared with the control cells
(Fig. 3G). Finally, western blot
analysis was performed to quantify the levels of VEGFR2 and PI3K
protein in MKN45 cells (Fig. 3H).
Levels of total VEGFR2 and PI3K were affected by apatinib
treatment. However, the levels of p-VEGFER2 and p-PI3K were lower
following apatinib treatment. Taken together, these results suggest
that aspirin and apatinib induce PPARα and attenuate PI3K/AKT
signaling in GC cells, respectively.
Aspirin and apatinib inhibit
tumorigenesis of GC cells
Next, the roles of aspirin and apatinib in
tumorigenesis were evaluated. First, to evaluate the effects of
these compounds on viability of gastric cancer cells, cell
proliferation assay was performed using MKN45 cells.
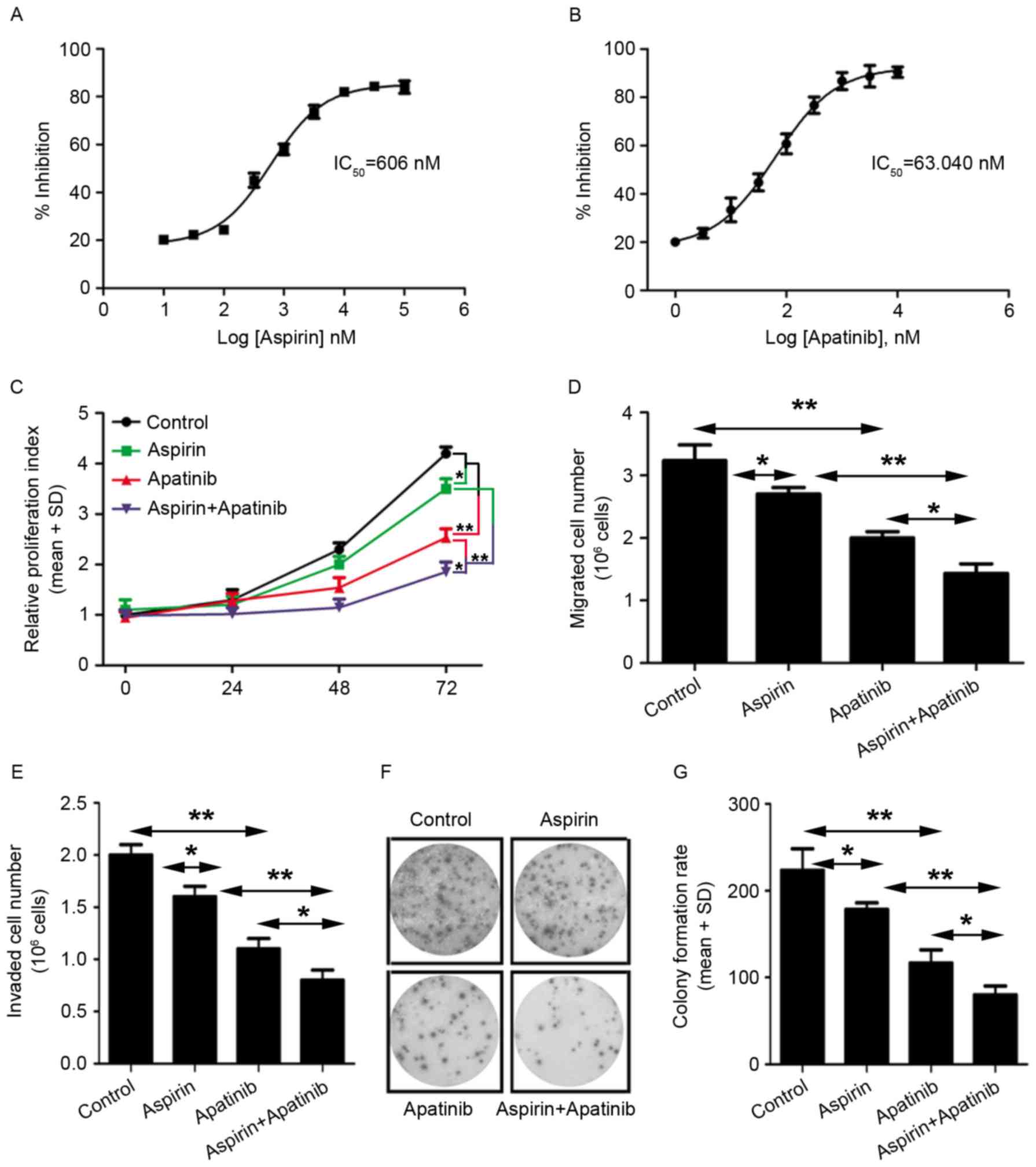
IC50 was calculated using GraphPad Prism 5 software. It
was observed that the cells were more sensitive to apatinib
treatment (IC50=63.04 nM) compared with aspirin
treatment (IC50=606 nM) (Fig.
4A and B). Next, the effect of aspirin and apatinib on MKN45
cell proliferation was assessed at various time-points.
Treatment of GC cells with a combination of aspirin
and apatinib inhibited cell proliferation, compared with treatment
with either aspirin or apatinib alone (Fig. 4C). Furthermore, the effect of aspirin
and apatinib on migration, colony formation and viability of MKN45
cells was evaluated (Fig. 4D-G). It
was observed that treatment with a combination of aspirin and
apatinib decreased migration (Fig.
4D), viability (Fig. 4E) and
colony formation (Fig. 4F and G) of
MKN45 cells. Taken together, these results reveal that apatinib
combined with aspirin is able to inhibit proliferation and
migration of gastric cancer cells, suggesting that these two agents
may have potential as a combination therapy in GC.
Discussion
In the present study, aspirin and apatinib were
demonstrated to exert antitumor effects in GC cells. Recently,
inhibition of angiogenesis has emerged as a potential therapeutic
strategy for GC (25). However,
anti-angiogenic agents, including bevacizumab, sunitinib and
sorafenib, have failed to increase patient survival (26). Apatinib, a novel tyrosine kinase
inhibitor that targets VEGFR2 kinase, has generated positive
results in initial preclinical and clinical studies involving GC
patients (26,27). There is dispute about the efficacy and
side effects of apatinib; it was associated with significant
survival prolongation compared with placebo in a chemorefractory
Chinese population (24). However,
contrasting results have been obtained in different populations
(28,29). Numerous studies have demonstrated that
p-VEGFR2 is able to activate the PI3K/AKT signaling pathway, which
regulates critical tumorigenic processes, including cellular
proliferation, survival, growth and motility (30). In addition, other studies have
indicated a role for miR-21 in cancer (11). Specifically, miR-21 was reported to
inhibit PPARα, a transcriptional activator and known regulator of
fatty acid metabolism (31). In
recent years, PPARα has also been demonstrated to mediate the
development of several types of cancer, including those of the
lung, liver and colon (19,32). Although VEGF and miR-21 serve
important functions in cancer development, the association between
them in GC remains to be fully understood. The present study
reported a novel treatment against gastric cancer that suggested
combining aspirin and apatinib might inhibit GC growth in
vitro. That aspirin, an inhibitor of the enzyme cyclooxygenase,
may be used as an antitumor drug is supported by the present study.
Aspirin may inhibit GC cell proliferation in vitro. Apatinib
may be used as a treatment for heavily pretreated patients with GC
by targeting VEGFR (33,34). However, VEGF, a ligand of VEGFR, may
promote GC cell proliferation by inhibiting PPARα. The present
study provided a novel strategy in which aspirin may be combined
with a certain specific antitumor drug to improve the curative
effect.
In the present study, miR-21expression and the
concentration of VEGF protein were higher in tissue from gastric
cancer patients, whereas the levels of PPARα were lower compared
with adjacent normal tissues. A correlation analysis revealed that
miR-21 levels were positively correlated with the levels of VEGF,
and negatively correlated with the levels of PPARα. In addition,
treatment of MKN45 GC cells with VEGF (8 ng/ml) significantly
increased the expression of miR-21. The level of PPARα protein was
significantly decreased by VEGF treatment, and this inhibitory
effect was attenuated by treatment with a miR-21 inhibitor.
Furthermore, two miR-21 binding sequences were identified in the
3′UTR of the PPARα gene. It was demonstrated that miR-21 was able
to directly suppress PPARα expression by binding to these sites.
Notably, miR-21 expression was decreased when PPARα was
overexpressed in MKN45 cells, whereas the levels of total and p-AKT
protein were significantly reduced. These results indicate the
presence of a signaling loop consisting of miR-21 and PPARα.
Specifically, miR-21 may repress PPARα by directly targeting its
3′UTR. In turn, decreased PPARα expression reduces the inhibition
of AKT activation and increases the expression of miR-21.
Ultimately, miR-21 promotes AKT signaling and increases the
inhibitory effects of miR-21 on PPARα.
To investigate the role of PPARα in gastric cancer,
MKN45 cells were treated with aspirin. In the clinic, aspirin is
widely used to treat fever, pain and inflammation (35), and may effectively prevent certain
types of cancer (36–38). However, the molecular mechanisms by
which aspirin alters cancer risk remain unknown. Recently, aspirin
was reported to be a potential PPARα activator (19). In agreement with these findings, the
results of the present study demonstrated that aspirin was able to
increase PPARα protein expression and reduce levels of p-AKT in
MKN45 cells. Additionally, the levels of p-VEGFR2 and p-PI3K were
decreased in apatinib-treated MKN45 cells. These results suggest
that treatment with aspirin and apatinib was able to increase PPARα
protein expression and reduce the levels of p-VEGFR2 and p-PI3K,
respectively, thereby blocking PI3K/AKT signaling. Finally, to
observe the effects of aspirin and apatinib on MKN45 cell
tumorigenesis, MKN45 cells were treated with aspirin or apatinib
alone, or the two compounds in combination. Following treatment
with aspirin and apatinib together, proliferation, migration,
invasion and colony formation of MKN45 cells were inhibited.
In summary, the results of the present study
indicate that aspirin and apatinib are able to stimulate PPARα and
inhibit VEGFR2 phosphorylation, respectively. Activated PPARα
reduced the levels of phosphorylated and total AKT, and decreased
the levels of miR-21 in GC cells. These effects attenuated the
inhibitory effect of miR-21 on PPARα, ultimately leading to
persistent PPARα-mediated inhibition of p-AKT. Treatment with
apatinib suppressed VEGFR2 phosphorylation, which further reduced
the protein levels of p-PI3K and p-AKT. In conclusion, aspirin and
apatinib exert antitumor effects by blocking PI3K/AKT signaling in
GC cells.
Acknowledgements
The authors would like to thank Dr Hanyang Hu (Wuhan
University, Wuhan, China) for providing the pGL3-PPARα-3′UTR
plasmid and analyzing miRNA targets and transfection, Dr Yushan Ren
(Lunan Pharmaceutical Group Co., Linyi, China) for discussion and
Dr Jie Li (Lunan Pharmaceutical Group Co.) for technical
support.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagner AD, Unverzagt S, Grothe W, Kleber
G, Grothey A, Haerting J and Fleig WE: Chemotherapy for advanced
gastric cancer. Cochrane Database Syst Rev. 17:CD0040642010.
|
|
3
|
Petrioli R, Francini E, Roviello F,
Marrelli D, Fiaschi AI, Laera L, Rossi G, Bianco V, Brozzetti S and
Roviello G: Sequential treatment with epirubicin, oxaliplatin and
5FU (EOF) followed by docetaxel, oxaliplatin and 5FU (DOF) in
patients with advanced gastric or gastroesophageal cancer: A
single-institution experience. Cancer Chemother Pharmacol.
75:941–947. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ebos JM, Bocci G, Man S, Thorpe PE,
Hicklin DJ, Zhou D, Jia X and Kerbel RS: A naturally occurring
soluble form of vascular endothelial growth factor receptor 2
detected in mouse and human plasma. Mol Cancer Res. 2:315–326.
2004.PubMed/NCBI
|
|
5
|
Hicklin DJ and Ellis LM: Role of the
vascular endothelial growth factor pathway in tumor growth and
angiogenesis. J Clin Oncol. 23:1011–1027. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: Structure,
function, intracellular signaling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Qin S, Xu J, Xiong J, Wu C, Bai Y,
Liu W, Tong J, Liu Y, Xu R, et al: Randomized, double-blind,
placebo-controlled phase III trial of apatinib in patients with
chemotherapy-refractory advanced or metastatic adenocarcinoma of
the stomach or gastroesophageal junction. J Clin Oncol.
34:1448–1454. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ilson DH: Targeting the vascular
endothelial growth factor pathway in gastric cancer: A hit or a
miss? J Clin Oncol. 34:1431–1432. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang H, Kim N, Park JH, Nam RH, Choi YJ,
Lee HS, Yoon H, Shin CM, Park YS, Kim JM and Lee DH: Different
microRNA expression levels in gastric cancer depending on
Helicobacter pylori infection. Gut Liver. 9:188–196. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suárez Y and Sessa WC: MicroRNAs as novel
regulators of angiogenesis. Circ Res. 104:442–454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu LZ, Li C, Chen Q, Jing Y, Carpenter R,
Jiang Y, Kung HF, Lai L and Jiang BH: miR-21 induced angiogenesis
through AKT and ERK activation and HIF-1α expression. PLoS One.
6:e191392011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang FS, Tian SS, Lu JJ, Ding XH, Qian
CD, Ding B, Ding ZS and Jin B: Cardamonin Regulates miR-21
expression and suppresses angiogenesis induced by vascular
endothelial growth factor. Biomed Res Int. 2015:5015812015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou J, Wang KC, Wu W, Subramaniam S, Shyy
JY, Chiu JJ, Li JY and Chien S: MicroRNA-21 targets peroxisome
proliferators-activated receptor alpha in an autoregulatory loop to
modulate flow-induced endothelial inflammation. Proc Natl Acad Sci
USA. 108:10355–10360. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kersten S, Desvergne B and Wahli W: Roles
of PPARs in health and disease. Nature. 405:421–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim JH, Kim T, Qu A and Gonzalez FJ:
Abstract 23: Nuclear receptor PPARα activation triggers hepatic
cell death in Ikkβ-deficient mice. Cancer Res. 75:232015.
View Article : Google Scholar
|
|
17
|
Aboud Abu O, Donohoe D, Bultman S, Fitch
M, Riiff T, Hellerstein M and Weiss RH: PPARα inhibition modulates
multiple reprogrammed metabolic pathways in kidney cancer and
attenuates tumor growth. Am J Physiol Cell Physiol. 308:C890–C898.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chandran K, Goswami S and Sharma-walia N:
Implications of a peroxisome proliferator-activated receptor alpha
(PPARα) ligand clofibrate in breast cancer. Oncotarget.
7:15577–15599. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Skrypnyk N, Chen X, Hu W, Su Y, Mont S,
Yang S, Gangadhariah M, Wei S, Falck JR, Jat JL, et al: PPARα
activation can help prevent and treat non-small cell lung cancer.
Cancer Res. 74:621–631. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Planagumà A, Titos E, López-Parra M, Gaya
J, Pueyo G, Arroyo V and Clària J: Aspirin (ASA) regulates
5-lipoxygenase activity and peroxisome proliferator-activated
receptor alpha-mediated CINC-1 release in rat liver cells: Novel
actions of lipoxin A4 (LXA4) and ASA-triggered 15-epi-LXA4. FASEB
J. 16:1937–1939. 2002.PubMed/NCBI
|
|
21
|
Søgaard KK, Farkas DK, Pedersen L, Lund
JL, Thomsen RW and Sørensen HT: Long-term risk of gastrointestinal
cancers in persons with gastric or duodenal ulcers. Cancer Med.
5:1341–1351. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SS, Nam JS, Cho HJ, Won JH, Kim JW, Ji
JH, Yang MJ, Park JH, Noh CK, Shin SJ, et al: Plasma microRNA-122
as a predictive marker for treatment response following
transarterial chemoembolization in patients with hepatocellular
carcinoma. J Gastroenterol Hepatol. 32:199–207. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang W and Li Y: miR-148a downregulates
the expression of transforming growth factor-β2 and SMAD2 in
gastric cancer. Int J Oncol. 48:1877–1885. 2016.PubMed/NCBI
|
|
24
|
Fontana E and Smyth EC: Novel targets in
the treatment of advanced gastric cancer: A perspective review.
Ther Adv Med Oncol. 8:113–125. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cancer Genome Atlas Research Network:
Comprehensive molecular characterization of gastric adenocarcinoma.
Nature. 513:202–209. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roviello G, Petrioli R, Marano L, Polom K,
Marrelli D, Perrella A and Roviello F: Angiogenesis inhibitors in
gastric and gastroesophageal junction cancer. Gastric Cancer.
19:31–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y,
Li J and Lou L: YN968D1 is a novel and selective inhibitor of
vascular endothelial growth factor receptor-2 tyrosine kinase
withpotent activity in vitro and in vivo. Cancer Sci.
102:1374–1380. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lazăr DC, Tăban S, Cornianu M, Faur A and
Goldiş A: New advances in targeted gastric cancer treatment. World
J Gastroenterol. 22:6776–6799. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fornaro L, Vasile E and Falcone A:
Apatinib in advanced gastric cancer: A doubtful step forward. J
Clin Oncol. Aug 15–2016.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: Rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmed W, Ziouzenkova O, Brown J, Devchand
P, Francis S, Kadakia M, Kanda T, Orasanu G, Sharlach M, Zandbergen
F and Plutzky J: PPARs and their metabolic modulation: New
mechanisms for transcriptional regulation? J Intern Med.
262:184–198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gonzalez FJ and Shah YM: PPARalpha:
Mechanism of species differences and hepatocarcinogenesis of
peroxisome proliferators. Toxicology. 246:2–8. 2012. View Article : Google Scholar
|
|
33
|
Hu X, Zhang J, Xu B, Jiang Z, Ragaz J,
Tong Z, Zhang Q, Wang X, Feng J, Pang D, et al: Multicenter phase
II study of apatinib, a novel VEGFR inhibitor in heavily pretreated
patients with metastatic triple-negative breast cancer. Int J
Cancer. 135:1961–1969. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li J, Qin S, Xu J, Guo W, Xiong J, Bai Y,
Sun G, Yang Y, Wang L, Xu N, et al: Apatinib for
chemotherapy-refractory advanced metastatic gastric cancer: results
from a randomized, placebo-controlled, parallel-arm, phase II
trial. J Clin Oncol. 31:3219–3225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sweetman SC: Martindale: The Complete Drug
Reference. 37th. Pharmaceutical Press; London, UK: 2011
|
|
36
|
Algra AM and Rothwell PM: Effects of
regular aspirin on long-term cancer incidence and metastasis: A
systematic comparison of evidence from observational studies versus
randomised trials. Lancet Oncol. 13:518–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rothwell PM, Price JF, Fowkes FG,
Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M,
Mehta Z and Meade TW: Short-term effects of daily aspirin on cancer
incidence, mortality, and non-vascular death: analysis of the time
course of risks and benefits in 51 randomised controlled trials.
Lancet. 379:1602–1612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: A study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|