Introduction
Microsatellite instability (MSI) status has drawn
attention as a guide to immunotherapy against different types of
tumor (1). Immune checkpoint
inhibitors represent a significant advance in precision medicine,
inducing durable tumor responses even in patients with late-stage
cancer who have failed to respond to multiple previous lines of
therapy (1,2). Anti-programmed death (PD)-1 inhibitors,
including pembrolizumab, are humanized monoclonal antibodies which
block the interaction between PD-1 and its ligands, PD-L1 and
PD-L2, and allow T cells to kill the tumor cells (1).
Notably, a phase II study (NCT01876511) in
metastatic carcinomas demonstrated that the MSI phenotype
constituted an important biomarker for patient response to
immunotherapy (1). Most notably, the
study revealed that immune checkpoint proteins, including PD-1 and
PD-L1, were significantly upregulated in tumors with MSI, enabling
them to survive. In MSI colorectal cancer (CRC), PD-L1 is expressed
on tumor-infiltrating lymphocytes and/or myeloid cells as opposed
to tumor cells (1,2).
MSI is characterized by widespread somatic
alterations in the length of nucleotide repeat sequences, which are
known as microsatellites (3). The MSI
phenotype is a marker of defects in the DNA mismatch repair (MMR)
system during DNA replication (3,4). The MSI
phenotype is present in all cases of hereditary nonpolyposis CRC
syndrome, as well as ~15% of sporadic CRC, while it is less
frequently observed in other tumors, including gastric, biliary
tract, pancreas, ovary, prostate and small intestine tumors
(1,5).
In CRC, the presence of MSI is also associated with a number of
clinicopathological features, including proximal location,
poorly-differentiated tumors, low frequency of distant metastases
and an improved prognosis (6).
Gastrointestinal stromal tumors (GISTs) are the most
common mesenchymal neoplasms of the gastrointestinal tract
(7), with a global annual incidence
of 11–18 per million (8,9). GISTs are considered to originate from
the interstitial cells of Cajal, or a common stem/precursor cell
(8,10), and usually arise in the stomach
(40–70%) or small intestine (20–40%), and less frequently in the
esophagus, colon and rectum (8,11). GISTs
also occur elsewhere within the abdominal cavity, primarily in the
omentum, mesentery or retroperitoneum (<5% of all GISTs), and
these are referred to as extra-gastrointestinal tract tumors
(12,13). Histologically, the spectrum of
morphology includes spindle, epithelioid or mixed cells (14).
The malignant potential of GISTs ranges from
entirely benign to aggressive tumors. However, ~40% of GISTs that
are localized at the time of diagnosis eventually metastasize
(13). The metastatic dissemination
has a predilection to the liver, omentum, peritoneum and other
intra-abdominal sites (13). The
prognosis of patients with GISTs is based on criteria established
by the Armed Forces Institutes of Pathology (AFIP) criteria
(15), including tumor location, size
and mitotic index. This criterion ranks the patients as benign,
very low, low, intermediate and high risk (8).
The majority of GISTs are positive for the
proto-oncogene receptor tyrosine kinase (KIT) protein (anti-CD117
is used to identify KIT), and this positivity acts as a crucial
diagnostic marker for these tumors (8,16). KIT is
a member of the type III receptor tyrosine kinase family, and the
binding of its growth factor, stem cell factor (SCF), to the
extracellular domain results in dimerization of the receptor and
downstream activation of mitogen-activated protein kinase,
phosphatidylinositol 3-kinase and Janus kinase/signal transducers
and activators of transcription pathways (13).
KIT gene mutations are present in 70–80% of
GIST cases (17). These oncogenic
mutations result in the constitutive activation of the receptor and
consequently, the activation of intracellular pathways (17). KIT mutations typically affect
the juxtamembrane domain encoded by exon 11 (70% of cases), the
extracellular domain encoded by exon 9 (6–15%) and the kinase I and
II domains encoded by exons 13 and 17 (2%) (17,18). In
particular, deletions have been associated with a worse clinical
outcome compared with other types of exon 11 mutation, with shorter
progression-free and overall survival times (9). In addition, GISTs harboring KIT
exon 9 mutations are characterized by small bowel location,
aggressive clinical characteristics (9,19) and
decreased sensitivity to first line therapy compared with
KIT exon 11 mutant tumors (9).
Another member of the tyrosine kinase receptor
family, platelet-derived growth factor receptor α (PDGFRA), is also
involved in GIST pathogenesis (16,20).
Mutations in the PDGFRA gene occur in 5–7% of cases, in
domains which are similar to those in the KIT gene (16,21). GISTs
harbor mutations in the PDGFRA juxtamembrane domain (encoded
by exon 12), the ATP-binding domain (encoded by exon 14) or the
activation loop (encoded by exon 18) (21). The majority of GISTs with
mutated-PDGFRA have a distinct phenotype, including gastric
location, epithelioid morphology, variable/absent KIT expression as
determined by immunohistochemistry and an indolent clinical course
(22). In addition, mutations in exon
18 of PDGFRA are associated with a lack of response to
imatinib therapy (21). Consistent
with their functional overlap, KIT and PDGFRA
mutations are mutually exclusive in GISTs (8,16).
Between 10–15% of GISTs are KIT or
PDGFRA wild-type (22). These
tumors form a heterogeneous group, a number of which are driven by
oncogenic mutations acting downstream of the receptor kinases, such
as B-Raf proto-oncogene, serine/threonine kinase (BRAF)
mutations (described in 1.3% of all tumors) (23,24). A
previous study demonstrated that wild-type GISTs exhibit a
different genetic background, including mutation in succinate
dehydrogenase (21). In either of
these cases, there is poor response to first line therapy (9).
Molecular-targeted agents are being utilized as
first line treatment for GISTs, including imatinib mesylate and
sunitinib maleate. These two agents are KIT/PDGFRA competitive
inhibitors that stabilize the inactivated form of the receptors,
inhibiting downstream signaling activation (25–27). The
median survival time for patients with advanced disease treated
with imatinib is 5 years, with 34% of patients surviving >9
years (8). Despite this, the vast
majority (≥80%) of patients eventually develop secondary resistance
(13). Acquired mutations in
KIT or PDGFRA usually occur in the kinase domain and
interfere with drug binding, causing resistance (9,21). The
majority of mutations in exon 9 are 6-nucleotide duplications
encoding Ala502-Tyr503, which require twice the normal dose of
imatinib (800 mg/day) for optimal clinical results. In the
PDGFRA gene, the most common mutation is a missense mutation
in exon 18, which leads to substitution of Asp to Val (termed
D842V) (19,28). This mutation is usually resistant to
treatment with imatinib (19,28).
In GISTs, the characterization of MSI is limited and
the results are controversial (29–31).
Therefore, the present study aimed to assess the presence and
frequency of MSI using an accurate methodology in a series of 88
Brazilian GISTs, and investigated the association with
clinicopathological features of patients.
Patients and methods
Patient population and tissue
samples
The present study analyzed 88 patients submitted to
resection at Barretos Cancer Hospital (São Paulo, Brazil) between
January 1989 and December 2012. A total of 79 primary GISTs were
included in the KIT/PDGFRA molecular test and MSI analysis. The
other 9 cases were excluded due to poor DNA quality and lower
quantity. Clinicopathological data of patients were retrospectively
obtained, including age, sex, tumor localization and risk
classification (according to AFIP criteria), local disease
recurrence, metastasis, chemotherapy and follow-up status (as of
March 2015). In addition, information concerning GIST molecular
status (KIT, PDGFRA and BRAF mutations) was
previously reported for 60 cases (32,33). The
clinical and molecular data are summarized in Table I.
 | Table I.Clinicopathological features of
gastrointestinal stromal tumors. |
Table I.
Clinicopathological features of
gastrointestinal stromal tumors.
| Variable | Patients, n
(%) |
|---|
| Sex |
|
|
Female | 41 (46.6) |
|
Male | 47 (53.4) |
| Histological
subtype |
|
|
Spindle | 67 (81.7) |
|
Epithelioid | 12 (14.6) |
|
Mixed | 3 (3.7) |
| Primary
localization |
|
|
Esophagus | 1 (1.1) |
|
Stomach | 44 (50.0) |
| Small
intestine | 25 (28.4) |
|
Rectum | 6 (6.9) |
|
Mesentery | 1 (1.1) |
|
Retroperitoneum | 6 (6.9) |
|
Colon | 1 (1.1) |
|
Othersa | 4 (4.5) |
| Tumor size |
|
| ≤5
cm | 28 (37.3) |
| 5.1–10
cm | 22 (33.3) |
| >10
cm | 25 (29.3) |
| Mitotic index |
|
| ≤5 | 39 (58.2) |
|
>5 | 25 (37.3) |
|
6–10 | 3 (4.5) |
| AFIP risk
classification |
|
|
Benign | 7 (11.3) |
| Very
low | 7 (11.3) |
|
Low | 7 (11.3) |
|
Intermediate | 9 (14.5) |
|
High | 32 (51.6) |
| Imatinib |
|
|
Yes | 44 (95.7) |
| No | 2 (4.3) |
| Local disease
recurrence |
|
|
Absent | 66 (77.6) |
|
Present | 19 (22.4) |
| Metastasis |
|
|
Absent | 47 (54.7) |
|
Present | 39 (45.5) |
|
KIT/PDGFRA/BRAF mutation
status |
|
|
KIT | 66 (83.6) |
|
PDGFRA | 8 (10.1) |
|
BRAF | 0 (0.0) |
|
Wild-type | 5 (6.3) |
| Current status |
|
|
Mortality due to cancer | 28 (31.8) |
| Current status |
|
|
Mortality due to other
causes | 2 (2.3) |
| Alive
with cancer | 27 (30.7) |
| Alive
without cancer | 28 (31.8) |
The average age of the individuals was 57±12.4 years
old. The most common histological subtype was spindle cells, and
the most common primary localization was the stomach, followed by
small intestine, rectum and retroperitoneum, (Table I). The tumors were classified as high
risk in 51.6% of cases and the majority of patients were treated
with an oral administration of 400 mg of imatinib. Only 2 patients
were treated with 5-fluorouracil and/or etoposide. The majority of
patients (54.7%) did not experience local recurrence or metastasis.
Of those that did, liver (66.7%) and lung (7.7%) were the most
common sites of metastasis (Table
I).
The present study was approved by the local ethical
committees (approval no. 554/2011) of Barretos Cancer Hospital. The
ethics committee of our institution authorized that no patient
consent was required due to the retrospective nature of the
study.
DNA isolation
DNA from samples that had been fixed in 10% formalin
for 12–24 h at room temperature and then paraffin-embedded was
retrieved from 5-µm cuts, following careful macrodissection of the
tumor area and ensuring the presence of >75% of neoplastic
cells. DNA extraction was performed using the QIAamp DNA Micro kit
(Qiagen, Inc., Valencia, CA, USA), following the manufacturer's
protocol, quantified by NanoDropVR 2000 (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and stored at −20°C until subsequent
genetic analysis.
KIT/PDGFRA/BRAF mutations
KIT and PDGFRA mutational status was
analyzed by polymerase chain reaction (PCR) amplification and
subsequent DNA sequencing of exons 9, 11, 13 and 17 to KIT
and exon 12, 14 and 18 to PDGFRA, as previously described
(32,33).
Tumors with wild-type KIT and PDGFRA
mutations were analyzed for the presence of exon 15 BRAF
V600E mutations as previously described (24). The quality of PCR products was
confirmed with 1% agarose gel electrophoresis. DNA sequencing of
the PCR product was performed using the BigDye Terminator version
3.1 cycle sequencing kit (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and an ABI 3500XL Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.) in accordance with
manufacturer's protocol.
MSI analysis
The MSI evaluation was performed using a multiplex
PCR comprising five quasi-monomorphic mononucleotide repeat markers
(BAT-25, BAT-26, NR-21, NR-24 and NR-27) as previously reported
(34–36). The primer sequences used were
described in previous studies (34,35). Each
antisense primer was end labeled with a fluorescent dye:
6-carboxyfluorescein for BAT-26 and NR-21;
2′-chloro-7′-phenyl-1,4-dichloro-6-carboxyfluorescein for BAT-25
and NR-27; and
2,7,8-benzo-5-fluoro-2,4,7-trichloro-5-carboxyfluorescein for
NR-24. PCR was performed using the Qiagen Multiplex PCR kit
(Qiagen, Inc.), with 1 µl DNA at 50 ng/ml and the following
thermocycling conditions: 15 min at 95°C; 40 cycles of 95°C for 30
sec; 55°C for 90 sec and 72°C for 30 sec; and a final extension at
72°C for 40 min. PCR products were then submitted to capillary
electrophoresis on an ABI 3500XL Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and the results were analyzed using
GeneMapper v4.1 software (Applied Biosystems; Thermo Fisher
Scientific, Inc.). In all analyses, the DNA from the HCT-15 cell
line (ATCC® CCL-225™; American Type Culture Collection,
Manassas, VA, USA) (MSI-high) was used as a positive control for
MSI.
A previous study by our group determined the
quasimonomorphic variation range of each marker for the Brazilian
population (36). Accordingly,
samples were considered MSI-high when two or more markers were
altered, MSI-low when one marker was altered and microsatellite
stable (MSS) in the absence of instability. In the MSI-low cases,
validation by analysis of normal tissue or the immunohistochemistry
of the MMR enzymes in tumor tissue was recommended (36).
MMR immunohistochemistry
Briefly, 10% formalin fixed (for 12–24 h at room
temperature) paraffin-embedded tissue specimens were cut into 4-µm
sections, which were deparaffinized by heating (75°C for 4 min) and
then were transferred to Autostainer Link 48 equipment (Dako;
Agilent Technologies, Inc. Santa Clara, CA, USA) (37). The antigen retrieval process was
performed in Tris-EDTA buffer (pH 9.0) at 97°C for 20 min. The
EnVision™ FLEX Wash Buffer (Dako; Agilent Technologies, Inc.)
contained Tris with Tween-20 (pH 7.6). Endogenous peroxidases were
blocked at room temperature with EnVision™ FLEX Peroxidase-Blocking
reagent for 20 min. The primary rabbit polyclonal anti-human
antibodies used in the present study were as follows: Anti-mutL
homolog 1 (MLH1; dilution, 1:100; clone G168-728, ref. 285M-1);
anti-mutS homolog 2 (MSH2; dilution, 1:100; clone G219-1129, ref.
286M-1); anti-PMS1 homolog 2, mismatch repair system component
(PMS2; dilution, 1:25; clone EPR3947, ref. 288R-1); and anti-mutS
homolog 6 (MSH6; dilution, 1:600; clone 44, ref. 287M-1). All
primary antibodies were obtained from Dako (Agilent Technologies,
Inc.) and were incubated at room temperature for 20 min. The
secondary antibody was the EnVision™ FLEX/horseradish peroxidase
anti-rabbit and anti-mouse IgG (<10 µg/ml) in 10% animal serum
in TBS (ref. RE7111; Agilent Technologies, Inc.), which was
incubated at room temperature with the samples for 20 min. EnVision
DAB solution was used for immunostaining visualization, and was
incubated at room temperature with the samples for 10 min. Slides
were counterstained with Hematoxylin of Harris (EP-101071;
EasyPath, São Paulo, Brazil) at room temperature for 5 min,
according to manufacturer's protocol. A light microscope was used
to analyze all specimens at magnification, ×100–400.
Statistical analysis
Associations between molecular and clinical data
from patients were analyzed using the χ2 test or
Fisher's test. Cumulative survival probabilities were calculated
using the Kaplan-Meier method. Differences between survival rates
were tested with the log-rank test. SPSS 19.0 software (IBM SPSS,
Armonk, NJ, USA) was used for all statistical analysis. P<0.05
was considered to indicate a statistically significant
difference.
Results
Molecular and clinical profile of
GISTs
Of the 88 GISTs analyzed, 9 cases were excluded due
to poor DNA quality and lower quantity, leaving a total of 79 GIST
cases. KIT mutations were observed in 83.6% (66/79) of cases
and 10.1% (8/79) of cases exhibited PDGFRA mutations
(Table I). None of the remaining
cases (n=5) exhibited BRAF mutations, leading to a frequency
of 6.3% (5/79) wild-type cases. The KIT mutation was located
at exon 11 in 58 cases (87.9%), exon 9 in 6 cases (9.1%) and exon
17 in 2 cases (3.0%) Fig. 1 depicts a
representative electropherogram of a mutation in exon 11. Regarding
PDGFRA, 5 cases were mutated at exon 18 (62.5%), 1 case was
mutated at exon 12 (12.5%) and 2 cases were mutated at exons 12 and
18 (25.0%).
The associations between KIT/PDGFRA mutation
status and GIST clinicopathological features are listed in Table II. All PDGFRA-mutated GISTs
had a gastric location and PDGFRA-mutation status was
significantly associated with lower mitotic index (P=0.018;
Table II). The average follow-up
period was 4.3±3.2 years, and 87.5% of patients with PDGFRA
mutations were alive with no evidence of cancer, compared with
25.4% of patients with KIT-mutations (P=0.010). All
KIT exon 9-mutated cases exhibited tumor progression
following imatinib treatment, while 44.4% of the KIT exon
11-mutated cases had stable disease subsequent to chemotherapy
(data not shown).
| Table II.Association between KIT/PDGFRA
mutation status and clinicopathological features of
gastrointestinal stromal tumors. |
Table II.
Association between KIT/PDGFRA
mutation status and clinicopathological features of
gastrointestinal stromal tumors.
| Variable | KIT
mutation, n (%) | PDGFRA
mutation, n (%) | Wild-type, n
(%) |
P-valuea |
|---|
| Sex |
|
|
| 1.000 |
|
Female | 32 (48.5) | 4 (50.0) | 2 (40.0) |
|
|
Male | 34 (51.5) | 4 (50.0) | 3 (60.0) |
|
| Primary
localization |
|
|
| 0.398 |
|
Esophagus | 1 (1.5) | 0 | 0 |
|
|
Stomach | 29 (43.9) | 8 (100) | 4 (80) |
|
| Small
intestine | 20 (30.3) | 0 | 0 |
|
|
Rectum | 5 (7.6) | 0 | 1
(20) |
|
|
Mesentery | 1 (1.5) | 0 | 0 |
|
|
Retroperitoneum | 6 (9.1) | 0 | 0 |
|
|
Other | 4 (6.1) | 0 | 0 |
|
| Tumor size |
|
|
| 0.963 |
| ≤5
cm | 19 (37.3) | 4 (50.0) | 2 (50.0) |
|
| 5.1–10
cm | 13 (25.5) | 2 (25.0) | 1 (25.0) |
|
| >10
cm | 19 (37.3) | 2 (25.0) | 1 (25.0) |
|
| Mitotic index |
|
|
| 0.018 |
| ≤5 | 24 (51.1) | 6 (75.0) | 3 (60.0) |
|
| 5.1–10
cm | 1 (2.1) | 2 (25.0) | 0 (0.0) |
|
|
>10 | 22 (46.8) | 0 (0.0) | 2 (40.0) |
|
| AFIP risk
classification |
|
|
| 0.198 |
|
Benign | 3 (7.1) | 1 (12.5) | 1 (20.0) |
|
| Very
low | 5 (11.9) | 2 (25.0) | 0 (0.0) |
|
|
Low | 5 (11.9) | 1 (12.5) | 0 (0.0) |
|
|
Intermediate | 4 (9.5) | 2 (25.0) | 2 (40.0) |
|
|
High | 25 (59.5) | 2 (25.0) | 2 (40.0) |
|
| Metastasis |
|
|
| 0.097 |
|
Absent | 34 (52.3) | 7 (87.5) | 4 (80.0) |
|
|
Present | 31 (47.7) | 1 (12.5) | 1 (20.0) |
|
| Status at last
follow-up |
|
|
| 0.010 |
| Alive
without cancer | 16 (25.4) | 7 (87.5) | 3 (60.0) |
|
| Alive
with cancer | 24 (38.1) | 0 (0.0) | 0 (0.0) |
|
|
Mortality due to cancer | 21 (33.3) | 1 (12.5) | 2 (40.0) |
|
|
Mortality due to other
causes | 2 (3.2) | 0 (0.0) | 0 (0.0) |
|
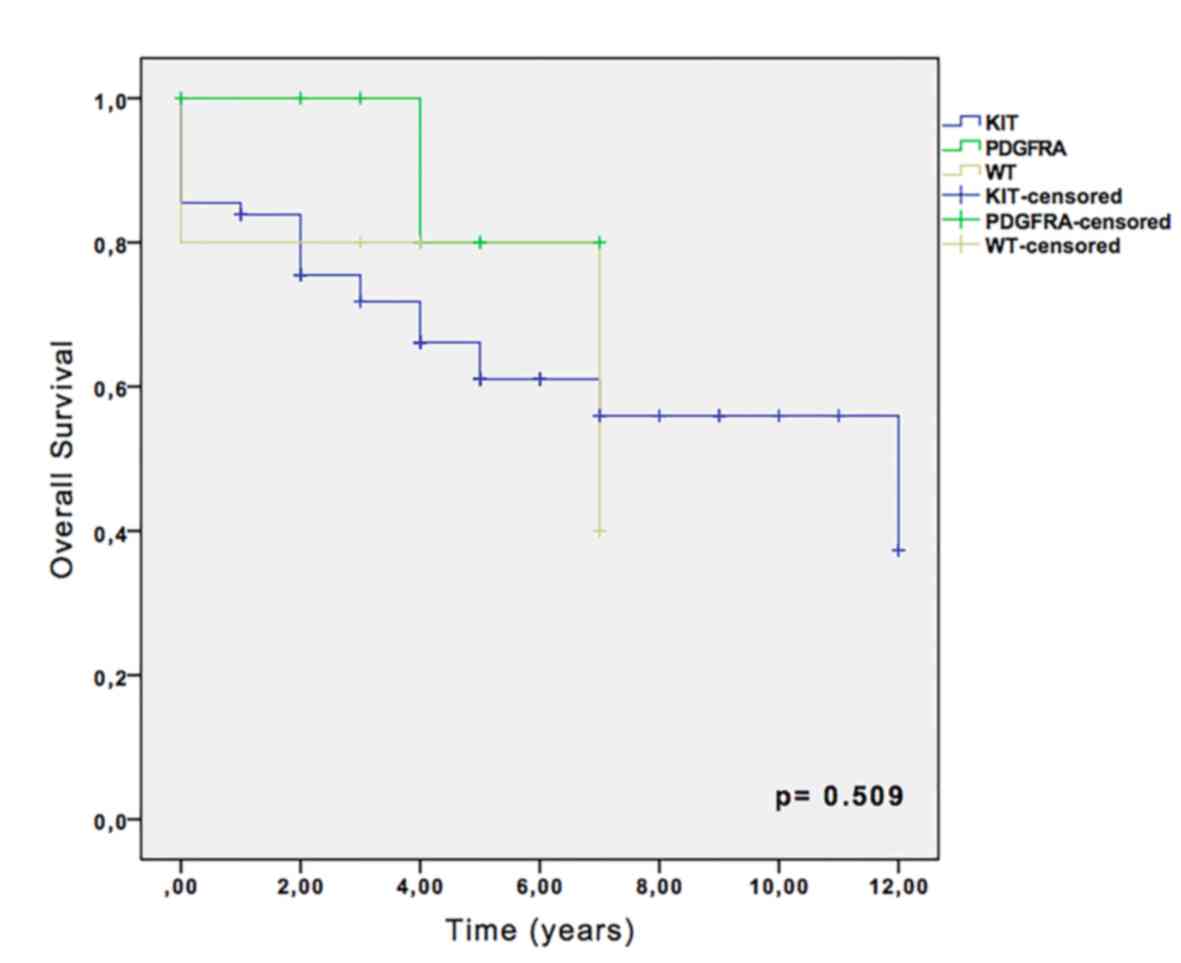
Kaplan-Meier survival analysis revealed that,
despite the absence of statistical significance, the 5-year overall
survival rate was 66.1% for KIT-mutated cases, and 80% for
PDGFRA and wild-type cases (Fig.
2). No significance was observed in recurrence-free survival
analysis among KIT, PDGFRA and wild-type groups (data
not shown).
MSI analysis
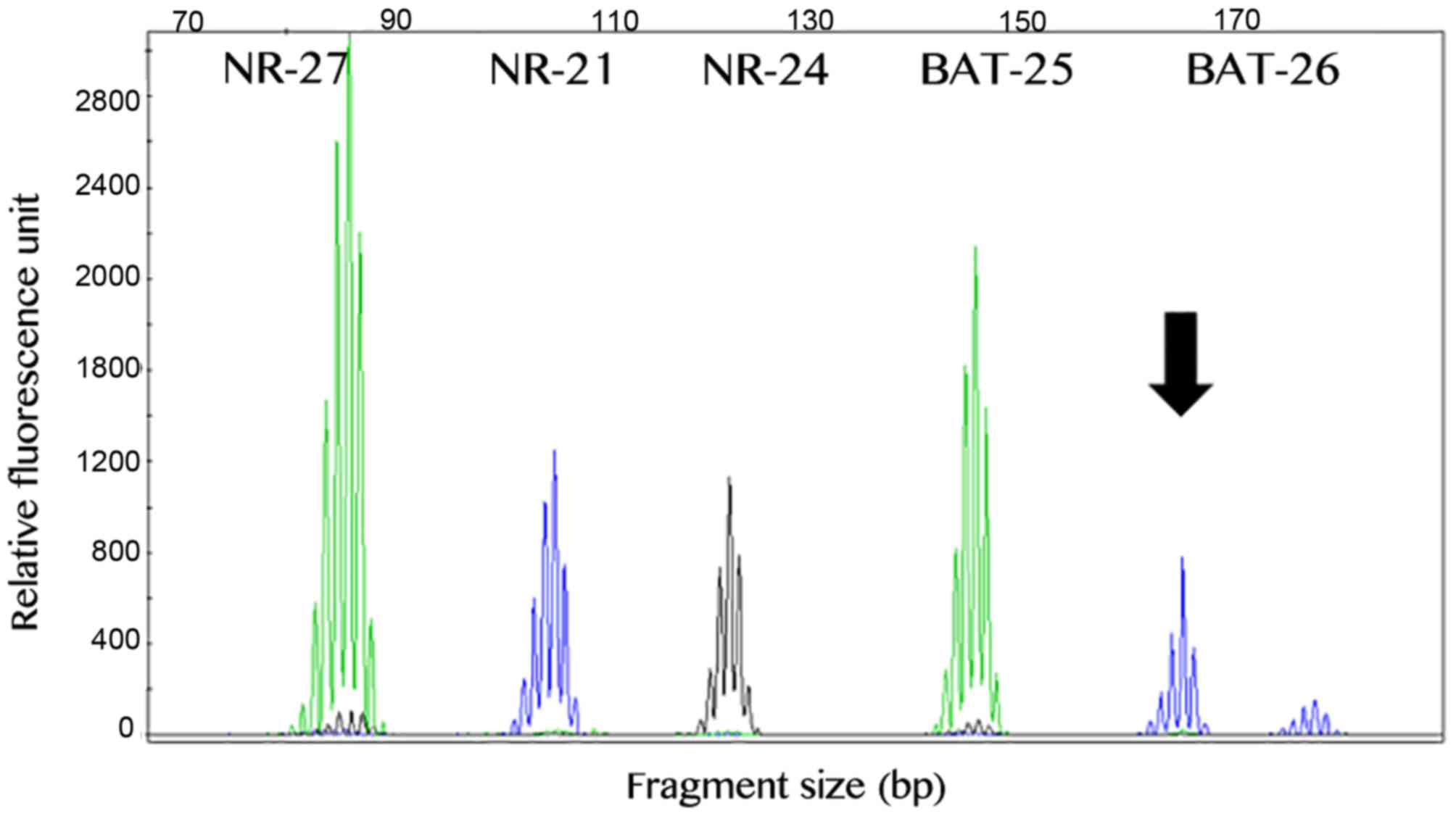
The MSI analysis was successful in all 79 GIST
cases. A total of 75 (~95%) samples exhibited a stable profile,
while 4 primary GISTs exhibited instability in one marker. In
total, 2 cases exhibited alteration of the BAT-26 marker, 1 case
exhibited alteration of the NR-21 marker and 1 case demonstrated
instability in the BAT-25 marker (Fig.
3). Our previous study reported that the presence of
instability in one marker in the Brazilian population may be due to
polymorphic variants (36).
Therefore, it was proposed that analysis of the MMR
immunohistochemistry or the MSI analysis of paired normal DNA
should be performed for these cases to accurately determine the MSI
status of these patients. The investigation of MMR
immunohistochemistry revealed positive staining for all MMR (MLH1,
MSH2, MSH6 and PMS2) proteins analyzed (Fig. 4). In addition, the MSI analysis of
paired normal DNA in all 4 cases revealed the same genotype in
normal and tumor DNA. Thus, these results indicated that all 4
cases were MSS.
Discussion
Determination of MSI status appears to be a marker
for novel treatments, and it may serve as a predictive marker for
the selection of patients who may benefit from pembrolizumab, an
anti-PD-1 immunotherapy (1). The data
from this phase II trial support the hypothesis that MMR-deficient
tumors are more responsive to PD-1 blockade compared with
MMR-proficient tumors (1). However,
there is still no data on clinical trials evaluating PD-1 agents in
GISTs, despite the growing interest.
The MSI phenotype in GISTs is poorly-characterized
and reports are not consensual. In the present study, MSI was
analyzed in 79 GIST samples using a multiplex PCR comprising five
quasi-monomorphic mononucleotide repeat markers. In the 4 cases
that exhibited alteration in only one marker, MSI analysis was
performed in paired normal DNA and MMR immunohistochemistry was
performed, which revealed the MSS nature of these samples.
Therefore, MSI was not present in the present series of GISTs.
These findings are in accordance with the first study addressing
the presence of MSI in GISTs by Lopes et al (31), which analyzed 33 GISTs. However, other
authors reported the presence of MSI in 5% (3/62) and 50% (10/22)
of cases (29,30).
It was proposed that these discrepant results may
have several causes. First, the number of cases analyzed in the
aforementioned two studies was too small for consistent results
(27,28). The present study examined 79 cases,
which is the largest series that has undergone MSI status
evaluation using molecular techniques. Secondly, distinct
methodologies for MSI assessment were used, and the accuracy of MSI
detection is known to be highly dependent on the techniques
selected. Kose et al (30)
used the BAT-26 marker in analysis of MSI, only in tumor DNA.
Fukasawa et al (29) evaluated
the loss of heterozygosity as well as MSI in paired normal and
tumor DNA using dinucleotide markers dispersed on several
chromosomes. Tissues were considered MSI-positive when one or more
markers were altered. Notably, the two studies evaluated MSI in
Japanese populations. This is particularly important due to the
quasimonomorphic nature and the effect of the ancestry of the MSI
markers. Buhard et al (34,35)
studied the global population and identified polymorphisms in the
BAT-26 marker in up to 3.3% of the Asiatic populations, whereas in
Caucasian populations this marker exhibited a monomorphic
nature.
In GISTs, the molecular profile serves as a
classification system that is useful for diagnostic, prognostic and
treatment planning purposes (19,22,38). In
the present study, the KIT and PDGFRA profiles of the
79 GIST cases and their clinicopathological associations were
similar to those previously reported in the literature (22). Mutations in KIT exon 11 were
the most common oncogenic mutations observed in GISTs, followed by
KIT exon 9. Exon 18 was also revealed to be the most
frequently mutated PDGFRA region. PDGFRA-mutant GISTs
frequently possessed characteristics of low-risk GIST, including a
gastric primary site and a low mitotic index, as previously
reported in the literature (19,22). In
addition, a tendency for patients with PDGFRA mutations and
those with wild-type GISTs to have a smaller risk of recurrence
compared with patients with KIT mutations was observed.
In conclusion, using accurate MSI methodologies
widely used for the assessment of CRC, a large series of confirmed
GISTs was analyzed for the presence of genetic instability
phenotypes. No cases with MSI were observed, and so it was
concluded that the MMR system is proficient in patients with GISTs,
and that MSI does not appear to be involved in GIST
tumorigenesis.
Acknowledgements
The present study was supported by The Brazilian
National Council for Scientific and Technological Development
(grant no. 476192/2013-7) and the São Paulo Research Foundation
Doctoral Fellowship (grant no. 2013/25787-3).
References
|
1
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dudley JC, Lin MT, Le DT and Eshleman JR:
Microsatellite instability as a biomarker for PD-1 blockade. Clin
Cancer Res. 22:813–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young J, Simms LA, Biden KG, Wynter C,
Whitehall V, Karamatic R, George J, Goldblatt J, Walpole I, Robin
SA, et al: Features of colorectal cancers with high-level
microsatellite instability occurring in familial and sporadic
settings: Parallel pathways of tumorigenesis. Am J Pathol.
159:2107–2116. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miquel C, Jacob S, Grandjouan S, Aimé A,
Viguier J, Sabourin JC, Sarasin A, Duval A and Praz F: Frequent
alteration of DNA damage signalling and repair pathways in human
colorectal cancers with microsatellite instability. Oncogene.
26:5919–5926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suraweera N, Duval A, Reperant M, Vaury C,
Furlan D, Leroy K, Seruca R, Iacopetta B and Hamelin R: Evaluation
of tumor microsatellite instability using five quasimonomorphic
mononucleotide repeats and pentaplex PCR. Gastroenterology.
123:1804–1811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sinicrope FA and Sargent DJ: Molecular
pathways: Microsatellite instability in colorectal cancer:
Prognostic, predictive, and therapeutic implications. Clin Cancer
Res. 18:1506–1512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McCarter MD, Antonescu CR, Ballman KV,
Maki RG, Pisters PW, Demetri GD, Blanke CD, von Mehren M, Brennan
MF, McCall L, et al: Microscopically positive margins for primary
gastrointestinal stromal tumors: Analysis of risk factors and tumor
recurrence. J Am Coll Surg. 215:53–60. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: Origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011.PubMed/NCBI
|
|
9
|
Doyle LA and Hornick JL: Gastrointestinal
stromal tumours: From KIT to succinate dehydrogenase.
Histopathology. 64:53–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Laurini JA and Carter JE: Gastrointestinal
stromal tumors: A review of the literature. Arch Pathol Lab Med.
134:134–141. 2010.PubMed/NCBI
|
|
11
|
Guller U, Tarantino I, Cerny T, Schmied BM
and Warschkow R: Population-based SEER trend analysis of overall
and cancer-specific survival in 5138 patients with gastrointestinal
stromal tumor. BMC Cancer. 15:5572015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fregnani JH, de Oliveira AT, de Lima
Vazquez V, Viana CR, Longatto-Filho A and Reis RM: Is the
gastrointestinal stromal tumor arising in the rectovaginal septum
an extragastrointestinal entity? A time for reflection. Int J
Colorectal Dis. 26:387–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Joensuu H, Hohenberger P and Corless CL:
Gastrointestinal stromal tumour. Lancet. 382:973–983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liegl-Atzwanger B, Fletcher JA and
Fletcher CD: Gastrointestinal stromal tumors. Virchows Arch.
456:111–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miettinen M and Lasota J: Gastrointestinal
stromal tumors: Pathology and prognosis at different sites. Semin
Diagn Pathol. 23:70–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stamatakos M, Douzinas E, Stefanaki C,
Safioleas P, Polyzou E, Levidou G and Safioleas M: Gastrointestinal
stromal tumor. World J Surg Oncol. 7:612009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Joensuu H, Martin-Broto J, Nishida T,
Reichardt P, Schöffski P and Maki RG: Follow-up strategies for
patients with gastrointestinal stromal tumour treated with or
without adjuvant imatinib after surgery. Eur J Cancer.
51:1611–1617. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joensuu H, Rutkowski P, Nishida T, Steigen
SE, Brabec P, Plank L, Nilsson B, Braconi C, Bordoni A, Magnusson
MK, et al: KIT and PDGFRA mutations and the risk of GI stromal
tumor recurrence. J Clin Oncol. 33:634–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Braggio E, Braggio Dde A, Small IA, Lopes
LF, Valadão M, Gouveia ME, Moreira Ados S, Linhares E, Romano S,
Bacchi CE, et al: Prognostic relevance of KIT and PDGFRA mutations
in gastrointestinal stromal tumors. Anticancer Res. 30:2407–2414.
2010.PubMed/NCBI
|
|
21
|
Rubin BP and Heinrich MC: Genotyping and
immunohistochemistry of gastrointestinal stromal tumors: An update.
Semin Diagn Pathol. 32:392–399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barnett CM, Corless CL and Heinrich MC:
Gastrointestinal stromal tumors: Molecular markers and genetic
subtypes. Hematol Oncol Clin North Am. 27:871–888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Agaimy A, Terracciano LM, Dirnhofer S,
Tornillo L, Foerster A, Hartmann A and Bihl MP: V600E BRAF
mutations are alternative early molecular events in a subset of
KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin
Pathol. 62:613–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martinho O, Gouveia A, Viana-Pereira M,
Silva P, Pimenta A, Reis RM and Lopes JM: Low frequency of MAP
kinase pathway alterations in KIT and PDGFRA wild-type GISTs.
Histopathology. 55:53–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Campanella NC, de Oliveira AT,
Scapulatempo-Neto C, Guimarães DP and Reis RM: Biomarkers and novel
therapeutic targets in gastrointestinal stromal tumors (GISTs).
Recent Pat Anticancer Drug Discov. 8:288–297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mol CD, Dougan DR, Schneider TR, Skene RJ,
Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC and Wilson KP:
Structural basis for the autoinhibition and STI-571 inhibition of
c-Kit tyrosine kinase. J Biol Chem. 279:31655–31663. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan CB, Zhi W, Shahzad G and Mustacchia P:
Gastrointestinal stromal tumors: A review of case reports,
diagnosis, treatment and future directions. ISRN Gastroenterol.
2012:5959682012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rossi S, Gasparotto D, Miceli R,
Toffolatti L, Gallina G, Scaramel E, Marzotto A, Boscato E,
Messerini L, Bearzi I, et al: KIT, PDGFRA, and BRAF mutational
spectrum impacts on the natural history of imatinib-naive localized
GIST: A population-based study. Am J Surg Pathol. 39:922–930. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fukasawa T, Chong JM, Sakurai S, Koshiishi
N, Ikeno R, Tanaka A, Matsumoto Y, Hayashi Y, Koike M and Fukayama
M: Allelic loss of 14q and 22q, NF2 mutation, and genetic
instability occur independently of c-kit mutation in
gastrointestinal stromal tumor. Jpn J Cancer Res. 91:1241–1249.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kose K, Hiyama T, Tanaka S, Yoshihara M,
Yasui W and Chayama K: Nuclear and mitochondrial DNA microsatellite
instability in gastrointestinal stromal tumors. Pathobiology.
73:93–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lopes JM, Silva P, Seixas M, Cirnes L and
Seruca R: Microsatellite instability is not associated with degree
of malignancy and p53 expression of gastrointestinal stromal
tumours. Histopathology. 33:579–581. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Oliveira AT, Pinheiro C, Longatto-Filho
A, Brito MJ, Martinho O, Matos D, Carvalho AL, Vazquez VL, Silva
TB, Scapulatempo C, et al: Co-expression of monocarboxylate
transporter 1 (MCT1) and its chaperone (CD147) is associated with
low survival in patients with gastrointestinal stromal tumors
(GISTs). J Bioenerg Biomembr. 44:171–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
de Oliveira AT, Reis RM, Afonso J,
Martinho O, Matos D, Carvalho AL, Vazquez VL, Silva TB,
Scapulatempo C, Saad SS and Longatto-Filho A: Lymphangiogenic
VEGF-C and VEGFR-3 expression in genetically characterised
gastrointestinal stromal tumours. Histol Histopathol. 26:1499–1507.
2011.PubMed/NCBI
|
|
34
|
Buhard O, Cattaneo F, Wong YF, Yim SF,
Friedman E, Flejou JF, Duval A and Hamelin R: Multipopulation
analysis of polymorphisms in five mononucleotide repeats used to
determine the microsatellite instability status of human tumors. J
Clin Oncol. 24:241–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Buhard O, Suraweera N, Lectard A, Duval A
and Hamelin R: Quasimonomorphic mononucleotide repeats for
high-level microsatellite instability analysis. Dis Markers.
20:251–257. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Campanella NC, Berardinelli GN,
Scapulatempo-Neto C, Viana D, Palmero EI, Pereira R and Reis RM:
Optimization of a pentaplex panel for MSI analysis without control
DNA in a Brazilian population: Correlation with ancestry markers.
Eur J Hum Genet. 22:875–880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Campanella NC, Penna V, Ribeiro G,
Abrahão-Machado LF, Scapulatempo-Neto C and Reis RM: Absence of
microsatellite instability in soft tissue sarcomas. Pathobiology.
82:36–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Corless CL: Gastrointestinal stromal
tumors: What do we know now? Mod Pathol. 27 (Suppl 1):S1–S16. 2014.
View Article : Google Scholar : PubMed/NCBI
|