Introduction
Baicalin is a flavonoid compound isolated from
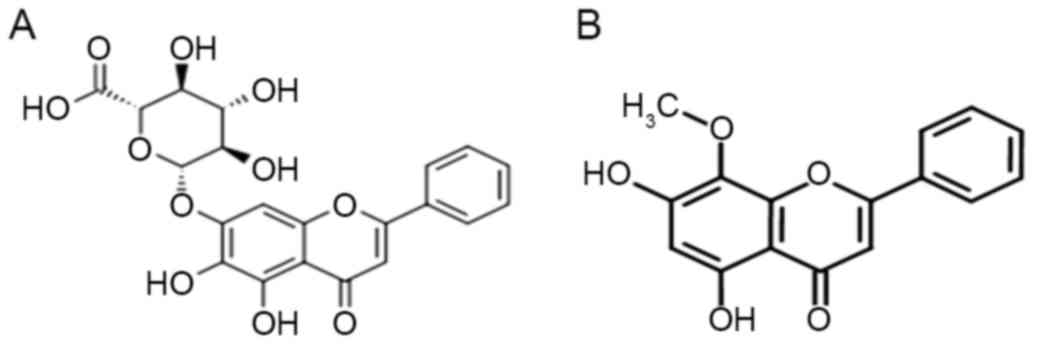
Scutellaria baicalensis (Fig.
1A), a Chinese traditional medicinal herb used as an
anti-inflammatory (1), antibacterial,
anxiolytic and hepatoprotective drug (2). Accumulating evidence has demonstrated
that baicalin exhibits potent antitumor properties by suppressing
cell growth, arresting the cell cycle and inducing differentiation
or apoptosis in leukemia cell lines (3,4), without
affecting primary, or normal cells (5), which raises the possibility of using
baicalin to treat patients with leukemia. Baicalin has been
documented to induce apoptosis of the Jurkat T-cell acute
lymphoblastic leukemia (T-ALL) cell line via the mitochondrial
pathway, which involves marginal generation of intracellular
reactive oxygen species (ROS), an increase in the cytosolic
fractions of cytochrome c and disruption of the
mitochondrial transmembrane potential (∆Ψm) (6). Another study revealed that baicalin
exhibited a remarkable cytotoxic effect in T-ALL cell line
CCRF-CEM, and triggered apoptosis regulator Bcl-2
(Bcl-2)-dependent, but not p53-dependent cell apoptosis (7). In addition, cyclin-dependent kinase
inhibitor p27 has been suggested to serve a role in
baicalin-induced apoptosis and cell cycle arrest (3). A study by Lu et al (8) confirmed that baicalin induced apoptosis
in human promyelocytic leukemia HL-60 cells through the
mitochondrial-involved pathway and endoplasmic reticulum-induced
apoptotic cell death. While to date, whether the extrinsic pathway
(death receptor pathway) is involved in apoptosis induced by
baicalin in leukemia cells has not been clarified, and the
mechanisms underlying the antitumor activity of baicalin remain
unclear.
The proliferation of HL-60 cells has also been
demonstrated to be inhibited by baicalin (8); however, its underlying mechanism
requires further elucidation. Myc proto-oncogene protein (c-Myc) is
a transcription factor that participates in various cellular
functions, including cell-cycle progression, proliferation,
apoptosis and terminal differentiation (9). The overexpression of c-Myc mRNA and its
encoded protein has been associated with neoplastic transformation
in a variety of tumors (10).
Therefore, perturbation of c-Myc levels is essential for
influencing the growth of malignant cells, particularly those of
hematopoietic origin (11). Wogonin,
another major bioactive flavonoid extracted from Scutellaria
baicalensis has been revealed to inhibit HL-60 cell growth via
telomerase inhibition through suppression of c-Myc (12). Due to the similar molecular structure
between baicalin and wogonin (Fig.
1B), it was hypothesized that baicalin may be able to inhibit
the growth of HL-60 cells via the same mechanism as wogonin.
In the present study, the effect of baicalin on the
expression of caspase-8, Fas cell surface death receptor (Fas), Fas
ligand (FasL), death receptor (DR)4 and DR5 in HL-60 cells was
assessed, and it was identified that the Fas-mediated extrinsic
pathway was involved in baicalin-triggered cell apoptosis, in
addition to the intrinsic pathway. Furthermore, baicalin was able
to inhibit the proliferation of HL-60 cells by arresting cells at
the G0/G1 phase and downregulating c-Myc
along with its target gene, human telomerase reverse transcriptase
(hTERT).
Materials and methods
Reagents
Baicalin was provided by Professor Xiao Wang
(Shandong Analysis and Test Center, Shandong Academy of Sciences,
Jinan, China). Baicalin was dissolved in DMSO at a stock
concentration of 10 mg/ml and stored at −20°C. Hoechst 33342 was
obtained from Beyotime Institute of Biotechnology (Nantong, China).
β-actin (cat. no. sc130065; dilution, 1:500) and c-Myc (cat. no.
sc-47694; dilution, 1:400) antibodies were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA). Antibodies directed
against apoptosis regulator Bax (Bax; cat. no. 2774; dilution,
1:1,000), Bcl-2 (cat. no. 2872; dilution, 1:1,000), cleaved
caspase-3 (cat. no. 9661; dilution, 1:1,000), caspase-9 (cat. no.
9502; dilution, 1:1,000), caspase-8 (cat. no. 9746; dilution,
1:1,000), Fas (cat. no. 4233; dilution, 1:1,000) and Fas ligand
(FasL; cat. no. 4273; dilution, 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase (HRP)-conjugated immunoglobulin G anti-mouse (cat. no.
SA00001-1; dilution, 1:10,000) and anti-rabbit (cat. no. SA00001-2;
dilution, 1:10,000) antibodies were purchased from ProteinTech
Group, Inc. (Chicago, IL, USA). Cell Counting Kit-8 (CCK-8) was
purchased from Dojindo Molecular Technologies, Inc. (Kumamoto,
Japan).
Cell culture and proliferation
assay
HL-60 cells were cultured at Key Laboratory for
Tumor Immunology and Chinese Medicine Immunology of Shandong
Province (Institute of Basic Medicine, Shandong Academy of Medical
Sciences, Jinan, China). The cells were cultured in RPMI-1640
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% newborn calf serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 IU/ml penicillin and 100 IU/ml streptomycin
in a 5% CO2 atmosphere at 37°C. Logarithmically growing
HL-60 cells were seeded on a 96-well plate at a density of
1×104 cells/well in 50 µl of medium in triplicate, and
then different concentrations of baicalin (5, 10, 20, 40 and 80
µg/ml) in 50 µl were added. Cells treated with 0.1% (v/v) DMSO were
used as the control. After incubation for 24 h, cell proliferation
was detected using CCK-8, according to the manufacturer's protocol
as previously described (13).
Cell cycle distribution
Following baicalin (20 µg/ml) treatment for
different durations, cells (3×105) were washed twice
with ice-cold PBS, and fixed in cold 75% ethanol at 4°C for at
least 24 h. Then, the cells were rinsed with PBS, and resuspended
using 0.5 mg/ml RNaseA and 20 µg/ml propidium iodide (PI) prior to
incubation for 20 min at room temperature in the dark.
Subsequently, the cells were analyzed using an Elite-ESP flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA).
Analysis of apoptosis
Hoechst 33342 staining was used to visualize the
change in nuclear morphology. Following baicalin (20 µg/ml)
treatment for 24 h, cells were washed twice with PBS and fixed with
4% paraformaldehyde for 15 min at room temperature. Then, washed
cells were stained with 1 µg/ml Hoechst 33342 for 15 min at room
temperature in the dark. Cells were observed under a fluorescence
microscope and imaged. Apoptotic cells were characterized by
condensed or fragmented chromatin, while normal nuclei were smooth,
symmetric and oval. To quantify the cells undergoing apoptosis,
Annexin V/PI (BD Pharmingen; BD Biosciences, San Jose, CA, USA)
double staining was performed according to the manufacturer's
protocol. Briefly, the cells were harvested by centrifugation for
10 min at 800 × g at 4°C. Subsequently, the cells were washed twice
with ice-cold PBS and re-suspended in 500 µl of 1X binding buffer,
stained with Annexin V and PI for 15 min at room temperature.
Apoptotic cells appeared as Annexin V-positive and PI-positive.
Assays for analysis of ∆Ψm
Fluorescent dye rhodamine 123 staining and flow
cytometric analysis were performed to assess the ∆Ψm. Briefly,
following baicalin treatment for 24 h, the cells were incubated
with 10 mg/ml Rh123 for 30 min at 37°C, and washed twice with PBS,
then analyzed using fluorescence-activated cell sorting.
Reverse
transcription-semi-quantitative polymerase chain reaction
(RT-sqPCR) assays
Total RNA was extracted from HL-60 cells that had
been cultured with baicalin (20 µg/ml) for different durations
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according
to the manufacturer's protocol. RT-PCR was performed as described
previously (13). The primers used in
the present study were as follows: Fas (238 bp) forward,
5′-GGACCCAGAATACCAAGTGC-3′ and reverse,
5′-GCCACTGTTTCAGGATTTAAGG-3′; hTERT (198 bp) forward,
5′-TCACCTCGAGGGTGAAGGCACTGTT-3′ and reverse,
5′-ATGCTGGCGATGACCTCCGTGA-3′; c-Myc (180 bp) forward,
5′-GATTCTCTGCTCTCCTCGAC-3′ and reverse, 5′-TCCAGACTCTGACTTTTGC-3′;
DR5 (389 bp) forward, 5′-TGCAGCCGTAGTCTTGATTG-3′ and reverse,
5′-GCACCAAGTCTGCAAAGTCA-3′; DR4 (307 bp) forward,
5′-CAGAGGGATGGTCAAGGTCAAG-3′ and reverse,
5′-TTGCTGCTCAGAGACGAAAGTG-3′; β-actin (540 bp) forward,
5′-GTGGGGCGCCCCAGGCAGGCACCA-3′ and reverse,
5′-CTCCTTAATGTCACGCACGATTTC-3′. The following thermocycling
conditions were maintained: 94°C for 5 min; 30 cycles of 94°C for
45 sec, 55°C for 45 sec and 72°C for 1 min; and 72°C for 10 min.
The PCR products were electrophoresed on 1.5% agarose gels. The
software used to quantify the bands was AlphaImager 2200 (version
0.2.1.0; Alpha Innotech, Tarzana, CA, USA).
Western blot analysis
For immunoblotting, HL-60 cells treated with 20
µg/ml baicalin for 12 and 24 h were washed twice with cold PBS and
lysed using extraction buffer (50 mM Tris-HCl pH 7.4, 1 mM
phenylmethylsulfonyl fluoride, 150 mM NaCl, 1 mM EDTA, 1% Triton
X-100, 0.5% deoxycholate, and 0.1% SDS) for 15 min on ice. Protein
concentrations were measured using the Bradford protein assay
(Beyotime Institute of Biotechnology). Protein samples (50 µg) were
electrophoresed using 12% SDS-PAGE, transferred onto a
polyvinylidene difluoride membrane. After blocking with Tris
buffered saline-Tween-20 containing 5% non-fat dry milk for 1 h at
room temperature, the membranes were incubated with primary
antibodies against β-actin, cleaved caspase-3, caspase-8,
caspase-9, Fas, FasL, c-Myc, Bcl-2 and Bax overnight at 4°C.
Subsequently, membranes were incubated with HRP-conjugated
secondary antibodies for 1 h at room temperature. The protein bands
were visualized using an enhanced chemiluminescence detection
system (EMD Millipore, Billerica, MA, USA) and imaged using a
LAS-4000 mini luminescent image analyzer (Fujifilm, Tokyo, Japan).
The software used to analyze the protein band density was Multi
Gauge (version 3.0; Fujifilm, Tokyo, Japan).
Statistical analysis
Results are expressed as the mean ± standard error
mean. The data were analyzed using one-way analysis of variance and
Tukey's honest significant test post hoc analysis using SPSS
(version 13.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Baicalin inhibits the proliferation of
HL-60 cells, and c-Myc together with hTERT participates in its
growth arrest
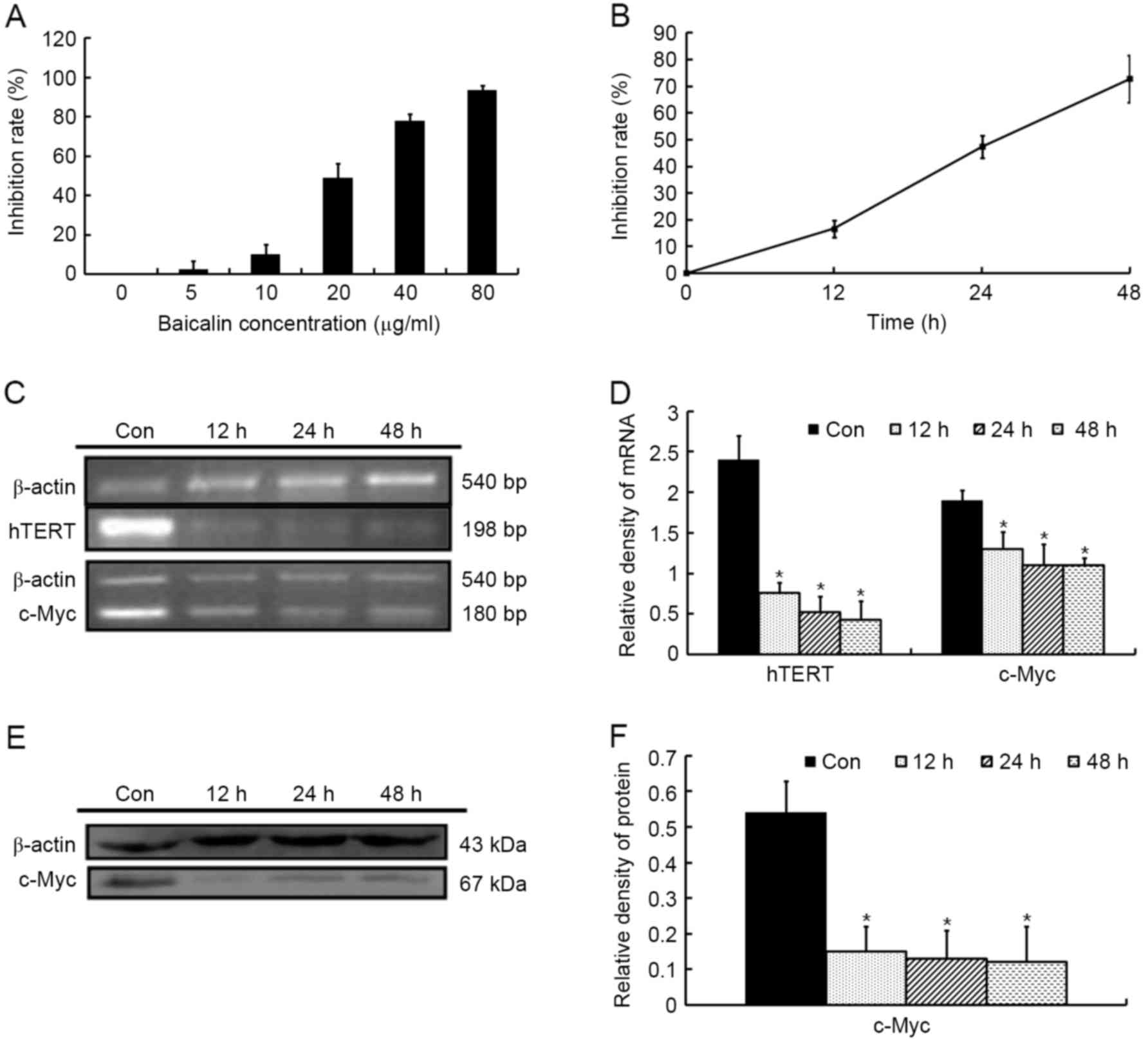
The effect of baicalin on the growth of HL-60 cells
was detected using a CCK-8 assay. As presented in Fig. 2A, baicalin inhibited the proliferation
of HL60 cells in a dose-dependent manner with an IC50
value of 21.8 µg/ml at 24 h. Therefore, a baicalin concentration of
20 µg/ml was selected to perform the majority of the following
experiments. At this concentration, baicalin arrested cell growth
in a time-dependent manner (Fig.
2B).
c-Myc possesses an essential function
in controlling cell growth
To investigate the roles of c-Myc in growth
inhibition, the mRNA and protein levels of c-Myc gene expression
were detected in HL-60 cells exposed to 20 µg/ml baicalin for
different durations. As presented in Fig.
2C-F, baicalin resulted in a significant downregulation of
c-Myc mRNA and protein levels compared with untreated cells. In
line with the aforementioned results, it was demonstrated that the
mRNA expression of hTERT, the catalytic subunit of the telomerase
holoenzyme complex, a direct transcriptional target of c-Myc
(14), consequently decreased
(Fig. 2C and D).
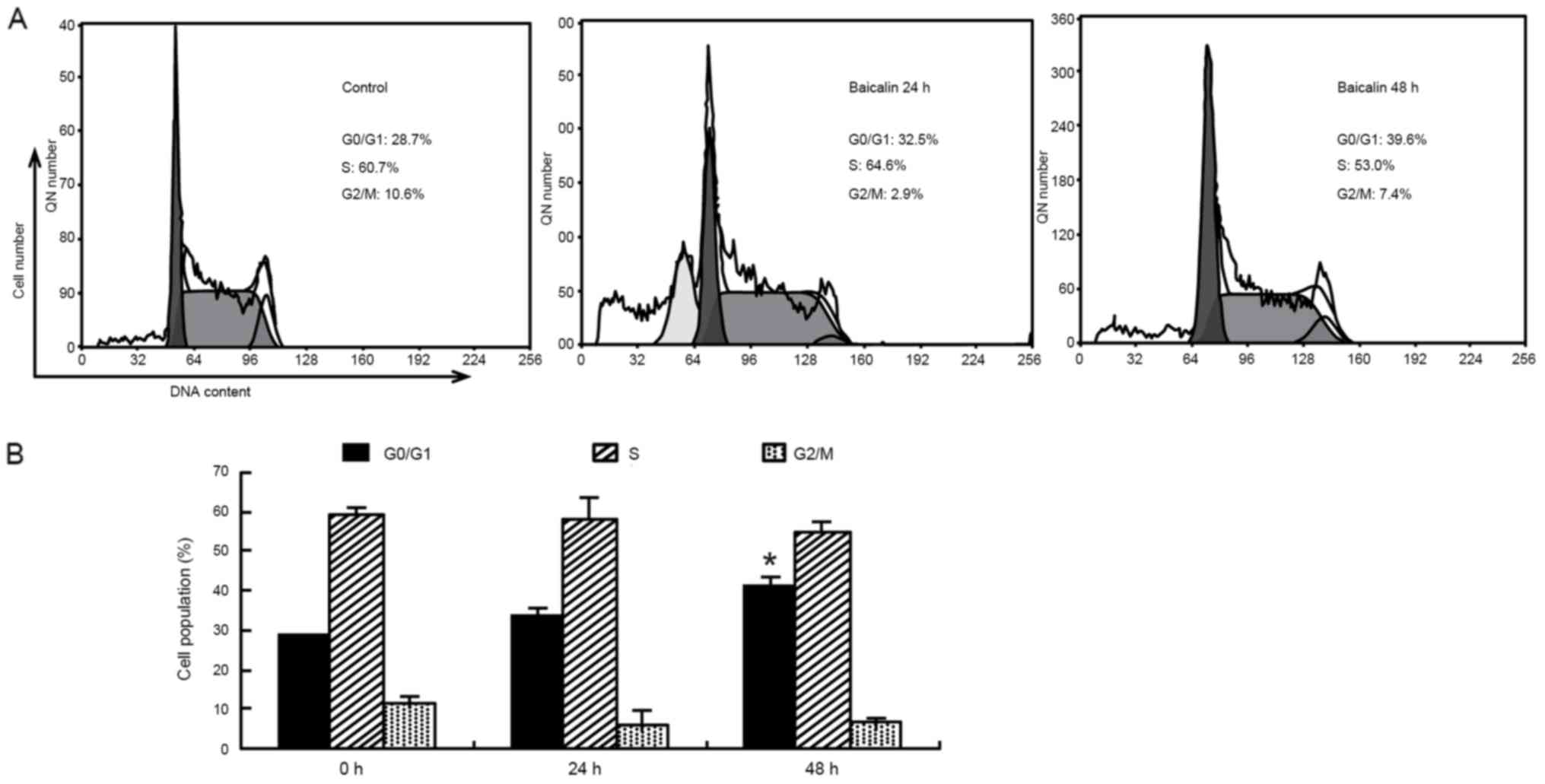
Baicalin causes G0/G1 arrest in HL-60
cells
To investigate the mechanisms underlying growth
inhibition by baicalin in HL-60 cells, cell cycle analysis was then
performed using flow cytometry. As presented in Fig. 3, the time-course study revealed that
exposure of HL-60 cells to baicalin significantly increased the
proportion of cells in G0/G1 phase from
(29.2±0.5%) in the vehicle treated cells to (37.6±3.2%; P<0.05),
accompanied by a reduction in the percentages of HL-60 cells in the
G2/M and S phases. In addition, cells in the
sub-G1 phase rapidly increased at 24 h and the
sub-G1 hypodiploid population reached 20.1±2.9% (data
not shown). The data indicated that inhibition in cell cycle
progression and induction of apoptosis may be associated with the
inhibitory effect of baicalin treatment on the proliferation of
HL-60 cells.
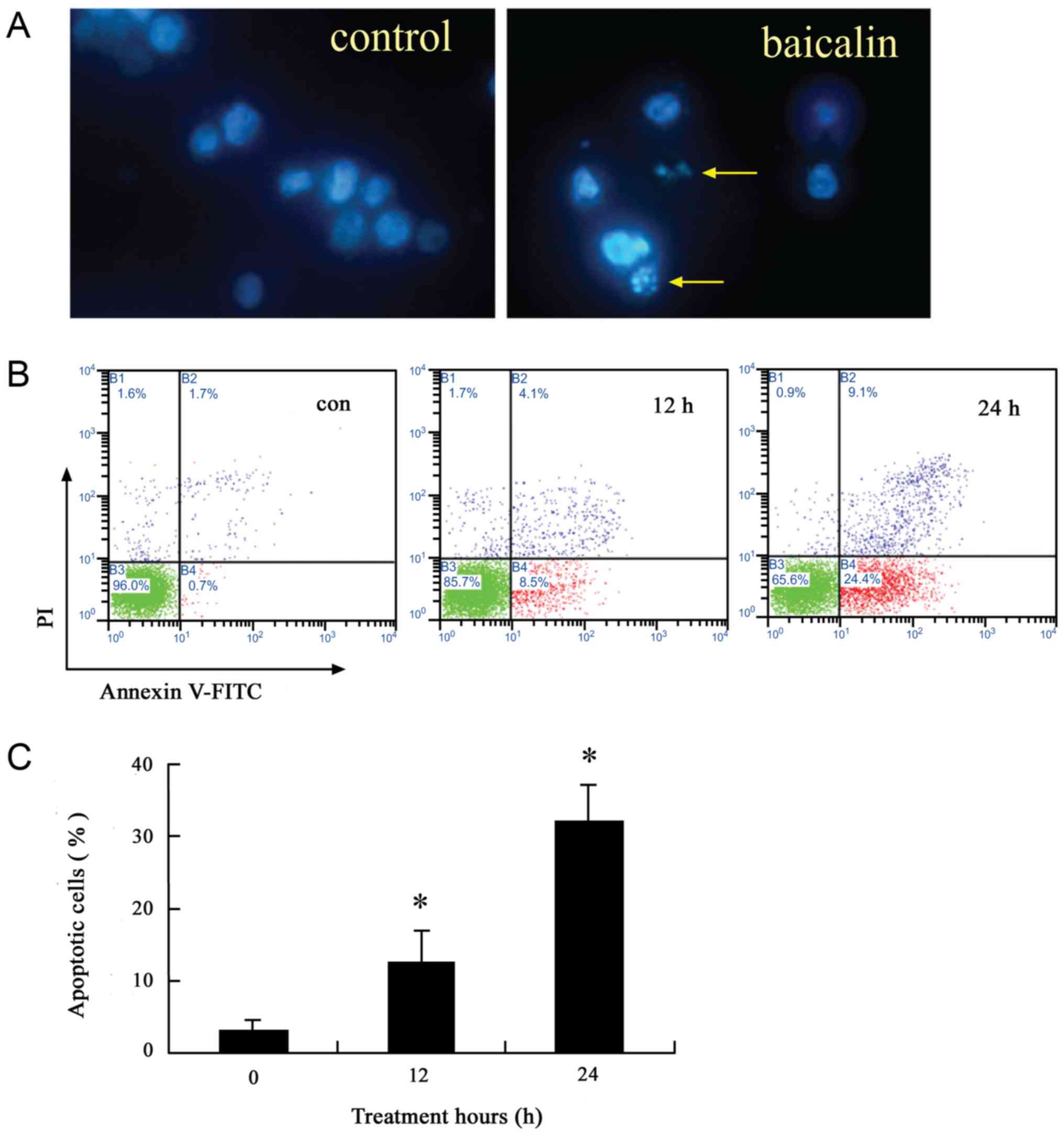
Baicalin induces apoptosis in HL-60
cells
To further characterize apoptosis induced by
baicalin, Hoechst 33342 staining and Annexin V/PI analysis was
performed. The nuclei of untreated cells were round and large in
size, exhibiting homogeneous blue fluorescence. In contrast, a
number of the cells treated with baicalin for 24 h were observed
with condensed or fragmented nuclei, which are characteristics of
cell apoptosis (Fig. 4A). In
addition, the induction of apoptosis was time-dependent (Fig. 4B and C). Baicalin induced apoptosis in
12.6±4.3% of HL-60 cells after 12 h treatment, while it induced
32.3±5.1% of cells after 24 h treatment.
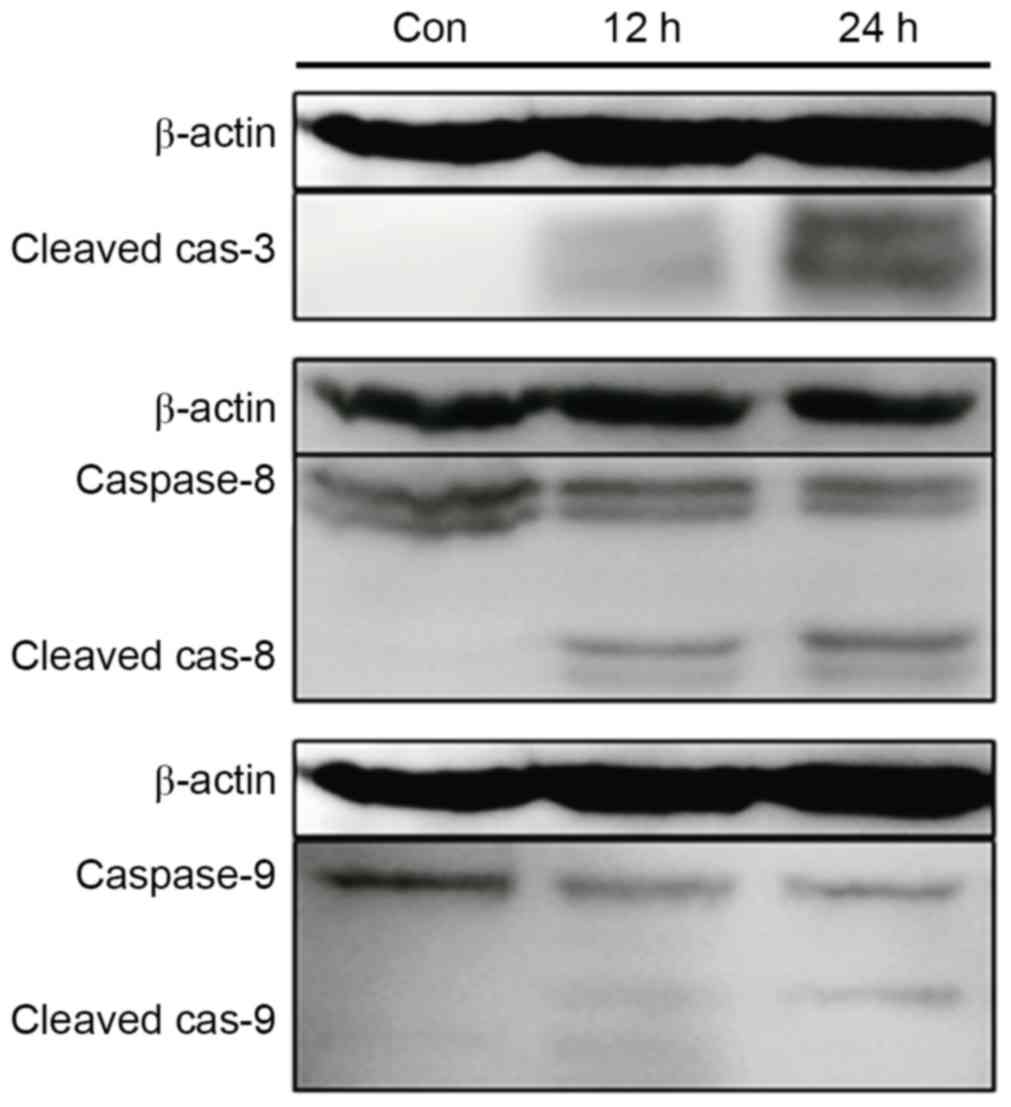
Baicalin induces the activation of
caspases
To determine the mechanism of apoptosis induction by
baicalin treatment, the activities of caspases, which have been
demonstrated to be essential mediators in the apoptosis pathway
(15), were analyzed using western
blotting. Treatment of cells with baicalin resulted in marked
time-dependent caspase-3, caspase-8 and caspase-9 activation, and
cleavage, which indicated that the extrinsic and intrinsic pathways
were associated with apoptosis induced by baicalin (Fig. 5).
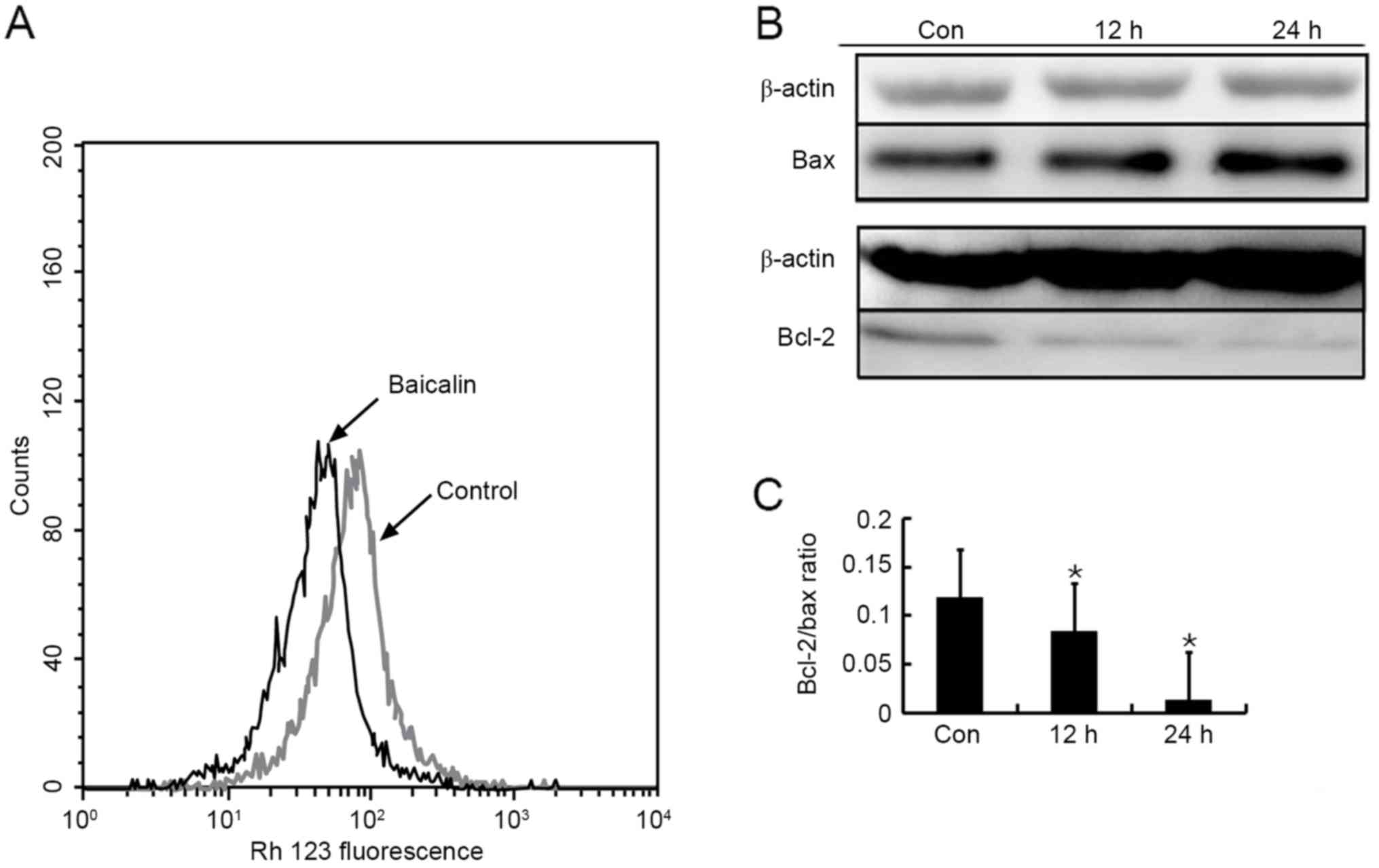
Baicalin-induced apoptosis is mediated
through intrinsic pathway
The decline in ΔΨm has been considered to be an
important event in intrinsic pathway, which leads to matrix
condensation and rapid cytochrome c release (16), followed by caspase-9 activation. Flow
cytometry analysis of Rh123-stained HL-60 cells was performed to
investigate the intrinsic pathway. As presented in Fig. 6A, treatment of HL-60 cells with
baicalin for 24 h led to a marked decrease in Rh123 fluorescence
compared with the control group, presenting dissipation of ΔΨm.
The Bcl-2 family of proteins is a key factor in the
regulation of cytochrome c release from mitochondria
(17). To further confirm the
involvement of the intrinsic pathway, anti-apoptotic factor Bcl-2
and pro-apoptotic factor Bax were detected using western blotting.
The protein expression of Bax was increased notably, but that of
Bcl-2 decreased following exposure of HL-60 cells to baicalin for
12 and 24 h (Fig. 6B). As a result,
the ratio of Bcl-2/Bax declined significantly (Fig. 6C). These results demonstrated that the
intrinsic signaling pathway was involved in baicalin-treated
apoptosis, in accordance with the previous studies (6,8).
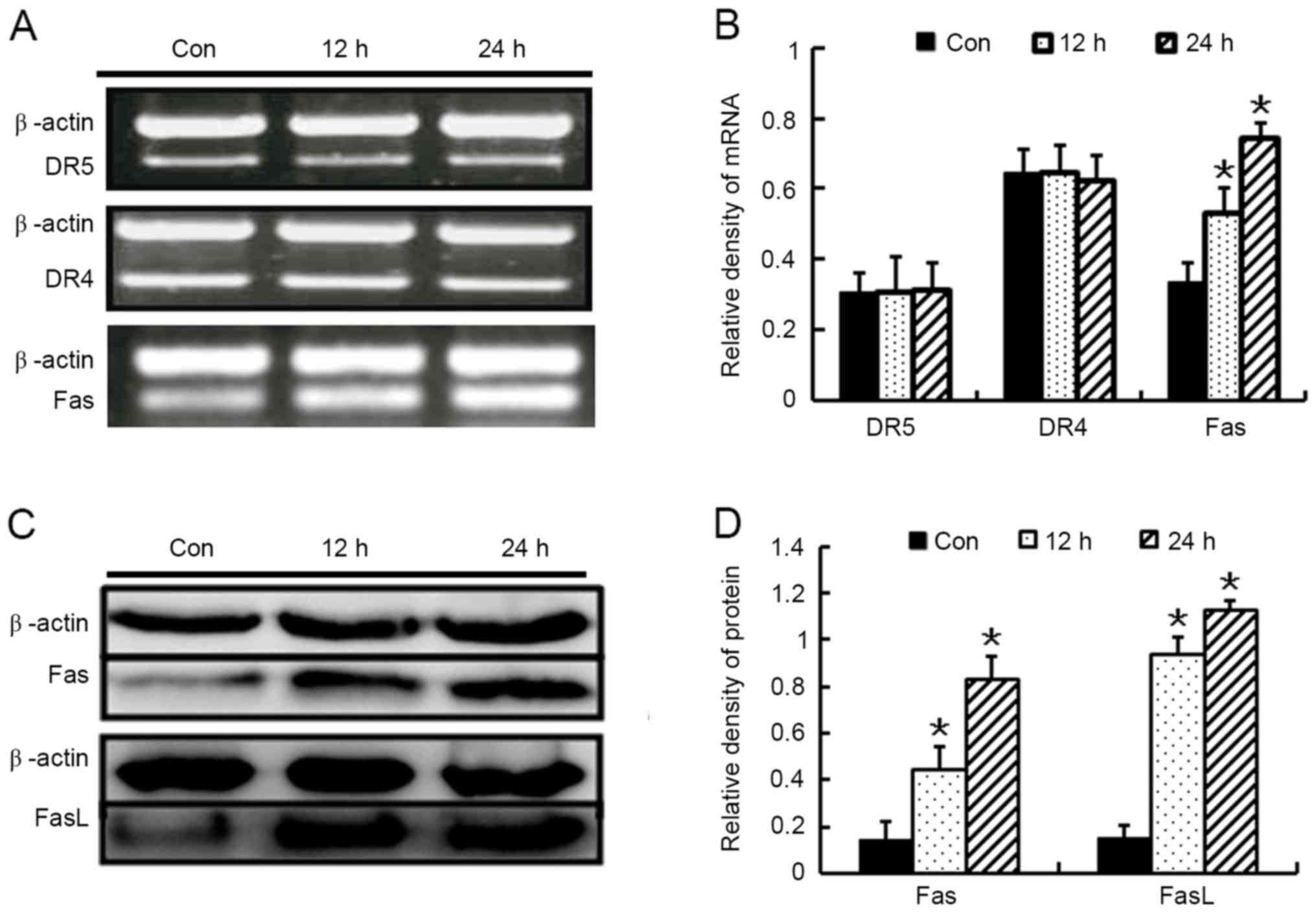
Baicalin induces apoptosis through the
Fas-mediated extrinsic pathway
To clarify which receptor mediated the extrinsic
pathway, the death receptors, including DR4, DR5 and Fas, were
detected using RT-PCR. The results revealed that exposure of HL-60
cells to baicalin for 24 h significantly triggered Fas expression,
but no significant effects of baicalin on the mRNA levels of DR4 or
DR5 were noted (Fig. 7A and B).
Western blot assays were then performed, which further confirmed
that baicalin induced FasL and Fas expression throughout the time
period examined (Fig. 7C and D). The
data presented in the present study suggest that activation of the
extrinsic Fas-associated pathway partially contributed to
baicalin-induced apoptosis.
Discussion
As previously reported (8), the present study confirmed that baicalin
exhibits high dose- and time-dependent anticancer activity. To
determine the molecular mechanisms of the inhibitory effect of
baicalin in HL-60 cells, cell cycle analysis was performed using
flow cytometry. The results demonstrated that baicalin induced
significant G0/G1 arrest 48 h later; however,
it was reported by Ikezoe et al (3) that baicalin arrested HL-60 cells at the
G2/M phase. Consistent with the
G0/G1 arrest, the results of our previous
study demonstrated the baicalin upregulated the expression of
cyclin-dependent kinase inhibitors, including p21 and p27 (13), which are involved in the G1
to S transition (18). It was
concluded that baicalin reduced cell viability partially through
arresting cells at the G0/G1 phase. However,
the appearance of the hypodiploid peak in the cell cycle
distribution demonstrated that apoptosis also contributed to cell
proliferation inhibition by baicalin. Furthermore, the cell
morphological changes observed and Annexin V/PI staining results
further confirmed this, and are concordant with preceding reports
(3,4,6,8).
It is well known that caspases, a conserved family
of cysteine proteases, serve a central role in the execution of
cell apoptosis (19). They are
generally divided into two classes: The initiator caspases, which
include caspase-2, −8, −9 and −10; and the effector caspases, which
include caspases-3, −6 and −7 (15).
Classical apoptosis occurs through the extrinsic and intrinsic
pathway in all mammalian cells. Both pathways converge on
activation of initiator and effector caspases (15). The former depends on death receptors,
including Fas or TNF-related apoptosis-inducing ligand (TRAIL)
receptors, DR4 and DR5 (20). When
bound by their specific death ligand, these receptors are
activated, resulting in the formation of a death-inducing signaling
complex and activation of the initiator caspase-8, which further
directly or indirectly activates effector caspase-3 (20). The latter depends on mitochondria.
Once the apoptotic stimuli are received, including heat shock,
oxidative stress, DNA damage or cytotoxic drugs, the permeability
of the inner mitochondrial membrane increases, and the
mitochondrial membrane potential is disrupted (21). Subsequently, cytochrome c is
released, triggering caspase-9, and downstream effectors caspase-3
and/or −7 activation (21).
To elucidate the mechanism of apoptosis elicited by
baicalin in HL-60 cells, the protein expression of caspase-3, −8,
and −9 was detected using western blotting. Consistent with
previous reports (6,8), the results of the present study
demonstrated that caspase-3 and −9 were cleaved, and activated
following baicalin treatment, demonstrating a role for mitochondria
in the process of ongoing apoptosis in HL-60 cells. Though
caspase-8 activation had been confirmed in cervical cancer HeLa
cells (22) and in human colorectal
carcinoma SW620 cells (23), the
roles of caspase-8 in baicalin-induced apoptosis in human leukemia
HL-60 cells remained to be clarified. In the present study,
increased expression of cleaved caspase-8 was detected following
exposure to baicalin for 12 h, and more notably after 24 h. Thus,
it was postulated that, in addition to the intrinsic signaling
pathway, the extrinsic pathway may also contribute to apoptosis
induced by baicalin in HL-60 cells.
To further confirm the convergence in apoptosis
signaling, molecular markers associated with the extrinsic and
intrinsic pathways were analyzed. Similar to the results acquired
from hepatoma cell lines (24) and
the leukemia-derived T cell line (6),
it was demonstrated that baicalin caused ΔΨm disruption of HL-60
cells and increased the expression of Bax, while it decreased
expression of Bcl-2, confirming that the intrinsic pathway
participated in apoptosis induced by baicalin.
It is well established that caspase-8 activation is
initiated by the activation of death receptors. Currently, six
distinct death receptors are known, including Fas (APO-1/CD95), the
TRAIL receptor (R)-1 (also named DR4), TRAIL-R2 (also named DR5),
tumor necrosis factor-R1, DR3 and DR6. Of these receptors, Fas and
the TRAIL receptors have been extensively investigated (25). A previous study validated that
modulation of Fas expression on the surface of tumor cells may
potentiate the induction of apoptosis in tumor cells in response to
chemo- and immuno-therapeutic agents (26). In addition, baicalin has been reported
to trigger Fas and FasL expression during HeLa cell apoptosis
(21). However, in the T-cell acute
lymphoblastic leukemia cell line CCRF-CEM, it appeared that
baicalin treatment exhibited no significant effect on the
expression of Fas (7). In the present
study, the upregulation of Fas and its ligand FasL was detected at
the mRNA, and protein level. However, there was no change in DR4
and DR5 mRNA expression. This discrepancy is likely attributed to
the specific cell type investigated. The data of the present study
implied that the Fas receptor, but not DR4 and DR5, was involved in
baicalin-induced caspase-8 activation.
An investigation regarding the involvement of
transcription factors in cell proliferation may be useful to
perform a more in-depth analysis of the exact mechanism by which
baicalin exhibits antitumor activity. c-Myc, as a transcription
factor, serves an important role in cell proliferation and is
overexpressed in numerous human tumors (27). Evidence has demonstrated that c-Myc
binds to thousands of promoters of its target genes, including
hTERT (28). It has been confirmed
that c-Myc is involved in the control of telomerase activity, which
is associated with cell proliferation in normal cells and tumors
(29), through its ability to induce
the transcriptional activation of hTERT. In the present study, the
downregulation of c-Myc was detected at the mRNA and protein level,
in addition, transcriptional activation of hTERT was also
decreased, in line with the inhibition of HL-60 cells.
In conclusion, the results of the current study
demonstrated that baicalin is able inhibit the proliferation of the
AML HL60 cell line through G0/G1 phase arrest
and significant apoptosis induction via the intrinsic, and
extrinsic pathways. In addition, the inhibition of HL-60 cell
growth was also mediated by telomerase inhibition through
suppression of c-Myc. These results raise the possibility that
baicalin may be a promising regimen for treatment of AML.
Acknowledgements
The authors would like to thank Dr Xiao Wang
(Shandong Analysis and Test Center, Shandong Academy of Sciences)
for providing purified baicalin. The present study was supported by
the National Natural Science Foundation of China (grant nos.
8117292 and 81573467), the ‘Twelfth Five-Year’ National Science and
Technology Support Program (grant no. 2013BAI07B02), the Natural
Science Foundation of Shandong Province of China (grant nos.
ZR2011HL045, ZR2015YL028, ZR2014HM085 and 2015ZRC03102), the Key
Science and Technology Program of Shandong (grant no. 2013YD18031),
the Project for Laureate of Taishan Scholar (grant no. ts201511075)
and The Innovation Project of Shandong Academy of Medical
Science.
References
|
1
|
Huang WH, Lee AR and Yang CH:
Antioxidative and anti-inflammatory activities of
polyhydroxyflavonoids of Scutellaria baicalensis GEORGI. Biosci
Biotechnol Biochem. 70:2371–2380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shang X, He X, He X, Li M, Zhang R, Fan P,
Zhang Q and Jia Z: The genus Scutellaria an ethnopharmacological
and phytochemical review. J Ethnopharmacol. 128:279–313. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ikezoe T, Chen SS, Heber D, Taguchi H and
Koeffler HP: Baicalin is a major component of PC-SPES which
inhibits the proliferation of human cancer cells via apoptosis and
cell cycle arrest. Prostate. 49:285–292. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zheng J, Hu JD, Chen YY, Chen BY, Huang Y,
Zheng ZH and Liu TB: Baicalin induces apoptosis in leukemia
HL-60/ADR cells via possible down-regulation of the PI3K/Akt
signaling pathway. Asian Pac J Cancer Prev. 13:1119–1124. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parajuli P, Joshee N, Rimando AM, Mittal S
and Yadav AK: In vitro antitumor mechanisms of various Scutellaria
extracts and constituent flavonoids. Planta Med. 275:41–48. 2009.
View Article : Google Scholar
|
|
6
|
Ueda S, Nakamura H, Masutani H, Sasada T,
Takabayashi A, Yamaoka Y and Yodoi J: Baicalin induces apoptosis
via mitochondrial pathway as prooxidant. Mol Immunol. 38:781–791.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shieh DE, Cheng HY, Yen MH, Chiang LC and
Lin CC: Baicalin-induced apoptosis is mediated by Bcl-2-dependent,
but not p53-dependent, pathway in human leukemia cell lines. Am J
Chin Med. 34:245–261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu HF, Hsueh SC, Ho YT, Kao MC, Yang JS,
Chiu TH, Huamg SY, Lin CC and Chung JG: ROS mediates
baicalin-induced apoptosis in human promyelocytic leukemia HL-60
cells through the expression of the Gadd153 and
mitochondrial-depedent pathway. Anticancer Res. 27:117–125.
2007.PubMed/NCBI
|
|
9
|
Grandori C, Cowley SM, James LP and
Eisenman RN: The Myc/Max/Mad network and the transcriptional
control of cell behavior. Annu Rev Cell Dev Biol. 16:653–699. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Felsher DW: MYC inactivation elicits
oncogene addiction through both tumor cell-intrinsic and
host-dependent mechanisms. Genes Cancer. 1:597–604. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sirinian MI, Pisegna S, Paroli M, Militi
S, Testa U and Peschle C: Zinc modulates c-Myc/Mad1 balance in
human leukemia cells. Leukemia. 17:272–274. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang ST, Wang CY, Yang RC, Chu CJ, Wu HT
and Pang JH: Wogonin, an active compound in Scutellaria
baicalensis, induces apoptosis and reduces telomerase activity in
the HL-60 leukemia cells. Phytomedicine. 17:47–54. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren X, Zhang Y, Li C, Wang H, Jiang Z,
Zhang Z, Guo Q, Song G, Bi K and Jiang G: Enhancement of baicalin
by hexamethylene bisacetamide on the induction of apoptosis
contributes to simultaneous activation of the intrinsic and
extrinsic apoptotic pathways in human leukemia cells. Oncol Rep.
30:2071–2080. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kyo S, Takakura M, Taira T, Kanaya T, Itoh
H, Yutsudo M, Ariga H and Inoue M: Sp1 cooperates with c-Myc to
activate transcription of the human telomerase reverse
transcriptase gene (hTERT). Nucleic Acids Res. 28:669–677. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ and
Los M: Apoptosis and cancer: Mutations within caspase genes. J Med
Genet. 46:497–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Steinman RA: Cell cycle regulators and
hematopoiesis. Oncogene. 21:3403–3413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lavrik IN, Golks A and Krammer PH:
Caspases: Pharmacological manipulation of cell death. J Clin
Invest. 115:2665–2672. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fulda S: Caspase 8 in cancer biology and
therapy. Cancer Lett. 281:128–133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arnoult D: Mitochondrial fragmentation in
apoptosis. Trends Cell Biol. 17:6–11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Peng Y, Fu ZZ, Guo CS, Zhang YX, Di Y,
Jiang B and Li QW: Effects and mechanism of baicalin on apoptosis
of cervical cancer HeLa cells In-vitro. Iran J Pharm Res.
14:251–261. 2015.PubMed/NCBI
|
|
23
|
Chen WC, Kuo TH, Tzeng YS and Tsai YC:
Baicalin induces apoptosis in SW620 human colorectal carcinoma
cells in vitro and suppresses tumor growth in vivo. Molecule.
17:3844–3857. 2012. View Article : Google Scholar
|
|
24
|
Chang WH, Chen CH and Lu FJ: Different
effects of baicalein, baicalin and wogonin on mitochondrial
function, glutathione content and cell cycle progression in human
hepatoma cell lines. Planta Med. 68:128–132. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thorburn A: Death receptor-induced cell
killing. Cell Signal. 16:139–144. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kolenko VM, Uzzo RG, Bukowski R and Finke
JH: Caspase-dependent and -independent death pathways in cancer
therapy. Apoptosis. 5:17–20. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meyer N and Penn LZ: Reflecting on 25
years with MYC. Nat Rev Cancer. 8:976–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch
W, Wang R, Green DR, Tessarollo L, Casellas R, et al: c-Myc is a
universal amplifier of expressed genes in lymphocytes and embryonic
stem cells. Cell. 151:68–79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Biroccio A, Amodei S, Benassi B, Scarsella
M, Cianciulli A, Mottolese M, Del Bufalo D, Leonetti C and Zupi G:
Reconstitution of hTERT restores tumorigenicity in melanoma-derived
c-Myc low-expressing clones. Oncogene. 21:3011–3019. 2002.
View Article : Google Scholar : PubMed/NCBI
|