Introduction
As the most malignant type of astrocytic tumors, the
recurrence and mortality rates of GBM are extremely high (1). Studies have found that the molecular
mechanisms of primary glioblastoma (GBM) and secondary GBM were
different (2). Primary GBM is caused
by the overexpression of epidermal growth factor receptor (EGFR),
while secondary GBM is caused by the mutations of p53 (3). Due to the differential expression of a
large number of genes in GBM, conventional biomolecular methods
cannot be used to demonstrate the pathogenesis of GBM. Gene
expression profile chip, which can measure the expression levels of
a large number of genes, is an ideal approach for the analysis of
molecular mechanism of GBM (4). In
recent years, more and more gene expression profile data become
available, and the use of bioinformatics to analyze gene expression
profile data has become a new research hotspot (5). In this study, bioinformatics methods
were used to analyze the data of gene expression profiles with an
expectation of analyzing the differentially expressed genes between
GBN and normal human brain cells, so as to provide new insights for
the studies on the pathogenesis of GBM.
Materials and methods
Gene expression profile data
Data of gene chip GSE12657 and GSE42656 were
obtained from GEO database. GSE12657 was from Neuropathology in the
Department of Medicine at Imperial College London with 7 cases of
GBM patients as experimental group and 5 cases of normal samples as
a control group. GSE42656 was from Neuroscience and Trauma at Barts
and the London School of Medicine and Dentistry with 5 cases of GBM
patients as experimental group and 8 cases of normal samples as a
control group. This study was approved by the Ethics Committee of
Xiangyang No. 1 People's Hospital, Hubei University of Medicine.
Signed written informed consents were obtained from the patients
and/or guardians.
Raw data preprocessing and screening and integration
of differentially expressed genes. Affymetrix Expression Console
and RMA algorithm were used for quality control, standardization
and log2 conversion for the raw data of gene chips. Microarray data
analysis package (Linear Models for Microarray Data, Limma) in ‘R’
software was used to screen the differentially expressed genes from
raw data of two gene chips. Gene integration of differentially
expressed genes identified from two gene chips was performed using
RobustRankAggreg.
Gene Ontology (GO) enrichment
analysis
DAVID and the plug-in unit ‘Bingo’ of Cytoscape
software (San Diego, CA, USA) were used for GO enrichment analysis
and functional annotation after gene integration. Database for
Annotation, Visualization and Integration Discovery (DAVID)
analysis, DAVID network software (NIH, Bethesda, MD, USA) contains
almost all major public bioinformatics resources. DAVID can be used
to annotate gene-related biological mechanisms using standardized
gene terminology.
DAVID knowledge base is designed to facilitate
high-throughput gene functional analysis. DAVID provides a wide
range of heterogeneous annotation data in a centralized location
for a given gene list. DAVID enriches the biological information
for individual genes. DAVID knowledge base can be downloaded from
the following website: https://david.ncifcrf.gov/.
KEGG pathway analysis. KEGG pathway analysis and
functional annotation for differentially expressed gene were
performed using KOBAS 3.0 software (Peking University, Beijing,
China).
KOBAS is the first software to use the
hypergeometric distribution method to determine the significance of
pathway enrichment. KOBAS has been successfully used in the study
of different organisms such as plants, animals and bacteria. KOBAS
server can be accessed at https://kobas.cbi.pku.edu.cn.
Protein interaction network
analysis
STRING software (STRING 10.0; European Molecular
Biology Laboratory, Heidelberg, Germany) was used to analyze the
protein-protein interaction (PPI) of differentially expressed
genes. PPI refers to the forming of protein complex by two or more
protein molecules through non-covalent bonds. STRING can be
accessed at https://string-db.org/.
Results
Screening of differentially expressed
genes
A total of 702 differentially expressed genes were
identified from gene chip GSE12657, and 548 genes were
significantly upregulated and 154 genes were significantly
downregulated (p<0.01, fold-change >1). In gene chip
GSE42656, 1,854 differentially expressed genes were identified, and
1,068 genes were significantly upregulated and 786 genes were
significantly downregulated (p<0.01, fold-change >1). After
gene integration, 167 differentially expressed genes including 67
downregulated genes and 100 upregulated genes were identified.
Those genes showed significantly different expression levels in GBM
compared with normal human brain cells (p<0.05).
GO enrichment analysis
The list of differentially expressed genes was
submitted to DAVID Bioinformatics Resource Network (https://david.ncifcrf.gov/) with OFFICIAL-GENE-SYMBOL
and Gene List were selected. All other parameters were default.
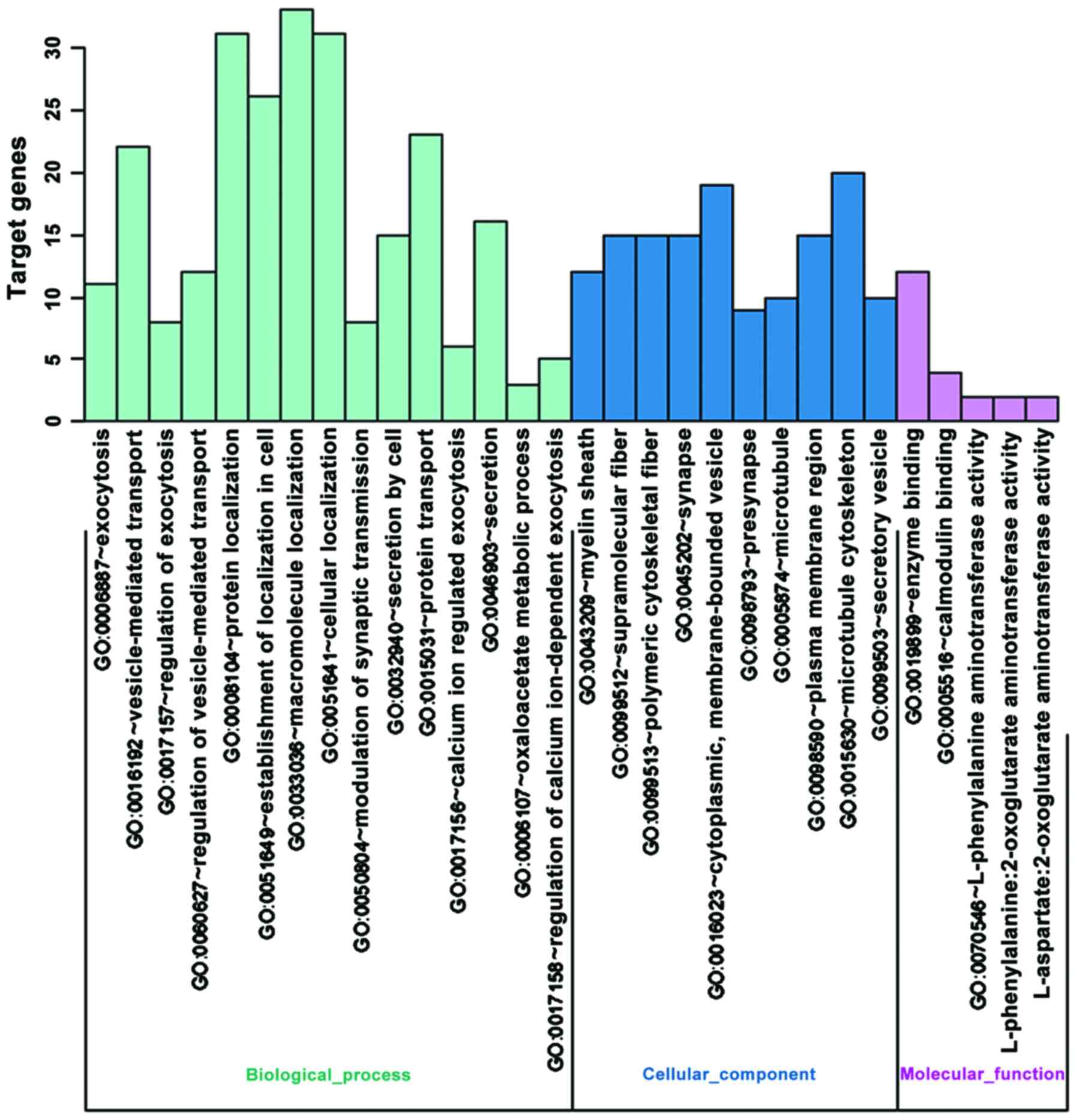
Differentially expressed genes were mainly enrich in
‘neurotransmitter:sodium symporter activity’ and ‘neurotransmitter
transporter activity’, which can affect the activity of
neurotransmitter transportation (Fig.
1).
KEGG pathway analysis
KEGG pathway analysis and functional annotation were
performed using KOBAS 3.0 software. Four key KEGG pathways
included: ‘Dopaminergic synapses’, ‘MAPK signaling pathway’,
‘Glyoxylate and dicarboxylate metabolism’ and ‘Protein processing
in endoplasmic reticulum’ (Table
I).
 | Table I.Results of KEGG pathway analysis. |
Table I.
Results of KEGG pathway analysis.
| Term | Count | P-value | FDR |
|---|
| hsa04141:Protein
processing in endoplasmic reticulum | 3 | 0.000309962 | 0.01239848 |
| hsa04728:Dopaminergic
synapse | 2 | 0.004768424 | 0.095368478 |
| hsa04010:MAPK
signaling pathway | 2 | 0.017081905 | 0.138117097 |
| hsa00630:Glyoxylate
and dicarboxylate metabolism | 1 | 0.022356127 | 0.138117097 |
| hsa03410:Base
excision repair | 1 | 0.026161426 | 0.138117097 |
| hsa04130:SNARE
interactions in vesicular transport | 1 | 0.026920764 | 0.138117097 |
|
hsa04962:Vasopressin-regulated water
reabsorption | 1 | 0.034482696 | 0.138117097 |
| hsa03420:Nucleotide
excision repair | 1 | 0.036740165 | 0.138117097 |
| hsa05030:Cocaine
addiction | 1 | 0.038242305 | 0.138117097 |
| hsa04978:Mineral
absorption | 1 | 0.040491267 | 0.138117097 |
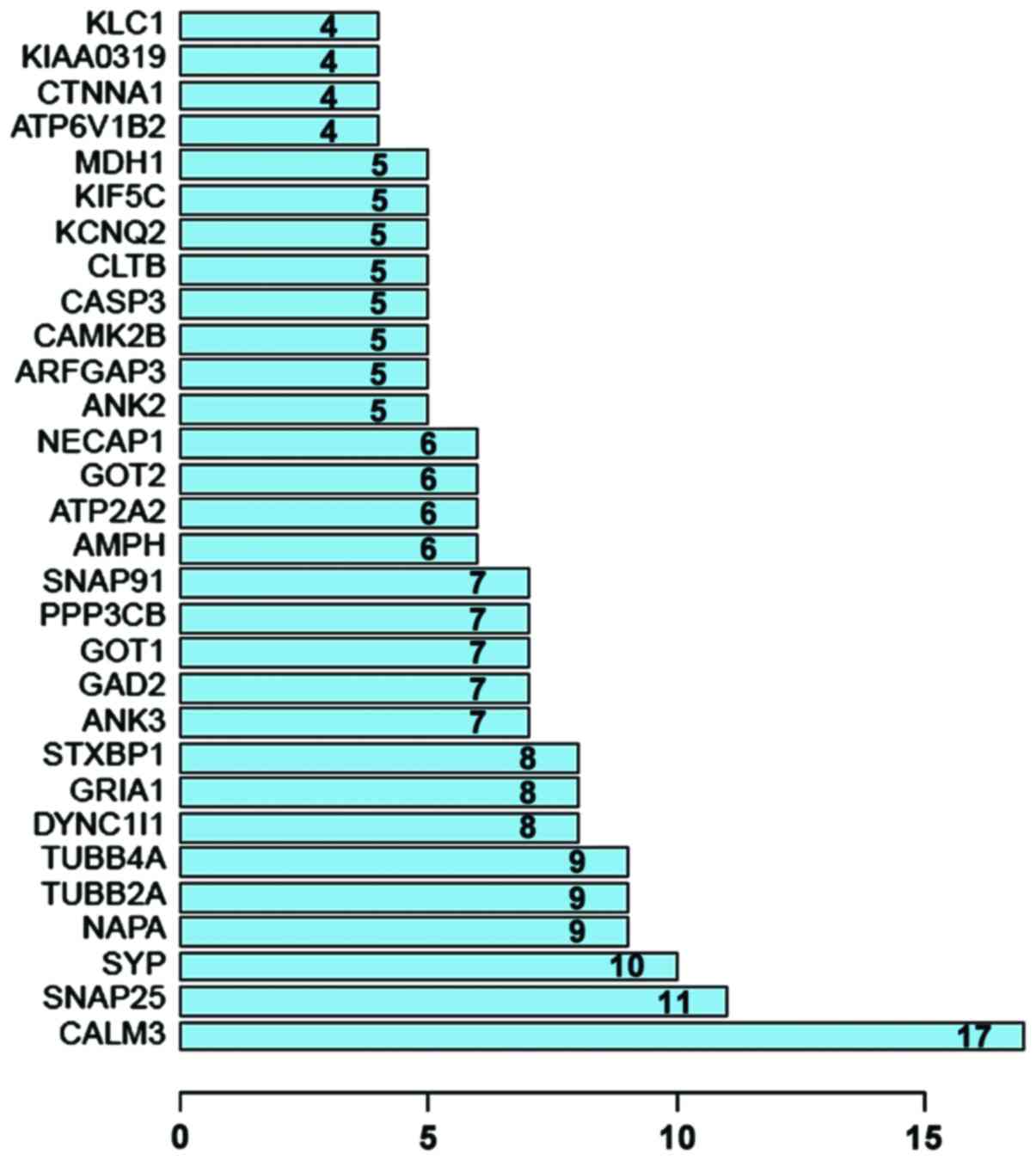
Protein interaction network analysis. Thirty
outstanding proteins were identified through PPI analysis of STRING
software. SNAP25, SYP, NAPA, TUBB2A and TUBB4A proteins were
relatively more important. As the most important protein, CALM3
connected 17 nodes (Figs. 2 and
3).
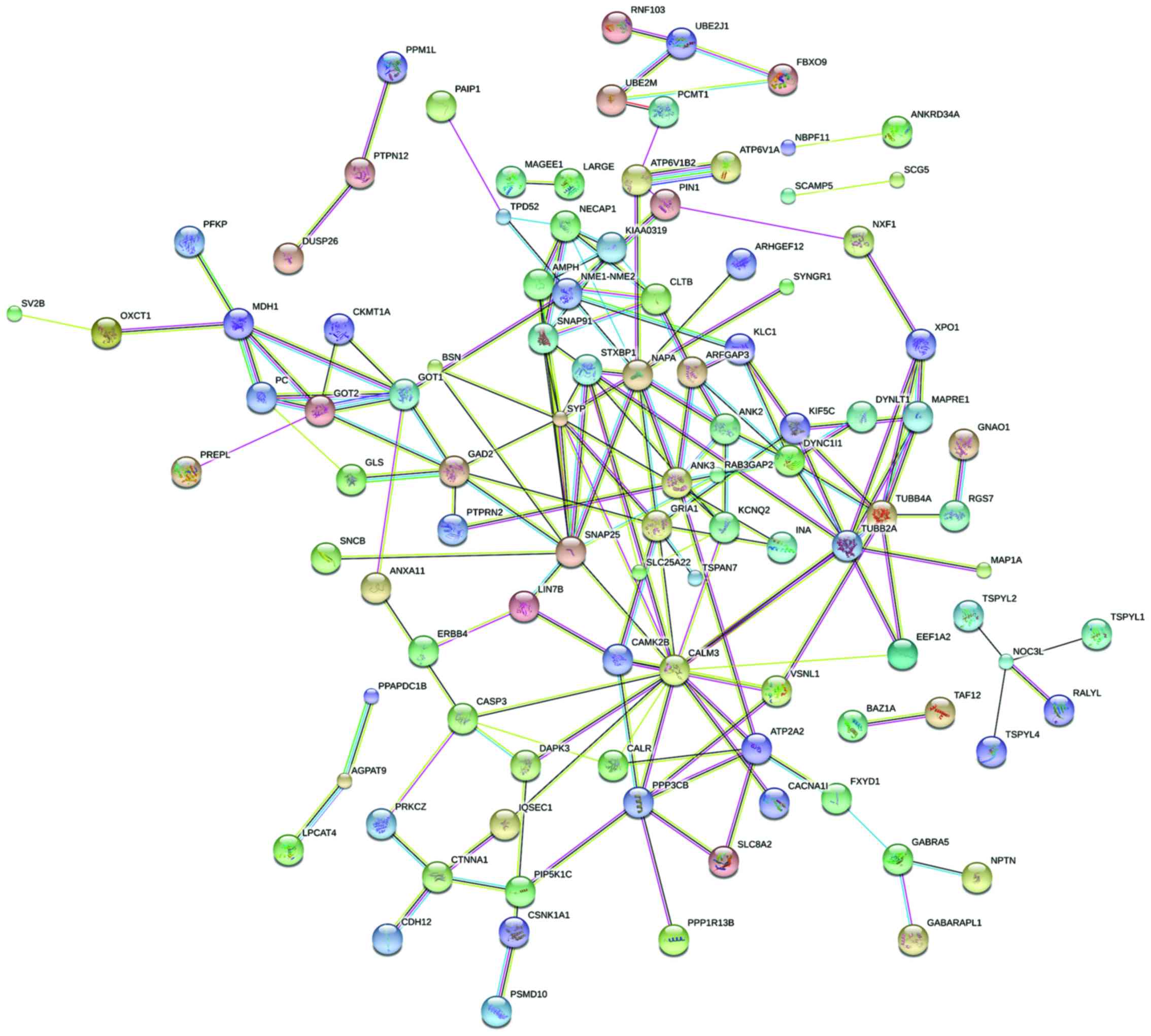 | Figure 2.The diagram of protein interaction
network. Circle represents the gene, and lines represent the
protein interaction between the genes, and the information inside
the circle describes protein structure: small nodes, protein of
unknown 3D structure; large nodes, some 3D structure is known or
predicted; a red line indicates the presence of fusion evidence; a
green line, neighborhood evidence; a blue line, coocurrence
evidence; a purple line, experimental evidence; a yellow line, text
mining evidence; a light blue line, database evidence; a black
line, coexpression evidence. |
Discussion
As the most common and most aggressive diffuse
glioma (6), GBM is the most malignant
type of astrocytic tumor. GBM develops from cortex and shows
infiltrative development. GBM can simultaneously affect several
lobes (7). GBM is characterized by
heterogeneity of morphology, genetics and gene expression (8). In recent years, with the rapid
development of bioinformatics, the use of gene expression profiles
to explore the relationship between gene differential expression
and disease development has attracted more and more attention.
However, those data have not been comprehensively investigated
(9). In this study, gene chip data
were used to identify the differentially expression genes between
GBM and normal human brain cells through enrichment analysis and
protein interaction analysis with the expectation of exploring the
pathogenesis of GBM. As the most common endogenous primary brain
tumor in adults, GBM represents the most common type of diffuse
glioma (10). Central Brain Tumor
Registry of the United States (CBTRUS) reported that GBM mainly
affect patients between 75 and 84 years, and the incidence of GBM
showing an increasing trend and incidence in white males is ever
higher (11). GBM is rare in children
and only account for ~3% of all primary brain and CNS tumors. The
five-year survival rate is about 12% for children and less than 5%
for adults (12). In spite of the
achievement in the treatment of GBM, the survival rate of patients
is still very low (13). GBM exists
in cerebral cortex and showed strong invasion ability, so the
course of disease is short and average survival period is only ~14
months (14). Without treatment, GBM
patients cannot survive more than 2 months, so the development of
effective diagnosis and treatment methods is always needed
(15). Previous studies have shown
that the development of GBM is very complex and is related to the
abnormal expression of proto-oncogenes or tumor suppressor genes,
which can lead to abnormal activation or dysregulation of
intracellular signaling pathways (16).
In this study, GEO public database was used. The
‘limma’ package was used to analyze and integrate data. A total of
167 differentially expressed genes were identified. GO enrichment
analysis and KEGG pathway analysis showed that the differentially
expressed genes were mainly involved in neurotransmitter
transporter activity, neurotransmitter:sodium symporter activity,
cellular processes, solute: sodium symporter activity, monoplast
processes, protein processing in endoplasmic reticulum,
dopaminergic synapses, MAPK signal transduction pathway and
glyoxylate and dicarboxylate metabolism. Neurotransmitter
transporter activity and protein processing in endoplasmic
reticulum are the two most outstanding pathways. However, CALM3
protein is the most influential protein on GBM in the network
formed by CALM3, SNAP25, SYP, NAPA, TUBB2A, TUBB4A, DYNC1I1, GRIA1,
STXBP1, ANK3, GOT1, GAD2, PPP3CB, SNAP91, AMPH, ATP2A2 and other
genes. CALM3 gene was reported to be closely associated with long
QT syndrome (17). More studies are
needed to investigate the mechanism of the roles of CALM3.
Bioinformatics is a new discipline that combines
biological science and computer science (18). Major bioinformatics tools were used in
this study to identify the differentially expressed genes. Public
database of gene chip data was used in this study, which
significantly reduced the use of financial and material resources.
Based on the strict inclusion criteria, the most reliable gene chip
data were selected to avoid errors.
This study is limited by the small sample size. Gene
expression in GBM can be altered by certain factors (19), and the small sample size failed to
cover different races and regions, which can affected the gene
expression in GBM (20).
In this study, CALM3 gene was proved to be related
to the protein processing and transporter activity in GBM. Our
future study will focus on those pathways. Studies on GBM at gene
level are rare. Therefore, more studies are needed to improve the
diagnosis, treatment and prognosis of GBM.
References
|
1
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jin Z, Jin RH, Ma C, Li HS and Xu HY:
Serum expression level of miR-504 can differentiate between
glioblastoma multiforme and solitary brain metastasis of non-small
cell lung carcinoma. J BUON. 22:474–480. 2017.PubMed/NCBI
|
|
3
|
Sharma V, Dixit D, Koul N, Mehta VS and
Sen E: Ras regulates interleukin-1β-induced HIF-1α transcriptional
activity in glioblastoma. J Mol Med (Berl). 89:123–136. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reed GJ, Boczek NJ, Etheridge SP and
Ackerman MJ: CALM3 mutation associated with long QT syndrome. Heart
Rhythm. 12:419–422. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Olar A and Aldape KD: Using the molecular
classification of glioblastoma to inform personalized treatment. J
Pathol. 232:165–177. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lun M, Lok E, Gautam S, Wu E and Wong ET:
The natural history of extracranial metastasis from glioblastoma
multiforme. J Neurooncol. 105:261–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Goodenberger ML and Jenkins RB: Genetics
of adult glioma. Cancer Genet. 205:613–621. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jones TS and Holland EC: Standard of care
therapy for malignant glioma and its effect on tumor and stromal
cells. Oncogene. 31:1995–2006. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: European Organisation for Research and Treatment
of Cancer Brain Tumour and Radiation Oncology Groups; National
Cancer Institute of Canada Clinical Trials Group: Effects of
radiotherapy with concomitant and adjuvant temozolomide versus
radiotherapy alone on survival in glioblastoma in a randomised
phase III study: Five-year analysis of the EORTC-NCIC trial. Lancet
Oncol. 10:459–466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bo LJ, Wei B, Li ZH, Wang ZF, Gao Z and
Miao Z: Bioinformatics analysis of miRNA expression profile between
primary and recurrent glioblastoma. Eur Rev Med Pharmacol Sci.
19:3579–3586. 2015.PubMed/NCBI
|
|
11
|
Carro MS, Lim WK, Alvarez MJ, Bollo RJ,
Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al:
The transcriptional network for mesenchymal transformation of brain
tumours. Nature. 463:318–325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lathia JD, Gallagher J, Myers JT, Li M,
Vasanji A, McLendon RE, Hjelmeland AB, Huang AY and Rich JN: Direct
in vivo evidence for tumor propagation by glioblastoma cancer stem
cells. PLoS One. 6:e248072011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie Q, Mittal S and Berens ME: Targeting
adaptive glioblastoma: An overview of proliferation and invasion.
Neuro-oncol. 16:1575–1584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Batchelor TT, Reardon DA, de Groot JF,
Wick W and Weller M: Antiangiogenic therapy for glioblastoma:
Current status and future prospects. Clin Cancer Res. 20:5612–5619.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie Y, Bergström T, Jiang Y, Johansson P,
Marinescu VD, Lindberg N, Segerman A, Wicher G, Niklasson M,
Baskaran S, et al: The human glioblastoma cell culture resource:
Validated cell models representing all molecular subtypes.
EBioMedicine. 2:1351–1363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo E, Wang Z and Wang S: miR-200c and
miR-141 inhibit ZEB1 synergistically and suppress glioma cell
growth and migration. Eur Rev Med Pharmacol Sci. 20:3385–3391.
2016.PubMed/NCBI
|
|
17
|
Joo KM, Kim J, Jin J, Kim M, Seol HJ,
Muradov J, Yang H, Choi YL, Park WY, Kong DS, et al:
Patient-specific orthotopic glioblastoma xenograft models
recapitulate the histopathology and biology of human glioblastomas
in situ. Cell Rep. 3:260–273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Meyer M, Reimand J, Lan X, Head R, Zhu X,
Kushida M, Bayani J, Pressey JC, Lionel AC, Clarke ID, et al:
Single cell-derived clonal analysis of human glioblastoma links
functional and genomic heterogeneity. Proc Natl Acad Sci USA.
112:851–856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ogden AT, Waziri AE, Lochhead RA, Fusco D,
Lopez K, Ellis JA, Kang J, Assanah M, McKhann GM, Sisti MB, et al:
Identification of A2B5+CD133−
tumor-initiating cells in adult human gliomas. Neurosurgery.
62:505–515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Savary K, Caglayan D, Caja L, Tzavlaki K,
Bin Nayeem S, Bergström T, Jiang Y, Uhrbom L, Forsberg-Nilsson K,
Westermark B, et al: Snail depletes the tumorigenic potential of
glioblastoma. Oncogene. 32:5409–5420. 2013. View Article : Google Scholar : PubMed/NCBI
|