Introduction
Pancreatic ductal adenocarcinoma (PDAC), one of the
most lethal of all types of human cancer, is a common cause of
cancer mortality in the USA and Japan (1). PDAC is the fourth most common cause of
cancer-associated mortality and its incidence is increasing
worldwide (2). PDAC displays local
invasion and distant metastasis during early disease stages and
this leads to an extremely poor prognosis, with an overall survival
rate of <5% (2). As such, there is
a requirement for novel and more effective treatment strategies.
Research has focused on the development of immunomodulatory
approaches (3).
Interactions between the immune system and malignant
cells serve an important role in tumorigenesis (4). Tumors have employed multiple mechanisms
of immune evasion. One such mechanism, particularly in high-grade
cancer, involves cytotoxic T lymphocyte (CTL) evasion through major
histocompatibility complex (MHC) class I downregulation (5). Tumor cells express cell-surface MHC
class I chain-related gene A/B (MICA/B), frequently induced under
cellular stress. MICA/B function as ligands for natural killer
group 2 member D (NKG2D), expressed on cytotoxic innate immune
cells, specifically γδT cells and natural killer (NK) cells
(6). Unlike conventional αβT cells,
γδT cells recognize antigens in an MHC-unrestricted manner, and may
provide a novel immunotherapeutic approach against tumor cells
(7,8).
Therefore, MICA/B expression in PDAC serve as targets for various
effector cells expressing NKG2D.
Histone deacetylase (HDAC) inhibitors, which alter
histone acetylation, are also promising anticancer agents (9,10). A
number of clinical studies are currently investigating HDAC
inhibitors, and certain HDAC inhibitors have already been approved
by the US Food and Drug Administration for the treatment of
cutaneous T cell lymphoma (11).
However, certain HDAC inhibitors are of limited therapeutic use
owing to toxic side effects at high doses. Valproic acid (VPA)
exhibits antitumor effects of HDAC inhibitors and has been
demonstrated to exert anticancer effects in various cancer models
(12,13). The therapeutic range of VPA is between
0.35 and 0.7 mM (14). A number of
studies have demonstrated that VPA stimulates the expression of
cell-surface MICA/B in a variety of tumors, enhancing the
susceptibility of tumor cells to cell-mediated cytotoxicity
(7,15–17).
Gemcitabine (GEM), a nucleoside analogue, is
commonly administered as an initial chemotherapy drug for the
treatment of pancreatic cancer (18).
GEM also induces MICA/B expression on the surface of pancreatic
cancer cells (19). In our previous
study, it was demonstrated that cell-surface MICA/B expression was
upregulated following low-dose GEM treatment, at a concentration
not affecting cell growth (20).
However, to the best of our knowledge, the combined
effects of VPA and GEM have not yet been investigated. In the
present study, the effect of VPA with GEM on the expression of
MICA/B was investigated in pancreatic cancer cell lines. It was
determined that the cytotoxic efficacy of γδT cells directed at
tumors is dependent on the ability of VPA and GEM to prevent tumor
immune evasion. This is facilitated by pancreatic cell-surface
MICA/B expression without cleavage or release of MIC molecules from
the tumor surface.
Materials and methods
Human pancreatic cancer cell
lines
Human pancreatic cancer cell lines (PANC-1, BxPC-3,
HPAF-II, MIA PaCa-2, Capan-1 and AsPC-1) were purchased from the
American Type Culture Collection (Manassas, VA, USA). These cell
lines were cultured in RPMI-1640 medium with 10% fetal bovine
serum, 10 mM HEPES, 1 mM sodium pyruvate, 1% non-essential amino
acid solution, 5×10−5 M 2-mercaptoethanol, 100 U/ml
penicillin and 100 µg/ml streptomycin (all from Thermo Fisher
Scientific, Inc., Waltham, MA, USA).
Antibodies and reagents
VPA was obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). GEM was obtained from Eli Lilly
(Indianapolis, IN, USA). Clear Back (human Fc receptor blocking
reagent; cat. no. MTG-001), anti-human MICA antibody (dilution,
1:100; cat. no. K0217-3), anti-human MICB antibody (dilution,
1:100; cat. no. K0220-3) and anti-mouse immunoglobulin
(Ig)G-fluorescein isothiocyanate (FITC) secondary antibody
(dilution, 1:160; cat. no. 238) were obtained from MBL
International Co. (Woburn, MA, USA). Phycoerythrin (PE)-conjugated
anti-human MICA/B antibody (dilution, 1:20; cat. no. 320906), mouse
IgG2a κ isotype control (dilution, 1:20; cat. no. 400211),
FITC-conjugated anti-human leukocyte antigen (HLA)-A, -B and -C
antibodies (dilution, 1:50; cat. no. 311403), and mouse IgG2a κ
isotype control (dilution, 1:50; cat. no. 400207) were purchased
from BioLegend, Inc. (San Diego, CA, USA). FITC-conjugated
anti-human Vγ9 antibody (dilution, 1:5; cat. no. IM1463) and
PE-cyanin 5.1-conjugated anti-human cluster of differentiation (CD)
3 antibody (dilution, 1:10; cat. no. A07749) were acquired from
Beckman Coulter, Inc. (Brea, CA, USA). ALyS-203, containing 1,000
IU/ml interleukin-2 (IL-2), was purchased from Cell Science &
Technology Institute (Sendai, Japan). Zoledronate
(Zometa®) was acquired from Novartis International AG
(Basel, Switzerland). Calcein-acetoxymethyl ester (AM) was obtained
from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan).
Anti-human NKG2D monoclonal antibody (mAb) (catalog No. MAB139) was
purchased from R&D Systems, Inc. (Minneapolis, MN, USA). WST-1
was acquired from Roche Diagnostics (Basel, Switzerland).
Expansion of γδT cells
γδT cells, obtained from healthy volunteers (n=3)
following provision of written informed consent, were expanded from
peripheral blood mononuclear cells (PBMCs), as described previously
(21). PBMCs were cultured with
ALys-203, containing 1,000 IU/ml IL-2 and 5 µM zoledronate. Cell
density was maintained at (0.5–2)x106 cells/ml.
Additional PBMCs were cultured with ALys-203, containing 1,000
IU/ml IL-2, in the absence of zoledronate. On day 12, cells were
harvested, and the frequency of γδT cells, identified by subsets
CD3+ and Vγ9+, were analyzed using flow
cytometry. These cells were used as effector cells (E) in the
cytotoxicity assay.
WST-1 assay
Human pancreatic cancer cell lines were treated with
various concentrations of VPA between 0 and 5 mM. After 48 h, the
viability of each cell line was analyzed using a WST-1 assay, as
described previously (22).
Immunofluorescence staining and flow
cytometry
Human pancreatic cancer cell lines were cultured in
a 10 mm tissue culture dish for 24 h. Each cell line was
subsequently treated with VPA alone, or VPA with GEM, for 48 h.
Prior to staining with fluorescent antibodies, the Fc receptor was
blocked with Clear Back. The expression of MICA/B, and HLA-A, -B
and -C on each pancreatic cancer cell line was determined by
immunofluorescence staining with PE-conjugated anti-MICA/B antibody
and FITC-conjugated HLA-A, -B and -C antibodies. To evaluate MICA/B
expression by each pancreatic cancer cell line, cells were first
stained with anti-MICA or -MICB antibody and subsequently stained
with anti-mouse IgG-FITC secondary antibody. Fluorescence was
analyzed using an EPICS XL flow cytometer (Beckman Coulter, Inc.,
Brea, CA, USA).
Cytotoxicity assay
γδT cell cytotoxicity against human pancreatic
cancer cell lines were evaluated using a calcein-AM release assay.
Pancreatic cancer cell lines were treated with 0.7 mM VPA, 0.001 µM
GEM or a combination of 0.7 mM VPA and 0.001 µM GEM for 48 h.
Subsequently, pancreatic cancer cell lines were labeled with 5 µM
calcein-AM for 30 min at 37°C. Following washing three times with
medium, calcein-AM-labeled cells were used as target cells (T). As
effector cells (E), γδT cells were expanded from PBMCs as
aforementioned. To evaluate γδT cell cytotoxicity, effector cells
were co-cultured with target cells at various E/T ratios (1,5 and
25) at 37°C for 3 h. In blocking experiments, anti-NKG2D mAb was
added to γδT cell suspension at 10 µg/ml, 30 min prior to
co-culturing with target cells. In order to measure the spontaneous
release and maximum release of fluorescence intensity from target
cells, medium or 6% Triton X-100 was added to target cells.
Fluorescence intensity was measured with Terascan VPC (Minerva
Tech, Tokyo, Japan) prior to and following culture. The percentage
of target cells killed by γδT cells was calculated using Calct-96l
software (Minerva Tech, Tokyo, Japan). Triplicate experiments were
performed.
ELISA
Following treatment of human pancreatic cancer cell
lines with 0.7 mM VPA, GEM (0.001, 0.1, 10 µM) or a combination of
0.7 mM VPA and GEM (0.001, 0.1, 10 µM) for 48 h, in order to detect
soluble MICA and MICB in the culture supernatant, sandwich ELISA.
An Ab-Match Universal kit (cat. no. 5310), Ab-Match ASSEMBLY Human
MICA kit (cat. no. 5330) and Ab-Match Assembly Human MICB kit (cat.
no. 5331, all MBL International Co., Woburn, MA, USA) were used
according to manufacturers' protocol.
Statistical analysis
Differences were analyzed for significance using
Student's t-test, Mann-Whitney rank sum test, χ2 test or
log-rank test, as appropriate. Data management and statistical
analysis were performed using SPSS software (version 15; SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
MICA/B, and HLA-A, -B and -C
expression by pancreatic cell lines
MICA/B, and HLA-A, -B, and -C expression by six
pancreatic cell lines were analyzed using flow cytometry. As
demonstrated in our previous study, the cell-surface MICA/B
expression was detected in four pancreatic cell lines (PANC-1,
BxPC-3, MIA PaCa-2 and Capan-1), but not in the remaining cell
lines (HPAF-II, AsPC-1) (20). The
HPAF-II cell line only expressed MICA but not MICB, and AsPC-1
expressed neither MICA nor MICB. Cell-surface expression of HLA-A,
-B and -C was detected in all pancreatic cell lines
investigated.
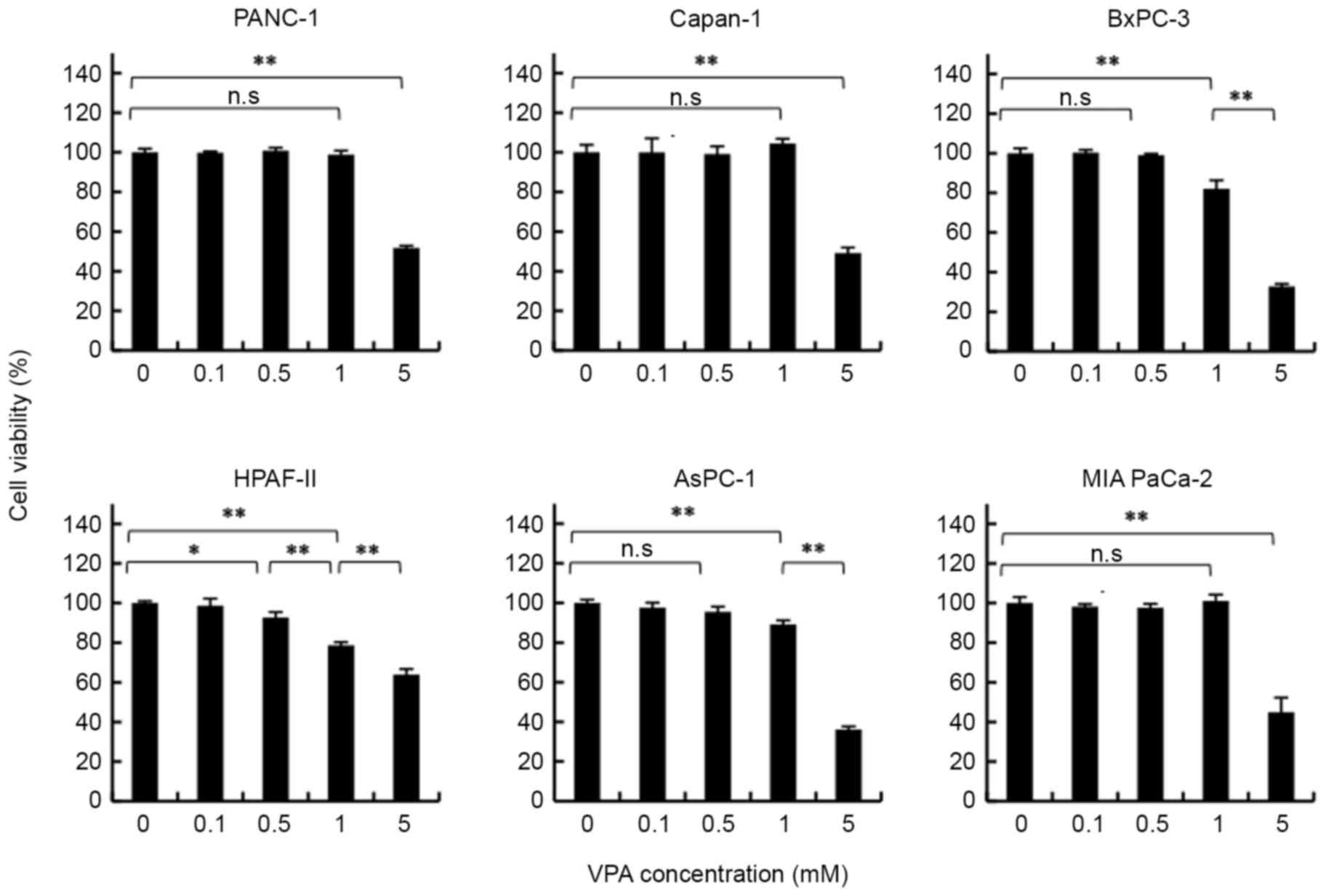
VPA concentration affects pancreatic
cancer cell growth
To determine whether the concentration of VPA had an
effect on the viability of each pancreatic cancer cell line, cell
lines were analyzed following treatment with various concentrations
of VPA for 48 h using a WST-1 assay. As presented in Fig. 1, VPA doses <1 mM did not affect the
viability of PANC-1, MIA PaCa-2, Capan-1 and AsPC-1 cells. However,
cell viability decreased with 1 mM VPA treatment in BxPC-3 and
HPAF-II cell lines. Following treatment with 5 mM VPA, cell
viability decreased in all pancreatic cancer cell lines. This
result indicated that VPA at a concentration of 5 mM has an
antineoplastic effect.
Effect of MICA/B, and HLA-A, -B and -C
expression on pancreatic cancer cell lines following VPA
treatment
MICA/B expression was quantified on pancreatic
cancer cell lines following treatment with various concentrations
of VPA. As presented in Fig. 2A, it
was determined that VPA treatment resulted in an alteration in
MICA/B expression in each pancreatic cancer cell line. MICA/B
expression was increased in VPA-treated MICA/B-positive cell lines
PANC-1, BxPC-3, HPAF-II, MIA PaCa-2 and Capan-1, but not in the
MICA/B-negative cell line AsPC-1. In MICA/B-positive cell lines,
the increase in cell-surface MICA/B expression was dependent on VPA
concentration. Following 5 mM VPA treatment, cell-surface MICA/B
expression increased in PANC-1 and BxPC-3 cells, and markedly in
MIA PaCa-2 cells, but not in AsPC-1 cells (Fig. 2B). This result demonstrates that
cell-surface MICA/B expression may be increased with high doses of
VPA in MICA/B-positive pancreatic cancer cell lines. As presented
in Fig. 2C and D, treatment with
various concentrations of VPA was able to alter HLA-A, -B and -C
expression levels, but not to the level observed with MICA/B. In
PANC-1 and Capan-1 cells, HLA-A, -B and -C expression was increased
following VPA treatment. However, HLA-A, -B and -C expression was
not observed to be increased in other pancreatic cancer cell
lines.
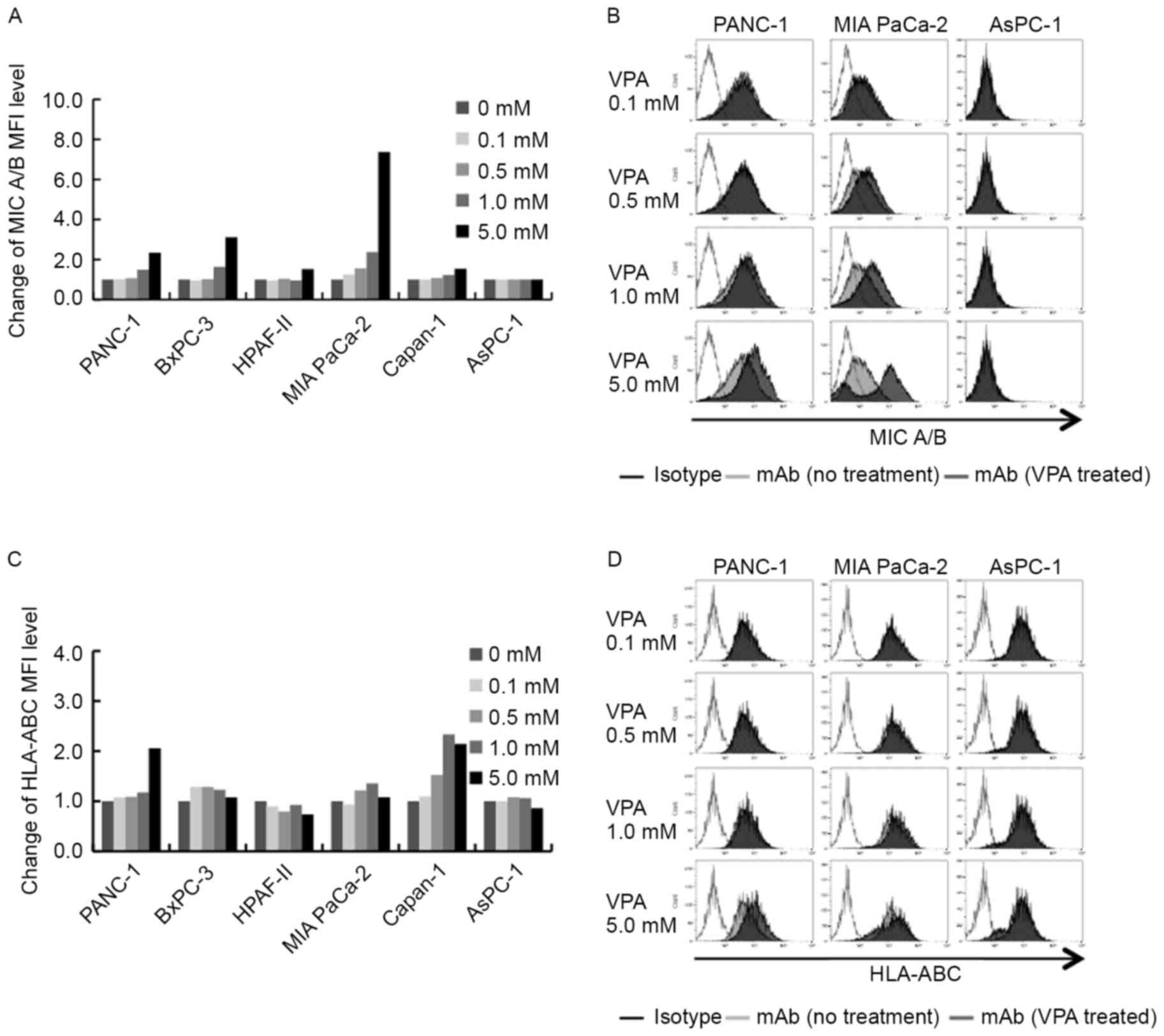 | Figure 2.Effects of VPA on the cell-surface
expression of MICA/B, and HLA-A, -B and -C. (A) Expression of
cell-surface MICA/B was increased in MICA/B-positive cell lines,
but not the MICA/B-negative cell line AsPC-1. (B) Representative
MICA/B expression profiles of PANC-1, MIA PaCa-2 and AsPC-1 cells
in the presence of VPA at between 0.1 and 5 mM. (C) Expression of
cell-surface HLA-A, -B and -C was increased in PANC-1 and Capan-1,
but not in the other cell lines. (D) Representative HLA-A, -B and
-C expression profiles of PANC-1, MIA PaCa-2 and AsPC-1 in the
presence of VPA at between 0.1 and 5 mM. VPA, valproic acid;
MICA/B, major histocompatibility complex class 1-related chain
A/B. |
MICA/B expression following treatment
with low-dose VPA and low-dose GEM
In an effort to increase MICA/B expression
effectively on the cell surface, MICA/B expression by pancreatic
cancer cell lines was investigated following treatment with
low-dose VPA combined with low-dose GEM.
Fig. 3A indicates that
the combination of 0.7 mM VPA and 0.001 mM GEM treatment resulted
in an alteration in the MICA/B expression level in each pancreatic
cancer cell line. VPA at 0.7 mM increased MICA/B expression on the
cell surface of the MICA/B-positive pancreatic cancer cell lines
PANC-1, BxPC-3, HPAF-II, MIA PaCa-2 and Capan-1. Likewise, MICA/B
expression on all MICA/B-positive pancreatic cancer cell lines,
except BxPC-3, was increased following low-dose GEM treatment. When
treated with a combination of VPA and GEM, MICA/B expression was
increased synergistically in pancreatic cancer cell lines to a
level greater than that with VPA or GEM treatment, individually.
Conversely, MICA/B expression was not increased in the
MICA/B-negative cell line AsPC-1 using VPA, GEM or a combination of
VPA and GEM treatment (Fig. 3B).
 | Figure 3.Effects of VPA, GEM and combination
of VPA and GEM on the cell-surface expression of MICA/B, and HLA-A,
-B and -C. (A) Expression of cell-surface MICA/B with the
combination of low-dose VPA and low-dose GEM treatment was
increased compared with that following VPA or GEM treatment alone,
in MICA/B-positive pancreatic cancer cell lines, but not the
MICA/B-negative pancreatic cell line AsPC-1. (B) Representative
MICA/B expression profiles for PANC-1 and AsPC-1 cells following
treatment with VPA, GEM, and the combination of VPA with GEM. (C)
Cell-surface expression of HLA-A, -B and -C was not increased in
pancreatic cancer cell lines. (D) Representative HLA-A, -B and -C
expression profiles for PANC-1 and AsPC-1 cells in the presence of
GEM, VPA, and the combination of VPA with GEM. VPA, valproic acid;
GEM, gemcitabine; MICA/B, MICA/B, major histocompatibility complex
class 1-related chain A/B. |
As presented in Fig. 3C
and D, the administration of low-dose VPA and low-dose GEM
resulted in an alteration in the HLA-A, -B and -C expression level
in each pancreatic cancer cell line. In all pancreatic cancer cell
lines, except HPAF-II, HLA-A, -B and -C expression was slightly
increased with a combination of VPA and GEM treatment.
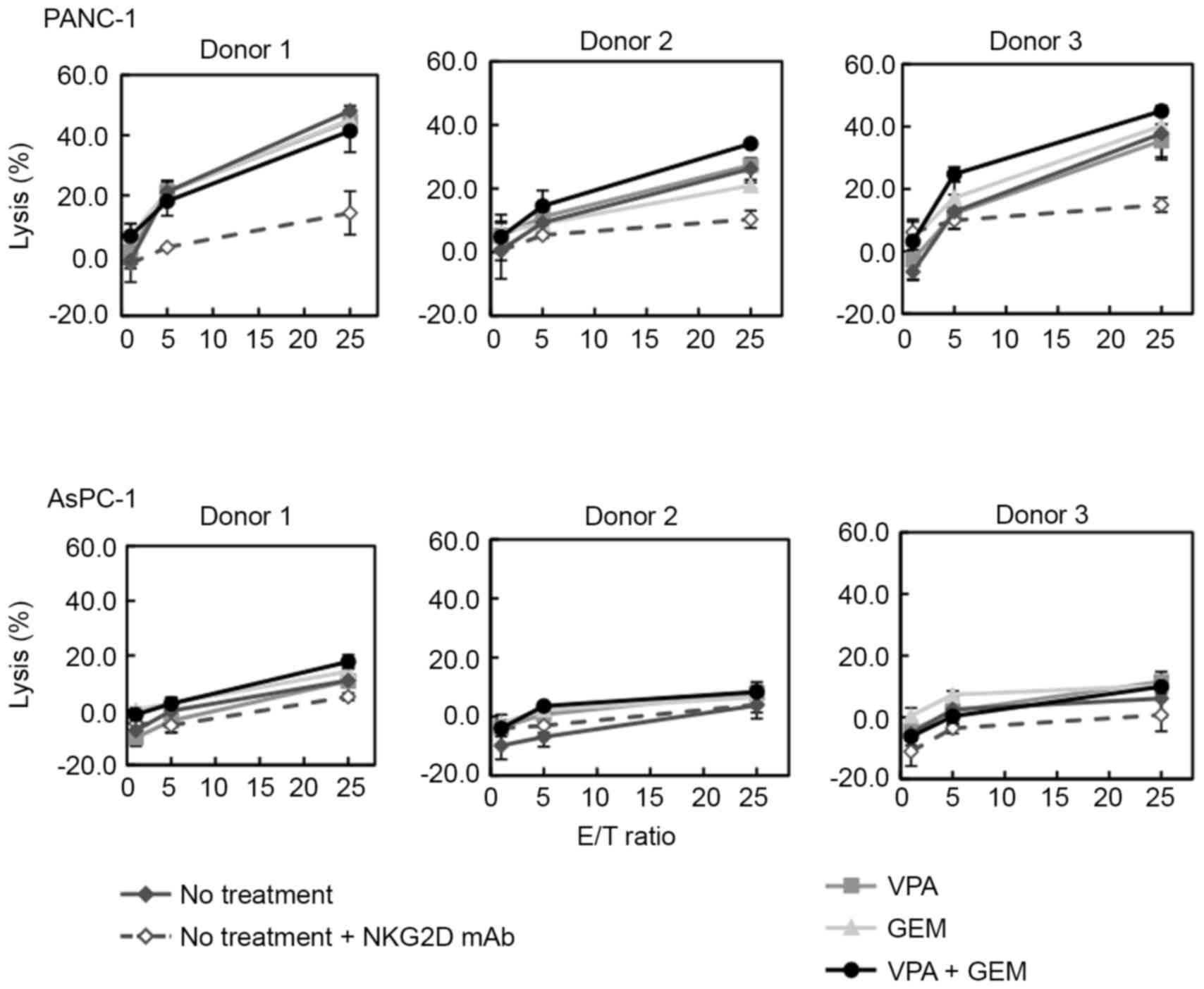
Cytotoxic activity of γδT cells
against pancreatic cancer cell lines treated with low-dose VPA and
low-dose GEM combination treatment
To assess whether NKG2D-dependent cytotoxic activity
was enhanced due to increased MICA/B expression on target cells,
γδT cell cytotoxicity against pancreatic cancer cell lines treated
with 0.7 mM VPA, 0.001 µM GEM or a combination of VPA and GEM was
evaluated. As presented in Fig. 4,
cytotoxic activity of γδT cells against PANC-1 was observed. In
addition, this cytotoxic activity was blocked using the anti-NKG2D
antibody. γδT cells killed PANC-1 cells, which was dependent on the
interaction of NKG2D with MICA/B. The cytotoxic activity of γδT
cells was enhanced with VPA or GEM, when administered individually.
However, when target cells were treated with a low-dose combination
of VPA and GEM, there was enhancement of γδT cell cytotoxicity
against tumor cells. In the MICA/B-negative cell line AsPC-1, the
cytotoxic activity of γδT cells was not detected with or without
VPA, GEM or a combination of VPA and GEM (Fig. 4).
Release of soluble MICA/B from
pancreatic cancer cell lines treated with GEM or VPA+GEM
The results of the present study demonstrate that
low-dose VPA (0.7 mM) with a cytostatic concentration of GEM (0.001
µM) most effectively increased MICA/B expression in PANC-1 cells.
To examine the value of the combination of VPA with GEM, the
presence of soluble MICA/B in the culture supernatant was
determined using ELISA. This would indicate that MICA/B was
released from pancreatic cancer cell lines treated with GEM, or
with a combination of VPA and GEM.
In PANC-1 cells (Fig.
5), a high concentration of soluble MICA/B was detected in the
culture supernatant. When PANC-1 cells were treated with various
doses of GEM, soluble MICA/B remained at high concentrations in the
culture supernatant. Soluble MICA/B decreased when treated with
low-dose GEM combined with low-dose VPA.
Soluble MICA/B was released, to a minimal degree, in
the culture supernatant of AsPC-1 (Fig.
5). Soluble MICB was detected, despite a lack of cell-surface
expression, in this cell line.
Discussion
In humans, MHC-associated molecules termed MICA/B,
are well known as NKG2D ligands. Rarely expressed in healthy cells,
MICA/B are frequently expressed in epithelial tumor cells, and in
cells under stress. These cells under stress include those
undergoing heat shock, viral infection and DNA damage (23,24). The
immunoreceptor that recognizes MICA/B, NKG2D, is expressed by γδT
and NK cells. Therefore, it may be possible that NKG2D-dependent
cytotoxic activity is enhanced with increased MICA/B
expression.
VPA has been used clinically to treat migraines and
as a mood stabilizer. The therapeutic dose of VPA ranges between 15
and 60 mg/kg/day, and results in a maximum plasma concentrations of
130 µg/ml (0.9 mM) (25). VPA levels
in epilepsy patients are typically <0.7 mM (26). VPA has been found to act as an HDAC
inhibitor (27). Concentrations of
VPA for histone acetylation range between 0.25 and 5 mM. In
addition, it has been reported that HDAC inhibitors display
antineoplastic activity (28,29) and induce NKG2D ligands, such as MICA/B
and UL16-binding proteins, on tumor cells (15,30).
Furthermore, in vivo and in vitro studies have
demonstrated that VPA enhances NK cell-mediated lysis and
upregulates MICA/B expression in pancreatic cancer through the
activation of the phosphoinositide 3-kinase/protein kinase B
signaling pathway, which has important implications in the
pancreatic cancer stroma microenvironment (31,32).
The results of the present study indicate that
increased cell-surface MICA/B expression on pancreatic cancer cell
lines was dependent on the VPA concentration. Furthermore, when
pancreatic cancer cell lines were treated with 5 mM VPA treatment,
the viability of those cell lines were decreased and cell-surface
MICA/B expression was increased markedly. Therefore, it may be
possible that high doses of VPA treatment combined with
immunotherapy are able to induce a marked antitumor response. This
is due to the enhanced NKG2D-dependent cytotoxicity of immune cells
and the antineoplastic effect of VPA. However, because the maximum
plasma concentration is 0.9 mM following VPA administration
(25), it is necessary to induce the
antineoplastic effect of VPA in low doses. Low-dose VPA, <1 mM,
was unable to kill pancreatic cancer cell lines with its
antineoplastic activity and slightly increased MICA/B expression.
Therefore, it was attempted to increase MICA/B expression
effectively with low-dose VPA combined with low-dose GEM, as
another immunomodulatory reagent.
GEM is a chemotherapeutic agent that has an
immunomodulatory effect. It functions by upregulating the
cell-surface MICA/B expression for various types of cancer. Results
of our previous study indicated that MICA/B expression on the cell
surface was increased effectively at low doses of GEM, with no
effect on cell viability (20). The
results of the present study indicate that the combination of
low-dose VPA with low-dose GEM is able to increase cell-surface
MICA/B expression on pancreatic cancer cell lines. In addition,
NKG2D-dependent cytotoxicity of γδT cells was enhanced with the
increase of MICA/B expression by pancreatic cancer cell lines. When
PANC-1 was treated with a combination of VPA and GEM, MICA/B
expression on the cell surface was increased 2.5-fold in comparison
with those untreated in the same cell line.
γδT cells are able to recognize and respond to
various stress-induced antigens, thereby developing innate immunity
(33). They also exhibit potent
effector functions, including cytotoxic activity, and the secretion
of cytokines/chemokines. The majority of γδT cells in peripheral
blood possess the Vγ9Vδ2 T cell receptor. The cytotoxicity of these
γδT cells is mediated by perforin-granzyme, CD95/CD95 ligands,
tumor necrosis factor (TNF)/TNF receptors and TNF-related
apoptosis-inducing ligand (TRAIL)/TRAIL receptor (TRAILR) systems
(34). It may be possible that the
cytotoxic activity of γδT cells was enhanced through the activation
of a number of effector functions. It was reported that HDAC
inhibitors were able to increase TRAIL sensitivity in target tumor
cells (35). In the present study, it
may be possible that cytotoxicity was enhanced by a combination of
effector functions, NKG2D-NKG2D ligand interactions and
TRAIL/TRAILR sensitivity following the treatment combination of VPA
and GEM. In TRAIL-resistant pancreatic cancer cell lines, it was
shown that the inhibition of anti-apoptotic B-cell lymphoma extra
large (Bcl-XL) protein promoted apoptosis with TRAIL (36). Therefore, the addition of another
agent that is able to inhibit Bcl-XL expression may enhance the
cytotoxic activity of γδT cells, when TRAIL-resistant tumor cells
are treated with VPA and GEM.
A number of clinical studies have emphasized the
therapeutic benefits of immunotherapy in combination with
chemotherapy (37,38). Therefore, to improve therapeutic
benefits, it may be required to combine another chemotherapeutic
agent with immunomodulatory agents. One effective approach to
improve the therapeutic efficacy of immunotherapy is to prevent
immune evasion mechanisms by tumor cells.
In a previous study, it was demonstrated that the
release of MIC molecules on the cell surface constitutes an immune
evasion mechanism in tumor cells (39). Soluble MICA/B, cleaved on tumor cells,
downregulate the cell-surface expression of NKG2D on T cells. This
induces the functional impairment of antitumor immune effector
cells. As such, proteolytic cleavage may decrease the expression of
NKG2D ligands on tumor cell surfaces and contribute to tumor
evasion from immunosurveillance. Soluble MIC is perceived as a
tumor immune evasion mechanism that may have an effect on the
expression of NKG2D, and functional activity of NK cells and T cell
subsets (40). MICA/B are cleaved by
a disintegrin and metalloproteinase (ADAM) 10 and ADAM17,
respectively (40). In the tumor
microenvironment, soluble MICA induces the internalization and
lysosomal degradation of the NKG2D receptor in CD8+ T
and NK cells (41). It may be
possible that the inhibition of these proteases enhances
NKG2D-dependent cytotoxicity. This is due to an increase in MICA/B
expression on target cell surfaces, and decrease in soluble MICA/B
induction through the prevention of MICA/B cleavage. In our
previous study, there was a problem with cleavage and release of
MIC molecules from the tumor surface, which resulted in tumors
evading immunosurveillance (20). In
the present study, soluble MICA/B expression decreased with the
combination of low-dose VPA and low-dose GEM, despite increased
expression in PANC-1 cells. On the surface of osteosarcoma cells,
VPA has been reported to be associated with the downregulation of
ADAM10, which cleaves MICA/B (42).
In addition, VPA downregulates the activity of matrix
metalloproteinases which produce soluble MICA/B (43,44).
In order to prevent the tumor cell immune evasion
mechanism and implement effective therapeutic efficacy in
immunotherapy, it may be essential to combine a number of
immunomodulatory agents and enhance effector functions on immune
cells.
The results of the present study indicate that the
combined administration of low-dose VPA and low-dose GEM is
valuable in enhancing the therapeutic efficacy of immunotherapy by
upregulating MICA/B without inducing soluble MIC from being
released in pancreatic cancer.
Acknowledgements
The present study was supported by the Japan Society
for the Promotion of Science (KAKENHI grant no. 25462106).
Glossary
Abbreviations
Abbreviations:
|
ADAM
|
a disintegrin and
metalloproteinase
|
|
CTL
|
cytotoxic T lymphocyte
|
|
GEM
|
gemcitabine
|
|
HDAC
|
histone deacetylase
|
|
HLA
|
human leukocyte antigen
|
|
MICA/B
|
major histocompatibility complex class
1-related chain A/B
|
|
NK
|
natural killer
|
|
NKG2D
|
natural killer group 2 member D
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
VPA
|
valproic acid
|
References
|
1
|
Lowenfels AB and Maisonneuve P:
Epidemiology and prevention of pancreatic cancer. Jpn J Clin Oncol.
34:238–244. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: The impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jaffee EM, Hruban RH, Biedrzycki B, Laheru
D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L,
Donehower RC, et al: Novel allogeneic granulocyte-macrophage
colony-stimulating factor-secreting tumor vaccine for pancreatic
cancer: A phase I trial of safety and immune activation. J Clin
Oncol. 19:145–156. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Igney FH and Krammer PH: Immune escape of
tumors: Apoptosis resistance and tumor counterattack. J Leukoc
Biol. 71:907–920. 2002.PubMed/NCBI
|
|
5
|
Bernal M, Garrido P, Jiménez P, Carretero
R, Almagro M, López P, Navarro P, Garrido F and Ruiz-Cabello F:
Changes in activatory and inhibitory natural killer (NK) receptors
may induce progression to multiple myeloma: Implications for tumor
evasion of T and NK cells. Hum Immunol. 70:854–857. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nausch N and Cerwenka A: NKG2D ligands in
tumor immunity. Oncogene. 27:5944–5958. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suzuki T, Terao S, Acharya B, Naoe M,
Yamamoto S, Okamura H and Gotoh A: The antitumour effect of
{gamma}{delta} T-cells is enhanced by valproic acid-induced
up-regulation of NKG2D ligands. Anticancer Res. 30:4509–4513.
2010.PubMed/NCBI
|
|
8
|
Groh V, Rhinehart R, Secrist H, Bauer S,
Grabstein KH and Spies T: Broad tumor-associated expression and
recognition by tumor-derived gamma delta T cells of MICA and MICB.
Proc Natl Acad Sci USA. 96:6879–6884. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Glozak MA and Seto E: Histone deacetylases
and cancer. Oncogene. 26:5420–5432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lane AA and Chabner BA: Histone
deacetylase inhibitors in cancer therapy. J Clin Oncol.
27:5459–5468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mann BS, Johnson JR, Cohen MH, Justice R
and Pazdur R: FDA approval summary: Vorinostat for treatment of
advanced primary cutaneous T-cell lymphoma. Oncologist.
12:1247–1252. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yagi Y, Fushida S, Harada S, Kinoshita J,
Makino I, Oyama K, Tajima H, Fujita H, Takamura H, Ninomiya I, et
al: Effects of valproic acid on the cell cycle and apoptosis
through acetylation of histone and tubulin in a scirrhous gastric
cancer cell line. J Exp Clin Cancer Res. 29:1492010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shoji M, Ninomiya I, Makino I, Kinoshita
J, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima H, Takamura
H, et al: Valproic acid, a histone deacetylase inhibitor, enhances
radiosensitivity in esophageal squamous cell carcinoma. Int J
Oncol. 40:2140–2146. 2012.PubMed/NCBI
|
|
14
|
Franssen EJ, van Essen GG, Portman AT, de
Jong J, Go G, Stegeman CA and Uges DR: Valproic acid
toxicokinetics: Serial hemodialysis and hemoperfusion. Ther Drug
Monit. 21:289–292. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Armeanu S, Bitzer M, Lauer UM, Venturelli
S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, et
al: Natural killer cell-mediated lysis of hepatoma cells via
specific induction of NKG2D ligands by the histone deacetylase
inhibitor sodium valproate. Cancer Res. 65:6321–6329. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Poggi A, Catellani S, Garuti A, Pierri I,
Gobbi M and Zocchi MR: Effective in vivo induction of NKG2D ligands
in acute myeloid leukaemias by all-trans-retinoic acid or sodium
valproate. Leukemia. 23:641–648. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamanegi K, Yamane J, Kobayashi K,
Kato-Kogoe N, Ohyama H, Nakasho K, Yamada N, Hata M, Nishioka T,
Fukunaga S, et al: Sodium valproate, a histone deacetylase
inhibitor, augments the expression of cell-surface NKG2D ligands,
MICA/B, without increasing their soluble forms to enhance
susceptibility of human osteosarcoma cells to NK cell-mediated
cytotoxicity. Oncol Rep. 24:1621–1627. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu X, Rao GS, Groh V, Spies T, Gattuso P,
Kaufman HL, Plate J and Prinz RA: Major histocompatibility complex
class I-related chain A/B (MICA/B) expression in tumor tissue and
serum of pancreatic cancer: Role of uric acid accumulation in
gemcitabine-induced MICA/B expression. BMC Cancer. 11:1942011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyashita T, Miki K, Kamigaki T, Makino I,
Nakagawara H, Tajima H, Takamura H, Kitagawa H, Fushida S, Ahmed
AK, et al: Low-dose gemcitabine induces major histocompatibility
complex class I-related chain A/B expression and enhances an
antitumor innate immune response in pancreatic cancer. Clin Exp
Med. 17:19–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kondo M, Izumi T, Fujieda N, Kondo A,
Morishita T, Matsushita H and Kakimi K: Expansion of human
peripheral blood γδ T cells using zoledronate. J Vis Exp. pii:3182.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tatsumi T, Takehara T, Yamaguchi S,
Sasakawa A, Sakamori R, Ohkawa K, Kohga K, Uemura A and Hayashi N:
Intrahepatic delivery of alpha-galactosylceramide-pulsed dendritic
cells suppresses liver tumor. Hepatology. 45:22–30. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu J, Aggarwal R, Kanji S, Das M, Joseph
M, Pompili V and Das H: Human ovarian tumor cells escape γδ T cell
recognition partly by down regulating surface expression of MICA
and limiting cell cycle related molecules. PLoS One. 6:e233482011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu J, Groh V and Spies T: T cell antigen
receptor engagement and specificity in the recognition of
stress-inducible MHC class I-related chains by human epithelial
gamma delta T cells. J Immunol. 169:1236–1240. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marchion D and Münster P: Development of
histone deacetylase inhibitors for cancer treatment. Expert Rev
Anticancer Ther. 7:583–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Catalano MG, Fortunati N, Pugliese M,
Costantino L, Poli R, Bosco O and Boccuzzi G: Valproic acid induces
apoptosis and cell cycle arrest in poorly differentiated thyroid
cancer cells. J Clin Endocrinol Metab. 90:1383–1389. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Phiel CJ, Zhang F, Huang EY, Guenther MG,
Lazar MA and Klein PS: Histone deacetylase is a direct target of
valproic acid, a potent anticonvulsant, mood stabilizer, and
teratogen. J Biol Chem. 276:36734–36741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blaheta RA and Cinatl J Jr: Anti-tumor
mechanisms of valproate: A novel role for an old drug. Med Res Rev.
22:492–511. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG
and Heinzel T: Valproic acid defines a novel class of HDAC
inhibitors inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang F, Shao Y, Yang F, Liu M, Huang J,
Zhu K, Guo C, Luo J, Li W, Yang B, et al: Valproic acid upregulates
NKG2D ligand expression and enhances susceptibility of human renal
carcinoma cells to NK cell-mediated cytotoxicity. Arch Med Sci.
9:323–331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shi P, Yin T, Zhou F, Cui P, Gou S and
Wang C: Valproic acid sensitizes pancreatic cancer cells to natural
killer cell-mediated lysis by upregulating MICA and MICB via the
PI3K/Akt signaling pathway. BMC Cancer. 14:3702014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takikawa T, Masamune A, Hamada S, Nakano
E, Yoshida N and Shimosegawa T: miR-210 regulates the interaction
between pancreatic cancer cells and stellate cells. Biochem Biophys
Res Commun. 437:433–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bonneville M, O'Brien RL and Born WK:
Gammadelta T cell effector functions: A blend of innate programming
and acquired plasticity. Nat Rev Immunol. 10:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Todaro M, Meraviglia S, Caccamo N, Stassi
G and Dieli F: Combining conventional chemotherapy and γδ T
cell-based immunotherapy to target cancer-initiating cells.
Oncoimmunology. 2:e258212013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schüler S, Fritsche P, Diersch S, Arlt A,
Schmid RM, Saur D and Schneider G: HDAC2 attenuates TRAIL-induced
apoptosis of pancreatic cancer cells. Mol Cancer. 9:802010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bai J, Sui J, Demirjian A, Vollmer CM Jr,
Marasco W and Callery MP: Predominant Bcl-XL knockdown disables
antiapoptotic mechanisms: Tumor necrosis factor-related
apoptosis-inducing ligand-based triple chemotherapy overcomes
chemoresistance in pancreatic cancer cells in vitro. Cancer Res.
65:2344–2352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laheru D, Lutz E, Burke J, Biedrzycki B,
Solt S, Onners B, Tartakovsky I, Nemunaitis J, Le D, Sugar E, et
al: Allogeneic granulocyte macrophage colony-stimulating
factor-secreting tumor immunotherapy alone or in sequence with
cyclophosphamide for metastatic pancreatic cancer: A pilot study of
safety, feasibility, and immune activation. Clin Cancer Res.
14:1455–1463. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen G and Emens LA: Chemoimmunotherapy:
Reengineering tumor immunity. Cancer Immunol Immunother.
62:203–216. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salih HR, Rammensee HG and Steinle A:
Cutting edge: Down-regulation of MICA on human tumors by
proteolytic shedding. J Immunol. 169:4098–4102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chitadze G, Lettau M, Bhat J, Wesch D,
Steinle A, Fürst D, Mytilineos J, Kalthoff H, Janssen O, Oberg HH
and Kabelitz D: Shedding of endogenous MHC class I-related chain
molecules A and B from different human tumor entities:
Heterogeneous involvement of the ‘a disintegrin and
metalloproteases’ 10 and 17. Int J Cancer. 133:1557–1566. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jinushi M, Takehara T, Tatsumi T,
Hiramatsu N, Sakamori R, Yamaguchi S and Hayashi N: Impairment of
natural killer cell and dendritic cell functions by the soluble
form of MHC class I-related chain A in advanced human
hepatocellular carcinomas. J Hepatol. 43:1013–1020. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yamanegi K, Yamane J, Kobayashi K, Ohyama
H, Nakasho K, Yamada N, Hata M, Fukunaga S, Futani H, Okamura H and
Terada N: Downregulation of matrix metalloproteinase-9 mRNA by
valproic acid plays a role in inhibiting the shedding of MHC class
I-related molecules A and B on the surface of human osteosarcoma
cells. Oncol Rep. 28:1585–1590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yamanegi K, Yamane J, Kobayashi K,
Kato-Kogoe N, Ohyama H, Nakasho K, Yamada N, Hata M, Fukunaga S,
Futani H, et al: Valproic acid cooperates with hydralazine to
augment the susceptibility of human osteosarcoma cells to Fas- and
NK cell-mediated cell death. Int J Oncol. 41:83–91. 2012.PubMed/NCBI
|
|
44
|
Lee KH, Choi EY, Kim MK, Kim KO, Jang BI,
Kim SW, Kim SW, Song SK and Kim JR: Inhibition of histone
deacetylase activity down-regulates urokinase plasminogen activator
and matrix metalloproteinase-9 expression in gastric cancer. Mol
Cell Biochem. 343:163–171. 2010. View Article : Google Scholar : PubMed/NCBI
|