Introduction
Epigenetics involves the regulation of gene
expression without a change in DNA sequence. Somatic cells retain
and transfer epigenetic information based on DNA methylation,
histone methylation, acetylation, ubiquitination, ADP ribosylation,
histone modification, small RNAs unrelated to genetic codes and
modification of chromatin structure through chromatin remodeling.
The term ‘chromatin remodeling’ refers to the alteration of
chromatin structure from a closed state to a loosened one, which is
termed ‘euchromatin’ (1). There are a
few types of chromatin remodeling complexes, including the
SWItch/sucrose non-fermentable (SWI/SNF) complex, which has several
subunits, including ARID1A and brahma homologue (BRM)-related gene
1 (BRG1; also referred to as SMARCA4) (2). Through the interaction between subunits,
chromatin remodeling complexes change chromatin structure, and this
determines gene expression levels via the regulation of the
interaction between proteins with double-stranded DNA (3). This change in accessibility may be
achieved by adenosine triphosphate (ATP)-dependent complexes
modulating histone-DNA association and by covalent modification of
core nucleosomal histones mediating the transcriptional activity
(4). Epigenetics is also associated
with intracellular communication (5).
These are key events in cell growth, and thus epigenetic
abnormalities may induce carcinogenesis, developmental defects and
multifactorial disease. The association between aberrant chromatin
remodeling with gynecological cancer is discussed in the present
review.
Chromatin remodeling
In eukaryotes, almost all genomic DNA is packaged by
core histones to form chromatin structures. These structures change
in events such as transcription, replication, modification and
recombination of genomic DNA (6). The
requirement for different chromatin structures is fulfilled by
chromatin remodeling, which is an important factor in the
regulation of gene expression.
Chromatin remodeling is performed by two enzyme
groups: Histone modifiers, which chemically alter histones; and
ATP-dependent chromatin remodeling factors, which bind to
nucleosome cores and surrounding DNA to change the chromatin
structure. Using energy from ATP dephosphorylation, remodeling
factors alter nucleosomal structure, transiently loosen binding
with DNA, and coordinate with specific chaperones, exchanging
specific or all nucleosome cores (4).
The nucleosomal structure is dynamically changed by remodeling
factors, resulting in prompt changes in the chromatin structure
(6). Several types of ATP-dependent
remodeling factors are known, including a number of
high-molecular-weight protein complexes with >10 subunits
(6). The activity of these complexes
is regulated and they are transferred to specific DNA sites to
regulate gene expression by changing the chromatin structure
(6,7).
ATP-dependent remodeling factors are classified into several
families: SWI/SNF, imitation SWI (ISWI), INO80, SWR1, nucleosome
remodeling deacetylase (NuRD)/Mi2/CHD and nucleosome remodeling
factor (7).
Aberrant chromatin remodeling and
cancer
Chromatin remodeling factors regulate epigenetic
gene expression, and aberrations in this process may induce
carcinogenesis. A large-scale study of genome sequences has
identified mutations of genes encoding remodeling factors in a
number of types of human cancer, including those for the SWI/SNF
complex, which has led to the suggestion that SWI/SNF complexes are
protective against cancer (7,8). Mutations in SWI/SNF-related,
matrix-associated, actin-dependent regulator of chromatin,
subfamily a, member 4 (SMRCA4/BRG1), which encodes the
ATPase subunit of the SWI/SNF complex, has been detected in >30%
of non-small cell lung carcinoma (NSCLC) (7). Similarly, mutations in the ARID1A, which
encodes an additional subunit of the SWI/SNF complex, has been
detected in 46–57% of clear cell carcinoma and 30% of endometrioid
carcinoma in ovarian cancer (9).
ARID1A mutations also occur in 13% of hepatocellular
carcinoma (HCC), 9.6% of gastrointestinal adenocarcinoma and 2.5%
of malignant melanoma (7).
Chromodomain helicase, DNA-binding protein 4 (CHD4), which forms
the nucleosome remodeling and deacetylase (NuRD) complex, is
overexpressed or mutated in serous endometrial cancer, and
metastasis-associated protein 1 overexpression has been detected in
breast cancer (10).
Deleted regions encoding mixed-lineage leukemia
protein 3 (MLL3) produce chromosomal aberrations that are
frequently associated with acute myeloid leukemia (AML) (11). Similar gene mutations are identified
in medulloblastoma, HCC (12),
bladder carcinoma (13), prostate
cancer (14), colorectal cancer
(15), gastric adenocarcinoma
(16), NSCLC (17), breast cancer (18) and pancreatic cancer (19) and in AML (11). Je et al (20) revealed mutations causing a frameshift
of MLL3 in 28.1% of cases of gastric cancer and 7.5% of
cases of colon cancer.
Chromatin remodeling-associated gene
mutations and carcinogenic mechanism
ARID1A is located at 1p35.3 and encodes an
~250-kD protein that is involved in interactions between numerous
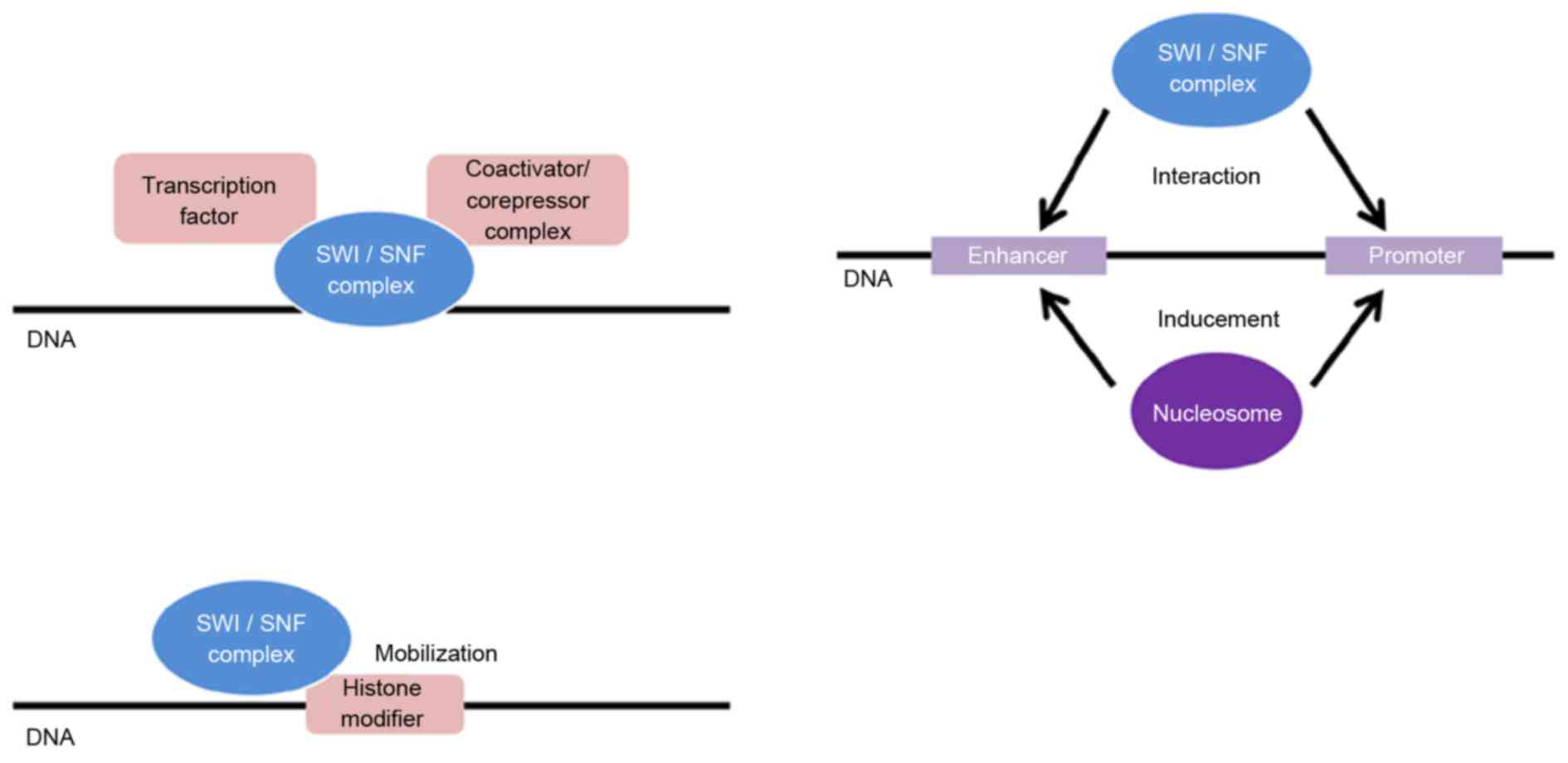
proteins, including the SWI/SNF complex. The SWI/SNF complex has
multiple activities, including the following: The promotion of
binding of transcription factors, coactivators and compressors;
mobilization of histone-modifying enzymes; promotion of binding of
nucleosomes with promoter and enhancer regions; and promotion of
chromatin loop formation to induce interactions of enhancers and
promoters (Fig. 1) (7,21). The
SWI/SNF complex and ARID1A also regulate transcription to induce
steroid hormones: It has been suggested that ARID1A may be involved
in recruiting SWI/SNF to regulate genes through its ability to
stimulate steroid hormone receptor-mediated transcriptional
activation (22,23). Wu and Roberts (21) proposed three activities of
ARID1A in the repression of tumors, namely, proliferation,
differentiation and apoptosis. Gastrointestinal and breast cancer
cells demonstrate a tendency to grow following
ARID1A-knockdown, and growth rates decrease subsequent to
re-expression of ARID1A. Ovarian epithelial cells and mouse
preosteoblast cells indicated similar proliferation behaviors
following ARID1A-knockdown. With regard to differentiation,
ARID1A-knockdown eliminated self-renewal of ES cells and
inhibited the differentiation of neurons and osteocytes in
vitro. For apoptosis, the Fas apoptotic pathway in Jurkat cells
was inhibited by knockdown of ARID1A. These results
demonstrate that an ARID1A deficit has those three effects
on tumor suppression (21). An
ARID1A deficit has also been associated with the activation
of the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT)
signaling pathway, and with the amplification of zinc-finger
protein 217 (ZNF217), which are involved in cancer development
(24).
Dynamic regulation of chromatin structures to allow
transcription factors to bind to DNA is necessary for gene
transcription, duplication and repair. Two complexes,
BRG1-associated factor (BAF) and polybromo-associated BAF (PBAF),
in the SWI/SNF family, perform this role in eukaryotes (22). BRG1 and BRM are subunits containing
ATPase domains that hydrolyze ATP to provide energy for
translocation of nucleosomes and changes in chromatin structure
(25). BRG1 binds to BRCA1 and
regulates cluster of differentiation 44 expression as part of the
epithelial-mesenchymal transition in cancer (25). BRG1 (also referred to as
SMARCA4) is located at p13.2 on the short arm of chromosome
19 (19p13.2). BRG1 regulates DNA transcription and serves a
role in tumor suppression due to remodeling of the chromatin
structure. Therefore, mutations and deletions of this gene are
identified in a number of cancer types, including ovarian small
cell carcinoma, rhabdoid tumors (kidney and brain),
medulloblastoma, lung adenocarcinoma, mantle cell lymphoma,
Burkitt's lymphoma, HCC, esophageal adenocarcinoma, melanoma,
non-melanoma skin cancer and intraductal papillary mucinous
neoplasms of the pancreas (26).
Chromodomain helicase DNA-binding protein 4 (CHD4)
is located on the short arm of chromosome 12 (12p13) and its
transcription product is a molecule in the SNF2/RAD54 helicase
family. CHD4 serves a key role in epigenetic transcription
suppression, as it acts in nucleosome remodeling in an
ATP-dependent manner, and is the major protein involved in the
formation of a deacetylase complex. CHD4 exhibits tandem
chromodomains in the N-terminal region and an ATPase-helicase
domain in the central region. The chromodomains recognize and bind
to nucleosomes and regulate interactions with chromatin, whereas
the ATPase-helicase domain is involved in DNA transcription,
duplication, recombination and repair (27). Mutations in these domains cause
hyposegmentation in cells, indicating that the two domains are
required for CHD4 function (27).
Mutations of CHD4 have been identified in
several cancer types, and particularly in serous endometrial
carcinoma: Zhao et al (28)
revealed that 11/52 patients exhibited a heterozygous somatic
CHD4 mutation. Le Gallo et al (29) also identified a somatic mutation in
CHD4 in 17% of patients with serous endometrial cancer. CHD4
is characterized by ‘signature’ motifs that contain important amino
acid residues required for ATP hydrolysis and helicase activity.
The normal function of CHD4 is eliminated by R957Q, R1127G and
R1162W mutations in these residues (30). In an immunohistological examination of
lesion tissues in gastric cancer and colorectal cancer, Kim et
al (30) identified no CHD4
expression in 56.4% of patients with gastric cancer and 55.7% with
colorectal cancer. Insertion or deletion of 1 to 2 bases caused a
somatic mutation in CHD4, with the resulting frameshift
causing elimination of normal CHD4 expression (30).
MLL3 belongs to a gene cluster of the MLL
family and is also called lysine N-methyltransferase 2C (KMT2C).
MLL3 exhibits a histone methyltransferase SET domain, a
HMG-binding domain, a nuclear receptor binding domain and 5 zinc
fingers, and acts as a nuclear receptor coactivator in mammals
(11). The MLL family transfers 1, 2
or 3 methyl groups to lysine K4 of methyl histone H3, and MLL3
particularly methylates H3K4 in enhancer regions (11). In a study of familial nasopharyngeal
carcinoma, Sasaki et al (31)
proposed that the mechanism of carcinogenesis involves the action
of acquired factors such as somatic mutation and Epstein-Barr virus
infection in regions containing germline mutations that frequently
cause a stop codon in MLL3. In an analysis of gene mutations
in patients with Lynch syndrome, Villacis et al (32) also suggested that a MLL3
mutation increases the risk of colorectal cancer.
Enhancer of zeste 2 polycomb repressive complex 2
subunit (EZH2) encodes proteins in the polycomb group (PcG)
family and is located on chromosome 7 (7q35-q36). PcG proteins
contribute to the epigenetic regulation of gene expression, for
example: EZH2 methylates histone H3 core protein lysine 27 and
inhibits gene transcription (33).
EZH2 demonstrates high expression in numerous types of
cancer, including breast cancer, melanoma and lung cancer (33). In gynecological cancer, high
EZH2 expression occurs in uterine fibroids and cervical
lesions. Yang et al (34)
proposed a mechanism in which EZH2 inhibits the expression of the
DNA mismatch repair gene Mutator S protein homolog 2 (MSH2)
and develops uterine fibroids. Cai et al (35) revealed that EZH2 was expressed
more frequently in cervical cancer tissues compared with normal
tissues, and that cisplatin resistance in cervical cancer was
increased by the inhibition of endogenous EZH2 expression
with short hairpin RNA. Furthermore, an overexpression of
EZH2 has been identified in 66% of tumors and 67% of
endothelial cells of tumor vessels in patients with ovarian cancer
(36). Patients with high expression
of EZH2 in tumors exhibited a significantly poorer prognosis
compared with those without high expression. The inactivation of
EZH2 expression increases apoptosis of cancer cells,
decreases the number of vessels in tumor tissues and reduces the
growth of ovarian cancer cells (36).
Aberrant chromatin remodeling and ovarian
cancer
Ovarian clear cell carcinoma (OCCC) is a
chemoresistant cancer due to delayed cell division (37). OCCC exhibits two carcinogenic
pathways, which are referred to as the adenofibroma-carcinoma and
endometriosis-carcinoma sequences (38,39). The
differences in the genetic backgrounds of these two pathways are
unclear, but the ARID1A mutation has been suggested to be
involved in the onset of OCCC via the endometriosis-carcinoma
sequence, rather than via the adenofibroma-carcinoma sequence
(38,39). Jones et al (9) detected ARID1A mutations in 24
(57%) of 42 patients with OCCC, and concluded that ARID1A is
a tumor suppressor gene and that ARID1A mutation inactivates
gene products through the aberrant chromatin remodeling associated
with OCCC pathogenesis. ARID1A encodes a component of the
SWI/SNF complex, which regulates cell growth, controls cell cycle
regulation and cell division and repairs DNA (40,41).
Wiegand et al (42) detected an ARID1A mutation in 55
(46%) of 119 patients with OCCC and identified a deficit in
BAF250a, a protein encoded by ARID1A, in 36% of these
patients (Table I). BAF250a gives
specificity to the SWI/SNF complex and enables regulation of gene
expression (22). Furthermore,
ARID1A mutations and BAF250a deficits were identified
in OCCC and adjacent endometriotic lesions, but not in distant
lesions, which suggests that this mutation and resultant
BAF250a deficit are events in the early stage of neoplastic
transformation of endometriosis (2,42). A
previous study confirmed that an ARID1A deficit was also an
early phenomenon in endometriosis-associated ovarian cancer (EAOC)
and endometriotic ovarian cysts, together with AKT protein
activation and a histone H2A variant (γH2AX) (43). An ARID1A deficit has also been
identified as a poor prognostic factor in patients with stage I/II
OCCC, and may be a useful biological marker for the prediction of
prognosis (42).
 | Table I.Aberrant chromatin
remodeling-associated genes in cancer. |
Table I.
Aberrant chromatin
remodeling-associated genes in cancer.
| Gene name | Mutation ratio,
% | Gene
abnormality | Type of cancer | (Refs.) |
|---|
| ARID1A | 46–57 | MS, NS, FS | Ovarian clear cell
carcinoma | (9) |
|
| 30 | MS, NS, FS | Ovarian
endometrioid carcinoma | (2) |
|
| 13 | MS, NS, FS | HCC | (7) |
|
| 9.6 | MS, NS, FS | Gastrointestinal
adenocarcinoma | (7) |
|
| 2.5 | MS, NS, FS | Malignant
melanoma | (7) |
|
| 36 | Mutation | Endometrial serous
carcinoma | (29) |
|
| 36 | Mutation | Uterine CS | (57) |
| CHD4 | 56.4 | FS | Gastric cancer | (30) |
|
| 55.7 | FS | Colorectal
cancer | (30) |
|
| 21 | Mutation | Endometrial serious
carcinoma | (28) |
|
| 7 | OE | Endometrial
carcinoma | (29) |
|
| 4 | OE | Endometrial clear
cell carcinoma | (29) |
| EZH2 | 66 | OE | Ovarian cancer | (36) |
|
| Unknown | OE | Melanoma, BC, lung
cancer, cervical cancer | (33,34) |
| MLL3 | <5 | MS, NS, FS | Bladder
carcinoma | (13) |
|
| 8 | MS, NS, FS | Prostate
cancer | (14) |
|
| 13 | MS, NS, FS | Gastric
adenocarcinoma | (16) |
|
| 27 | Mutation | Uterine CS | (57) |
|
| 14 | FS | Colorectal
cancer | (15) |
|
| 28.1 | FS | Gastric cancer | (20) |
|
| 7.5 | FS | Colon cancer | (20) |
|
| Unknown | Deletion | AML | (21) |
|
| Unknown | MS, NS, FS | BC,
medulloblastoma, pancreatic cancer, HCC, NSCLC | (12,17–19) |
|
SMARCA4/ | 10 | GM, NS, FS | Lung
adenocarcinoma | (49) |
| BRG1 | 31.3 | GM, NS, FS | Lung large cell
carcinoma | (49) |
|
| 36.4 | GM, NS, FS | Lung pleomorphic
carcinoma | (49) |
|
| 94 | GM, NS, FS | SCCOHT | (26,45–47) |
|
| <30 | Mutation | NSCLC | (7) |
|
| 10–20 | Mutation | Melanoma,
esophageal adenocarcinoma, intraductal papillary mucinous neoplasms
of the pancreas | (26) |
|
| Unknown | Mutation | OSCC, rhabdoid
tumor, mantle cell lymphoma, Burkitt lymphoma, non-melanoma skin
cancer | (26) |
Small cell carcinoma of the ovary, hypercalcemic
type (SCCOHT) associated with hypercalcemia is a rare disease and
is considered to be a rhabdoid tumor (26). SCCOHT is a poorly differentiated tumor
associated with a poor prognosis that develops in young females
(44). In an immunohistological
study, Conlon et al (44)
measured loss of SMARCA4 expression in 94% of patients with
SCCOHT, whereas loss of SMARCA4 expression is usually
identified in <5% of patients with ovarian cancer (44). Therefore, these data are considered to
be specific to SCCOHT (44). In
SCCOHT, germline mutations have been revealed in one allele of
SMARCA4, and expression is deleted due to an inactivating
germline mutation and frameshift and nonsense mutations in the
other allele (26,45–47).
Rhabdoid tumors that develop in organs other than the ovary,
including the kidney and brain, have germline and somatic
expression of SMARCA4 (48).
Immunostaining for the expression of SMARCA4 in tumor
tissues of patients with lung cancer revealed downregulation of
SMARCA4 in no patients with squamous cell carcinoma, in 10%
with adenocarcinoma, in 31.3% with large cell carcinoma and in
36.4% with pleomorphic carcinoma (49), and somatic mutation and deletion of
SMARCA4 are present in these types of cancer (26). SMARCA4 is a subunit of the BAF and
PBAF complexes, and mutation and deletion produces incomplete
complexes and abnormal subunits that may cause dysregulation of
genes and induce disease (50).
Aberrant chromatin remodeling and
endometrial cancer
Endometrial cancer includes endometrioid carcinoma
and serous carcinoma, which is less common compared with
endometrioid carcinoma and has a relatively poor prognosis
(51). Almost all serous carcinomas
are poorly differentiated type 2 endometrial cancer with
myometrial, vascular and extrauterine invasion (51). In exome sequencing of endometrial
serous carcinomas in 53 patients, Le Gallo et al (29) detected CHD4 mutations in 9
(17%) cases, and identified mutation of chromatin remodeling genes,
including ARID1A, in 19 (36%) (29) (Table I).
Similarly, Zhao et al (28)
identified CHD4 mutations in 11 (21%) of 52 patients with
endometrial serous carcinomas. CHD4 is a catalytic subunit
of the NuRD complex that inhibits transcription and repairs DNA
damage (52). CHD4
overexpression has also been revealed in 7% of endometrioid
carcinomas and 4% of endometrial clear cell carcinomas, with half
of CHD4 mutations affecting the ATPase/helicase domain or
helicase domain, which is suspected to be the cause of endometrial
cancer (29).
Carcinosarcoma (CS) is an extremely rare
gynecological disease with a poor prognosis (53). Histological results of CS demonstrate
mixed epithelial carcinoma and non-epithelial sarcoma (53). CS occurs commonly in the uterine body,
but has also been identified in the ovary, uterine cervix and
vagina (54–56). The incidence in the United States is 2
per 100,000, and the 5-year survival rates are 35–65% in the early
stage and ~10% in stage IV (53). In
22 patients with uterine CS, Jones et al (57) revealed ARID1A mutations in 8
(36%) cases, mutations of histone methyltransferase MLL3 in
6 (27%) cases, mutations of speckle-type POZ protein (SPOP), which
is involved in chromatin remodeling, in 3 (14%) cases, and
mutations of chromatin remodeling-associated genes in 14 (64%)
cases (57). ARID1A serves an
important role in the regulation of cell growth, and MLL3 is
a coactivator of tumor protein p53 (TP53), a tumor
suppressor p53 gene (2,58). SPOP is a transcriptional repressor of
p53 via the bric-a-brac/tramtrack/broad complex protein (59). Jones et al (57) suggested that a specific tissue-type of
uterine CS depends on aberrant chromatin remodeling. Therefore, a
complete understanding of genetic mutations in this cancer will be
useful for diagnosis, early detection and treatment.
Therapy targeting aberrant chromatin
remodeling
Cancer cells with an ARID1A deficit are
highly sensitive to small molecule inhibitors in the PI3K/AKT
signal transduction pathway. Therefore, drugs that inhibit this
pathway are effective in patients with cancer with an ARID1A
deficit (60). Therapy targeting
epigenetic regulatory mechanisms in cancer cells is also under
development. Bitler et al (61) focused on the activity of EZH2, a
methylation factor in cancer with ARID1A mutation, and
identified that proliferation of cells with an ARID1A
mutation was selectively inhibited by the administration of an EZH2
inhibitor. This suggests that EZH2 inactivation is a potential
therapy for cancer with ARID1A mutation, and EZH2 inhibition
has been demonstrated to reduce the number of ovarian tumors with
ARID1A mutations in vivo. Therefore, pharmacological
inhibition of EZH2 expression may be a therapeutic strategy
for cancer with an ARID1A mutation (61).
Guan et al (62) demonstrated that an ARID1A
in-frame mutation prevented ARID1A transport from the nucleus to
the cytoplasm (62). The ARID1A
protein was then degraded by the ubiquitin-proteasome system and
was not available downstream, resulting in the onset of cancer.
Thus, ARID1A degradation may be inhibited by targeting the
ubiquitin-proteasome system in cells with an ARID1A
mutation, with potential recovery of the original cancer inhibitory
effect (62).
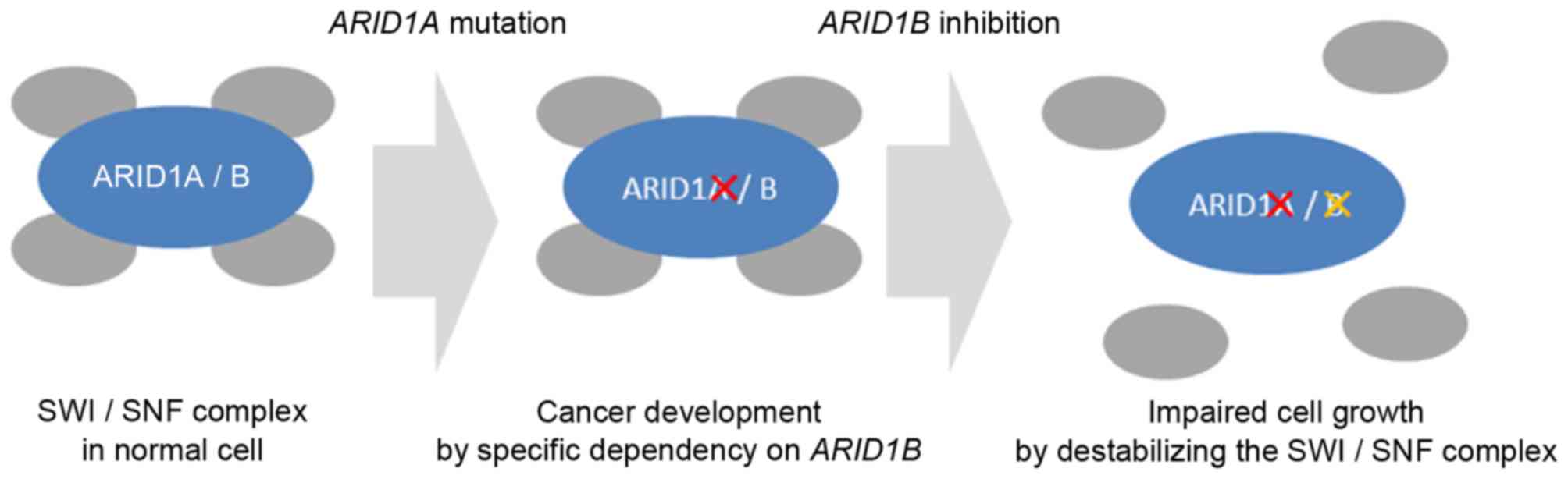
ARID1B has recently been identified as an
ARID1A homolog (63). In cells
with an ARID1A deficit, ARID1B is independently
expressed and its proliferation is enhanced, which suggests that
ARID1A and ARID1B may interact in promoting
carcinogenesis. However, blocking the mechanism of ARID1B in
cells with an ARID1A deficit destabilizes the SWI/SNF
complex and inhibits cell proliferation. Therefore, ARID1B
is also a therapeutic target in cancer with ARID1A mutation
(Fig. 2) (63). Immunohistochemical detection of
ARID1A expression may be a useful marker for the evaluation
of malignancy, prognosis and treatment effect (64).
Conclusion
ARID1A mutation is involved in gynecological
cancer types such as OCCC and uterine cancer through the induction
of aberrant chromatin remodeling and promotion of tumorigenesis.
Germline mutations and epigenetic regulatory mechanisms, including
chromatin remodeling, are involved in carcinogenesis. Therefore,
there is a requirement for methods for identifying chromatin
remodeling-associated gene mutations, including ARID1A and
BRG1, and for therapy targeting the carcinogenic mechanisms
of aberrant chromatin remodeling.
Acknowledgements
The authors would like to thank Dr S. Fujiwara and
Dr K. Hoshi (Keio University School of Medicine, Tokyo, Japan) for
their assistance, and are grateful for support from the Keio Gijuku
Academic Development Fund.
References
|
1
|
Weaver IC, Korgan AC, Lee K, Wheeler RV,
Hundert AS and Goguen D: Stress and the emerging roles of chromatin
remodeling in signal integration and stable transmission of
reversible phenotypes. Front Behav Neurosci. 11:412017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Takeda T, Banno K, Okawa R, Yanokura M,
Iijima M, Irie-Kunitomi H, Nakamura K, Iida M, Adachi M, Umene K,
et al: ARID1A gene mutation in ovarian and endometrial cancers
(Review). Oncol Rep. 35:607–613. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clapier CR and Cairns BR: The biology of
chromatin remodeling complexes. Annu Rev Biochem. 78:273–304. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ronan JL, Wu W and Crabtree GR: From
neural development to cognition: Unexpected roles for chromatin.
Nat Rev Genet. 14:347–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang B, Jiang C and Zhang R: Epigenetics:
The language of the cell? Epigenomics. 6:73–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alberts B, Johnson A, Lewis J, Raff M,
Roberts K and Walter P: Molecular biology of the cell, 5th edition.
Science. 215–216. 2008.PubMed/NCBI
|
|
7
|
Wilson BG and Roberts CW: SWI/SNF
nucleosome remodellers and cancer. Nat Rev Cancer. 11:481–492.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oike T, Ogiwara H, Nakano T, Yokota J and
Kohno T: Inactivating mutations in SWI/SNF chromatin remodeling
genes in human cancer. Jpn J Clin Oncol. 43:849–855. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones S, Wang TL, Shih IeM, Mao TL,
Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, et
al: Frequent mutations of chromatin remodeling gene ARID1A in
ovarian clear cell carcinoma. Science. 330:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mayes K, Qiu Z, Alhazmi A and Landry JW:
ATP-dependent chromatin remodeling complexes as novel targets for
cancer therapy. Adv Cancer Res. 121:183–233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li WD, Li QR, Xu SN, Wei FJ, Ye ZJ, Cheng
JK and Chen JP: Exome sequencing identifies an MLL3 gene germ line
mutation in a pedigree of colorectal cancer and acute myeloid
leukemia. Blood. 121:1478–1479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao
S, Wu R, Chen C, Li X, Zhou L, et al: Frequent mutations of
chromatin remodeling genes in transitional cell carcinoma of the
bladder. Nat Genet. 43:875–878. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lindberg J, Mills IG, Klevebring D, Liu W,
Neiman M, Xu J, Wikström P, Wiklund P, Wiklund F, Egevad L and
Grönberg H: The mitochondrial and autosomal mutation landscapes of
prostate cancer. Eur Urol. 63:702–708. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watanabe Y, Castoro RJ, Kim HS, North B,
Oikawa R, Hiraishi T, Ahmed SS, Chung W, Cho MY, Toyota M, et al:
Frequent alteration of MLL3 frameshift mutations in microsatellite
deficient colorectal cancer. PLoS One. 6:e233202011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zang ZJ, Cutcutache I, Poon SL, Zhang SL,
McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al:
Exome sequencing of gastric adenocarcinoma identifies recurrent
somatic mutations in cell adhesion and chromatin remodeling genes.
Nat Genet. 44:570–574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu P, Morrison C, Wang L, Xiong D, Vedell
P, Cui P, Hua X, Ding F, Lu Y, James M, et al: Identification of
somatic mutations in non-small cell lung carcinomas using
whole-exome sequencing. Carcinogenesis. 33:1270–1276. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ,
Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al:
Whole-genome analysis informs breast cancer response to aromatase
inhibition. Nature. 486:353–360. 2012.PubMed/NCBI
|
|
19
|
Biankin AV, Waddell N, Kassahn KS, Gingras
MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J,
et al: Pancreatic cancer genomes reveal aberrations in axon
guidance pathway genes. Nature. 491:399–405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Je EM and Lee SH, Yoo NJ and Lee SH:
Mutational and expressional analysis of MLL genes in gastric and
colorectal cancers with microsatellite instability. Neoplasma.
60:188–195. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu JN and Roberts CW: ARID1A mutations in
cancer: Another epigenetic tumor suppressor? Cancer Discov.
3:35–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Trotter KW, Fan HY, Ivey ML, Kingston RE
and Archer TK: The HSA domain of BRG1 mediates critical
interactions required for glucocorticoid receptor-dependent
transcriptional activation in vivo. Mol Cell Biol. 28:1413–1426.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inoue H, Furukawa T, Giannakopoulos S,
Zhou S, King DS and Tanese N: Largest subunits of the human SWI/SNF
chromatin-remodeling complex promote transcriptional activation by
steroid hormone receptors. J Biol Chem. 277:41674–41685. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang HN, Lin MC, Huang WC, Chiang YC and
Kuo KT: Loss of ARID1A expression and its relationship with
PI3K-Akt pathway alterations and ZNF217 amplification in ovarian
clear cell carcinoma. Mod Pathol. 27:983–990. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Biegel JA, Busse TM and Weissman BE:
SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C
Semin Med Genet. 166C:350–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reisman D, Glaros S and Thompson EA: The
SWI/SNF complex and cancer. Oncogene. 28:1653–1668. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yamada M, Sato N, Ikeda S, Arai T, Sawabe
M, Mori S, Yamada Y, Muramatsu M and Tanaka M: Association of the
chromodomain helicase DNA-binding protein 4 (CHD4) missense
variation p.D140E with cancer: Potential interaction with smoking.
Genes Chromosomes Cancer. 54:122–128. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao S, Choi M, Overton JD, Bellone S,
Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S,
et al: Landscape of somatic single-nucleotide and copy-number
mutations in uterine serous carcinoma. Proc Natl Acad Sci USA.
110:2916–2921. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Le Gallo M, O'Hara AJ, Rudd ML, Urick ME,
Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et
al: Exome sequencing of serous endometrial tumors identifies
recurrent somatic mutations in chromatin-remodeling and ubiquitin
ligase complex genes. Nat Genet. 44:1310–1315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim MS, Chung NG, Kang MR, Yoo NJ and Lee
SH: Genetic and expressional alterations of CHD genes in gastric
and colorectal cancers. Histopathology. 58:660–668. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sasaki MM, Skol AD, Bao R, Rhodes LV,
Chambers R, Vokes EE, Cohen EE and Onel K: Integrated genomic
analysis suggests MLL3 is a novel candidate susceptibility gene for
familial nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers
Prev. 24:1222–1228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Villacis RA, Miranda PM, Gomy I, Santos
EM, Carraro DM, Achatz MI, Rossi BM and Rogatto SR: Contribution of
rare germline copy number variations and common susceptibility loci
in Lynch syndrome patients negative for mutations in the mismatch
repair genes. Int J Cancer. 138:1928–1935. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim KH and Roberts CW: Targeting EZH2 in
cancer. Nat Med. 22:128–134. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang Q, Laknaur A, Elam L, Ismail N,
Gavrilova-Jordan L, Lue J, Diamond MP and Al-Hendy A:
Identification of polycomb group protein EZH2-mediated DNA mismatch
repair gene MSH2 in human uterine fibroids. Reprod Sci.
23:1314–1325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cai L, Wang Z and Liu D: Interference with
endogenous EZH2 reverses the chemotherapy drug resistance in
cervical cancer cells partly by up-regulating Dicer expression.
Tumour Biol. 37:6359–6369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu C, Han HD, Mangala LS, Ali-Fehmi R,
Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A, et
al: Regulation of tumor angiogenesis by EZH2. Cancer Cell.
18:185–197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Crotzer DR, Sun CC, Coleman RL, Wolf JK,
Levenback CF and Gershenson DM: Lack of effective systemic therapy
for recurrent clear cell carcinoma of the ovary. Gynecol Oncol.
105:404–408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Viganó P, Somigliana E, Chiodo I, Abbiati
A and Vercellini P: Molecular mechanisms and biological
plausibility underlying the malignant transformation of
endometriosis: A critical analysis. Hum Reprod Update. 12:77–89.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nishikimi K, Kiyokawa T, Tate S, Iwamoto M
and Shozu M: ARID1A expression in ovarian clear cell carcinoma with
an adenofibromatous component. Histopathology. 67:866–871. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nagl NG Jr, Wang X, Patsialou A, Van Scoy
M and Moran E: Distinct mammalian SWI/SNF chromatin remodeling
complexes with opposing roles in cell-cycle control. EMBO J.
26:752–763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Weissman B and Knudsen KE: Hijacking the
chromatin remodeling machinery: Impact of SWI/SNF perturbations in
cancer. Cancer Res. 69:8223–8230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y,
Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et
al: ARID1A mutations in endometriosis-associated ovarian
carcinomas. N Engl J Med. 363:1532–1543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Itamochi H, Oumi N, Oishi T, Shoji T,
Fujiwara H, Sugiyama T, Suzuki M, Kigawa J and Harada T: Loss of
ARID1A expression is associated with poor prognosis in patients
with stage I/II clear cell carcinoma of the ovary. Int J Clin
Oncol. 20:967–973. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Conlon N, Silva A, Guerra E, Jelinic P,
Schlappe BA, Olvera N, Mueller JJ, Tornos C, Jungbluth AA, Young
RH, et al: Loss of SMARCA4 expression is both sensitive and
specific for the diagnosis of small cell carcinoma of ovary,
hypercalcemic type. Am J Surg Pathol. 40:395–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jelinic P, Mueller JJ, Olvera N, Dao F,
Scott SN, Shah R, Gao J, Schultz N, Gonen M, Soslow RA, et al:
Recurrent SMARCA4 mutations in small cell carcinoma of the ovary.
Nat Genet. 46:424–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ramos P, Karnezis AN, Craig DW, Sekulic A,
Russell ML, Hendricks WP, Corneveaux JJ, Barrett MT, Shumansky K,
Yang Y, et al: Small cell carcinoma of the ovary, hypercalcemic
type, displays frequent inactivating germline and somatic mutations
in SMARCA4. Nat Genet. 46:427–429. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Witkowski L, Carrot-Zhang J, Albrecht S,
Fahiminiya S, Hamel N, Tomiak E, Grynspan D, Saloustros E, Nadaf J,
Rivera B, et al: Germline and somatic SMARCA4 mutations
characterize small cell carcinoma of the ovary, hypercalcemic type.
Nat Genet. 46:438–443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Schneppenheim R, Frühwald MC, Gesk S,
Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Subero
Martin JI, Obser T, et al: Germline nonsense mutation and somatic
inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor
predisposition syndrome. Am J Hum Genet. 86:279–284. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yoshimoto T, Matsubara D, Nakano T, Tamura
T, Endo S, Sugiyama Y and Niki T: Frequent loss of the expression
of multiple subunits of the SWI/SNF complex in large cell carcinoma
and pleomorphic carcinoma of the lung. Pathol Int. 65:595–602.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Helming KC, Wang X and Roberts CW:
Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell.
26:309–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sherman ME: Theories of endometrial
carcinogenesis: A multidisciplinary approach. Mod Pathol.
13:295–308. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Polo SE, Kaidi A, Baskcomb L, Galanty Y
and Jackson SP: Regulation of DNA-damage responses and cell-cycle
progression by the chromatin remodelling factor CHD4. EMBO J.
29:3130–3139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Duman BB, Kara IO, Günaldi M and Ercolak
V: Malignant mixed Mullerian tumor of the ovary with two cases and
review of the literature. Arch Gynecol Obstet. 283:1363–1368. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sharma NK, Sorosky JI, Bender D, Fletcher
MS and Sood AK: Malignant mixed mullerian tumor (MMMT) of the
cervix. Gynecol Oncol. 97:442–445. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ahuja A, Safaya R, Prakash G, Kumar L and
Shukla NK: Primary mixed mullerian tumor of the vagina - a case
report with review of the literature. Pathol Res Pract.
207:253–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
George EM, Herzog TJ, Neugut AI, Lu YS,
Burke WM, Lewin SN, Hershman DL and Wright JD: Carcinosarcoma of
the ovary: Natural history, patterns of treatment, and outcome.
Gynecol Oncol. 131:42–45. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jones S, Stransky N, McCord CL, Cerami E,
Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, et
al: Genomic analyses of gynaecologic carcinosarcomas reveal
frequent mutations in chromatin remodelling genes. Nat Commun.
5:50062014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee
SK, Roeder RG and Lee JW: A tumor suppressive coactivator complex
of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase
MLL3 or its paralogue MLL4. Proc Natl Acad Sci USA. 106:8513–8518.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol
JH, Baek SH, Chiba T, Tanaka K, Bang OS, et al: BTB
domain-containing speckle-type POZ protein (SPOP) serves as an
adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase.
J Biol Chem. 281:12664–12672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Samartzis EP, Gutsche K, Dedes KJ, Fink D,
Stucki M and Imesch P: Loss of ARID1A expression sensitizes cancer
cells to PI3K- and AKT-inhibition. Oncotarget. 5:5295–5303. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bitler BG, Aird KM, Garipov A, Li H,
Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IeM,
Conejo-Garcia JR, et al: Synthetic lethality by targeting EZH2
methyltransferase activity in ARID1A-mutated cancers. Nat Med.
21:231–238. 2015.PubMed/NCBI
|
|
62
|
Guan B, Gao M, Wu CH, Wang TL and Shih
IeM: Functional analysis of in-frame indel ARID1A mutations reveals
new regulatory mechanisms of its tumor suppressor functions.
Neoplasia. 14:986–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Helming KC, Wang X, Wilson BG, Vazquez F,
Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ,
et al: ARID1B is a specific vulnerability in ARID1A-mutant cancers.
Nat Med. 20:251–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nagymanyoki Z, Mutter GL, Hornick JL and
Cibas ES: ARID1A is a useful marker of malignancy in peritoneal
washings for endometrial carcinoma. Cancer Cytopathol. 123:253–257.
2015. View Article : Google Scholar : PubMed/NCBI
|