Introduction
Colorectal cancer (CRC) is the third leading cause
of all tumor-associated mortalities and resulted in 693,900 in 2012
worldwide in 2012, accounting for 8% of all cancer-associated
mortalities (1). Although the
prognosis of CRC has improved due to the development of surgical
procedures and therapeutic agents, survival is rarely longer than
three years (2). Consequently, the
identification of novel diagnostic markers and the elucidation of
the underlying molecular mechanisms of tumorigenesis of CRC are
required.
Reprogramming metabolic pathways is the key
characteristic of cancer cells (3). A
number of types of cancer cell exhibit increased glucose
consumption and lactate production independent of oxygen
availability, a phenomenon termed the Warburg effect (4). Pyruvate kinase (PK), one of the
rate-limiting enzymes of glycolysis, serves functions in modulating
glucose usage and metabolic flux in adaption to different stresses
during tumorigenesis (5). In mammals,
four isoforms of pyruvate kinase (PKL, PKR, PKM1 and PKM2) have
been identified. PKL and PKR are expressed in the liver and red
blood cells, respectively (6). PKM1
is expressed in adult tissue, whereas PKM2 is expressed in
embryonic tissue and tumors. PKM1 and PKM2 are products of
alternative splicing of PKM gene, which contain a mutually
exclusive exon (6). Exon 9 is
specific for PKM1 and exon 10 exists in PKM2 (7). PKM1 is the constitutively active
isoform, which preferentially diverts glucose into the citric acid
cycle to generate adenosine triphosphate. However, PKM2 exhibits
relatively low pyruvate kinase activity compared with PKM1, which
causes glucose to be metabolized by glycolysis, rather than by
oxidative phosphorylation. Furthermore, PKM2 may function as a
protein kinase and has been demonstrated to be involved in multiple
signaling pathways (8,9). Therefore, PKM2 exhibits metabolic
advantages in cancer cells and participates in oncogenic signaling
networks. As a result, PKM2 is required for the rapid growth of
cancer cells and tumorigenesis (10).
During the development of cancer, PKM gene
expression is transformed from PKM1 to PKM2, which is controlled by
a group of splicing factors. Three heterogeneous nuclear
ribonucleoproteins (hnRNPs), including hnRNPA1, hnRNPA2 and PTBP1,
have been identified to exclude exon 9 in the PKM transcript, which
results in the expression of the PKM2 isoform rather than the PKM1
isoform (7,11). It has been previously demonstrated
that microRNA (miR-)124 causes a switch in PKM gene expression from
PKM2 to PKM1, by targeting polypyrimidine tract-binding protein
(PTB) and hnRNPA2, which subsequently transforms glucose flux from
glycolysis to oxidative phosphorylation (12). As a result, expressing miR-124
inhibits the growth of CRC cells by modulating glucose metabolism
(12).
S6 kinase (S6K)1 and S6K2 are members of the AGC
kinase super-family (13). The
mechanistic target of rapamycin/S6K signaling pathway has been
identified to be involved in a number of types of disease,
including cancer and metabolic syndromes (14). The principal focus has been placed on
S6K1 and S6K2 has been neglected due to their similarities in
structure (15). However, a previous
study indicated that these two isoforms have distinct biological
functions (15). The overexpression
of S6K2 in tumor tissues is more common, compared with that of S6K1
(16,17). Furthermore, S6K2, rather than S6K1,
mediates pro-survival signaling of cancer cells (18). S6K2 phosphorylates hnRNPA1 on Ser4/6
sites, which promotes the translation of B-cell
lymphoma-extra-large (BCL-xL) and X-linked inhibitor of apoptosis
(XIAP), and exhibits pro-survival effects on cells (19). However, the underlying molecular
mechanism of S6K2 in tumorigenesis remains unknown.
In the present study, the results indicated that
S6K2 regulated glucose metabolism by modulating alternative
splicing of the PKM gene. The mRNA ratio of PKM2/PKM1 was markedly
increased by S6K2 in a hnRNPA1-dependent manner. As a result,
glycolysis was induced by S6K2. Phosphorylation of Ser6,
rather than Ser4, in hnRNPA1 by S6K2 is required to
increase the PKM2 isoform and induce glycolysis in CRC cells.
Notably, the level of Ser6 phosphorylation of hnRNPA1
predicted poor prognosis for patients with CRC. Therefore, the
present study identified that S6K2-mediated regulation of glucose
metabolism is involved in the development of CRC. S6K2 represents a
novel metabolic target for CRC treatment.
Materials and methods
Cell culture and reagents
HCT116 cells were cells were purchased from the
American Type Culture Collection (Manassas, VA, USA) and cultured
in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (Invitrogen; Thermo Fisher
Scientific, Inc.), 2 mM l-glutamine (Invitrogen; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin (Invitrogen; Thermo Fisher
Scientific, Inc.) and 100 µg/ml streptomycin (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C in humidified air containing 5%
CO2. An S6K2 short interfering (si)RNA oligonucleotide,
5′-GGAAGAAAACCAUGGAUAAUU-3′ and its complementary RNA were
synthesized by Genepharm, Inc. (Sunnyvale, CA, USA). Non-silencing
control siRNA was synthesized by Genepharm using a scrambled
sequence as the negative control. For transfection, siRNA or a
plasmid were transfected into HCT116 cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to manufacturer's instructions. The
sequence of an antisense oligonucleotide (ASO) against exon 10 for
the PKM gene was 5′-TGAGGAACTCCGCCG−3′. DASA-58 was purchased from
Cayman Chemical Company (Ann Arbor, MI, USA). Dimethyl sulfoxide
(DMSO) was used as the vehicle control for DASA-58. Cells were
treated with DMSO or 50 µM DASA-58 for 3 h at 37°C in humidified
air containing 5% CO2 prior to analysis. The antibody of
serine 6 phosphorylation of hnRNPA1 was generated as described
previously (20).
Patients and tissue specimens
The present retrospective study was conducted with
the approval of the Ethics Committee of Zhongshan Hospital
(Shanghai, China). All colorectal cancer tissues and adjacent
non-cancerous tissues were obtained from 75 colorectal cancer
patients between January 2002 and December 2008 in Zhongshan
Hospital. Formalin-fixed, paraffin-embedded blocks of human tissues
were re-cut to a thickness of 5 µm and were subjected to
immunostaining. There were 43 male and 32 female patients, with
ages ranging between 26 and 83 years (median, 65 years). The median
follow-up data was 56 months (range, 0–97 months). For the analyses
of overall survival, survival time was defined as the period
between the date of diagnosis and the date of mortality or the date
of the last follow-up.
Immunohistochemical staining
Immunohistochemical staining on the
paraffin-embedded sections was performed using an LSAB kit (Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA), as described
previously (21). Briefly, tissue
sections were de-waxed, hydrated and washed. After microwave
antigen retrieval, the slides were treated with 3%
H2O2 for 10 min at room temperature to block
endogenous peroxidase activity and then incubated overnight with
the primary antibody of rabbit anti-serine 6 phosphorylation of
hnRNPA1 (dilution, 1:200; antibody epitope, MSKSES(p) PKEPEQLRKL)
at 4°C. The sections were then incubated with the horseradish
peroxidase-conjugated secondary antibody (dilution, 1:1,000; cat.
no. ab6721; Abcam, Cambridge, UK) for 1 h at room temperature.
Signals were visualized using diaminobenzidine as the chromogen and
counter-stained with hematoxylin (3 min at room temperature) and
eosin (1 min at room temperature) Scoring was performed with the
H-score on the basis of the proportion of tumor cells staining at
various intensities as follows: 0×(% tumor cells with no
staining)+1×(% tumor cells with faint expression)+2×(% tumor cells
with moderate expression)+3×(% tumor cells with strong expression).
H-scores for specimens with multiple cores were averaged. High
p-hnRNPA1-S6 level is defined as H-score equal or greater than the
average score. Low p-hnRNPA1-S6 level staining is defined as
H-score less than the average score.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from HCT116 cells using the
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. Briefly, cells were lysed
in a culture dish by adding TRIzol reagent. The RNA was
precipitated from the aqueous phase by mixing with 0.2 ml isopropyl
alcohol per 1 ml TRIzol reagent. RNA was precipitated by
centrifugation at 12,000 × g for 15 min at 4°C. The RNA pellet was
washed once with 75% ethanol. Subsequently, RNA was dissolved in
RNase-free water. RNA concentration was measured using a Nanodrop
spectrophotometer (Thermo Fisher Scientific, Inc.). CDNA was
synthesized using the First-Strand cDNA Synthesis kit (GE
Healthcare, Chicago, IL, USA), according to the manufacturer's
protocol. The sequences of the primers used for the real-time PCR
analysis were as follows. S6K2 sense, CGG GCT GAG AGG AAC ATT CTA
and antisense, CTG GAA AGC ATA GGC CAG TTC T; PKM1 sense, CTG GAG
AAA CAG CCA AAG G and antisense, GCC AGA CTC CGT CAG AAC TA; PKM2
sense, GGG TTC GGA GGT TTG ATG and antisense, ACG GCG GTG GCT TCT
GT; β-actin sense, ATG GAT GAC GAT ATC GCT GCG C and antisense, GCA
GCA CAG GGT GCT CCT CA. Real-time PCR was performed with the
Sequence Detection system 7900HT (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Thermal cycling conditions were designed as
follows: Initial denaturation at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec and 60°C for 1 min. Threshold cycle (ΔCT)
values were calculated by subtracting the CT value from the
corresponding β-actin CT (internal control) value. The comparative
Cq (ΔΔCq) method was used to determine the expression level of mRNA
(22).
Apoptosis analysis
Cell apoptosis was analyzed using the Annexin
V-fluorescein isothiocyanate Apoptosis Detection kit
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
harvested by treating with trypsin. As described previously
(23), cells were washed with
ice-cold PBS, resuspended in the Annexin V binding buffer and,
following incubation, cells were analyzed using a FACStarPlus flow
cytometer (BD Biosciences, Franklin lakes, NJ, USA). Data were
collected and analyzed using CellQuest software (version 5.1; BD
Biosciences).
Western blot analysis
As described in detail previously (24), cells were washed twice in PBS,
suspended in lysis buffer (50 mM Tris at pH 8.0, 150 mM NaCl, 0.1%
SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40,
phenylmethylsulfonyl fluoride at 100 µg/ml, aprotinin at 2 µg/ml,
pepstatin at 1 µg/ml and leupetin at 10 µg/ml) and placed on ice
for 30 min. Following centrifugation at 15,000 × g for 20 min at
4°C, the suspension was collected. Protein concentration was
estimated using the BCA protein assay kit (Thermo Fisher
Scientific, Inc.). Proteins (20 µg) were resolved using 12%
SDS-PAGE and transferred to polyvinylidne difluoride membranes. The
membranes were blocked in 5% non-fat milk in TBST (0.1% Tween in
TBS) for 1 h at room temperature. Proteins were probed with mouse
anti-flag antibody (1:1,000 dilution, cat. no. F3165;
Sigma-Aldrich; Merck KGaA), rabbit anti-phoshposerine (1:1,000
dilution, cat. no. ab9332; Abcam) for 3 h at room temperature and
followed by incubation with a horseradish peroxidase-conjugated
secondary antibody (1:10,000 dilution; cat. no. ab9332; Abcam) for
1 h at room temperature. The bands were visualized using enhanced
chemiluminescence reagents (Thermo Fisher Scientific, Inc.).
Chromatin immunoprecipitation assay
(ChIP)
ChIP was performed using the EZ-ChIP Assay kit (EMD
Millipore, Billerica, MA, USA), according to manufacturer's
instructions. HCT116 cells (2×106) were transfected with
siRNA against S6K2 or non-silencing control for 48 h at 37°C. The
cells were cross-linked with 1% formaldehyde for 10 min at room
temperature and the reaction was quenched using 0.125 M glycine.
Cells were washed twice using ice cold PBS containing protease
inhibitors. Cells were pelleted for 4 min at 500 × g and 4°C. Cell
pellets were resuspended in SDS Lysis Buffer (cat. no. 20-163; EMD
Millipore). The lysate was sonicated to obtain DNA fragments
between 200 and 1,000 basepairs in length and validated using
electrophoresis on 1% agarose gels. The sheared DNA was centrifuged
at 12,000 × g at 4°C for 10 min. Soluble chromatin was
immunoprecipitated overnight with mouse anti- hnRNP A1 antibody (5
µg; cat. no. sc-32301; Santa Cruz Biotechnology, CA, USA) overnight
at 4°C. Immunocomplexes were washed once in 1 ml Low Salt Immune
Complex Wash Buffer (cat. no. 20-154; EMD Millipore), once in 1 ml
High Salt Immune Complex Wash Buffer (cat. no. 20-155; EMD
Millipore), once in 1 ml LiCl Immune Complex Wash Buffer (cat. no.
20-156; EMD Millipore), twice in 1 ml TE Buffer (cat. no. 20-157;
EMD Millipore). Following reversal and recovery of the
immunoprecipitated chromatin DNA, the final DNA pellets were
dissolved in 50 µl H2O. The purified DNA samples were
used as templates for semi-quantitative and qPCR to determine the
relative enrichment.
qPCR was performed using SYBR Green Real-Time PCR
Master mix (Thermo Fisher Scientific, Inc.). The comparative Cq
(ΔΔCq) method was used to determine the expression level of mRNA
(22). Thermal cycling conditions
were designed as follows: Initial denaturation at 95°C for 10 min,
followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The
sequences of the primers were: EI9 sense, TCC TAG AGC AGG TGG AGC
AA and anti-sense, CTG ACG GAG TCT GGC AGG TA; β-actin sense, ATG
GAT GAC GAT ATC GCT GCG C and antisense, GCA GCA CAG GGT GCT CCT
CA. β-actin was used as a reference gene.
Immunoprecipitation
HCT116 cells were transfected with wild-type
Flag-hnRNPA1, S6A or S4A mutant of Flag-hnRNPA1 for 48 h at 37°C in
humidified air containing 5% CO2. Cells were collected
and protein were extracted as described previously. Subsequently,
cell lysates were incubated with anti-FLAG-M2 beads (Sigma-Aldrich;
Merck KGaA) for 3 h at 4°C with constant rotation. The beads were
washed 5 times with buffer (50 mM Tris HCl, 150 mM NaCl, 1 mM EDTA
and 1% Triton X-100). The immunocomplexes were then eluted with 3X
Flag peptide (Sigma-Aldrich; Merck KGaA) in 0.5 M Tris HCl (pH 7.5)
with 1 M NaCl for 30 min at 4°C. The beads were centrifuged for 30
sec at 8,000 × g at 4°C to collect supernatants for western blot
analysis. Western blot analysis was performed as described
previously.
Glucose uptake assay
Glucose uptake in cells was quantified using a
Glucose Uptake Assay kit (BioVision, Inc., Milpitas, CA, USA)
Briefly, cells were cultured in glucose-free DMEM for 16 h at 37°C
and then incubated with high-glucose DMEM for an additional 24 h at
37°C. The culture medium was removed, and the intracellular glucose
levels were evaluated according to the kit manufacturer's
instructions.
Statistical analysis
All experiments were performed in triplicate and the
results are presented as the mean ± standard deviation. The
significance of differences between experimental groups was
analyzed using the unpaired Student's t-test or one-way analysis of
variance followed by Student Newman Keuls post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
S6K2 regulates alternative splicing of
PKM gene in a hnRNPA1-dependent manner
To obtain an insight into the function of S6K2 in
alternative splicing of the PKM gene, three siRNAs were used to
knockdown S6K2 expression in the CRC cell line HCT116 (Fig. 1A). The mRNA ratio of PKM2/PKM1 was
downregulated by S6K2 knockdown (Fig.
1B). Binding of hnRNPA1, hnRNPA2 and PTB to the flanking
regions of exon 9 result in the exclusion of exon 9 and subsequent
expression of the PKM2 isoform (7).
Therefore, it was investigated whether S6K2 may affect the binding
of hnRNPA1, hnRNPA2 and PTB to splicing sites of the PKM gene. ChIP
analysis was applied to determine the abundance of hnRNPA1, hnRNPA2
and PTB on the PKM gene in HCT116 cells. Compared with
non-silencing control, S6K2 knockdown decreased hnRNPA1 binding to
the downstream exon 9 splice site (EI9; Fig. 1C), whereas binding of hnRNPA2 to the
corresponding region was unaffected (Fig.
1D). PTB binding to the upstream exon 9 splicing site (EI8) was
unchanged following S6K2 knockdown (Fig.
1E). Therefore, the results of the present study indicated that
S6K2 was involved in alternative splicing of PKM gene by modulating
hnRNPA1 binding to the splicing site.
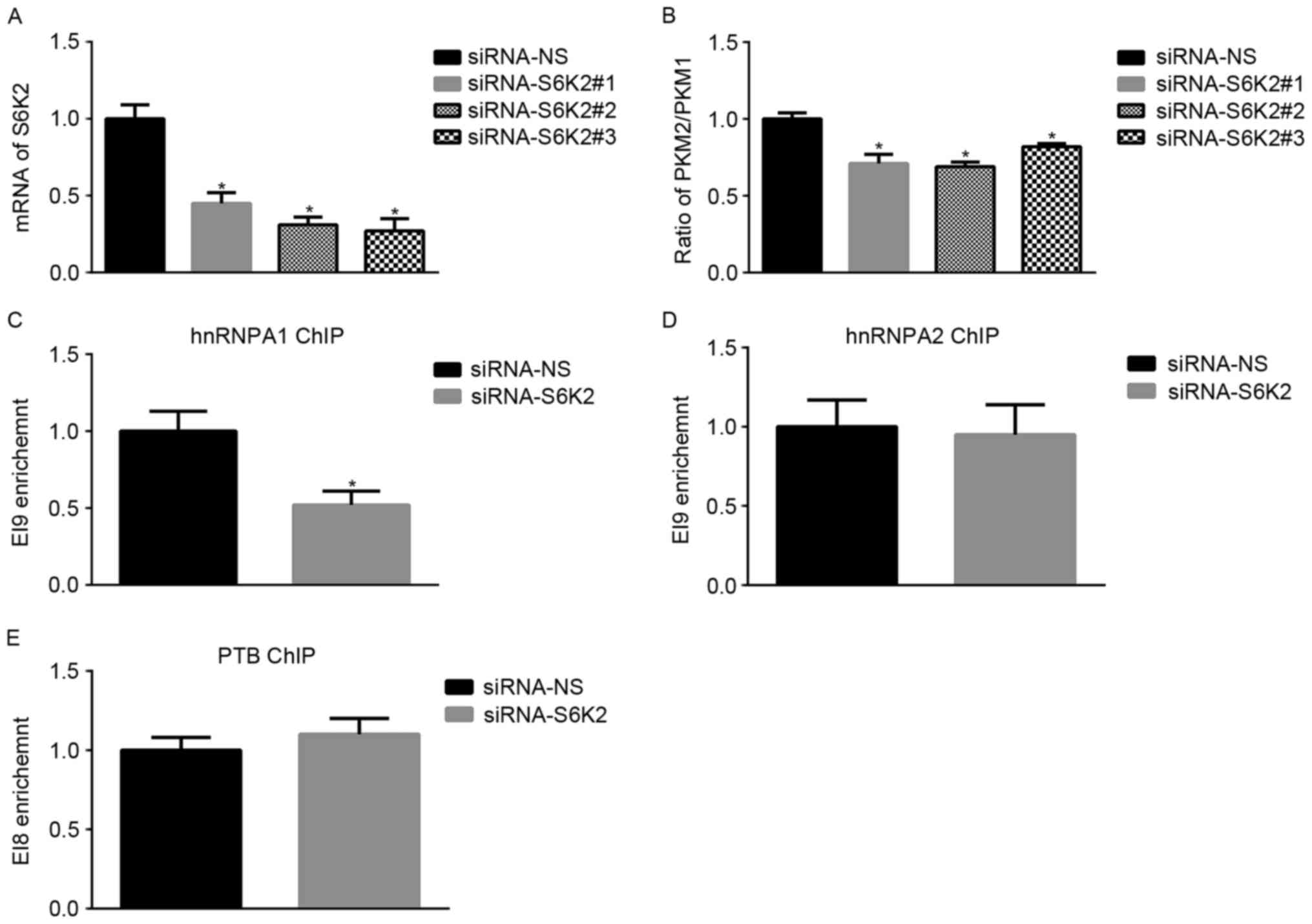 | Figure 1.S6K2 regulates alternative splicing
of the PKM gene. (A) HCT116 cells were transfected with siRNAs
against S6K2 or non-silencing control. The mRNA levels of S6K2 were
analyzed using qPCR. (B) HCT116 cells were transfected with siRNAs
against S6K2 or NS control. The mRNA levels of PKM1 or PKM2 were
analyzed using qPCR. The ratio of PKM2/PKM1 was calculated
according to their mRNA levels. HCT116 cells were transfected with
siRNA against S6K2 or non-silencing control. (C) hnRNPA1, (D)
hnRNPA2 and (E) PTB antibodies were used for chromatin
immunoprecipitation analysis. The primers recognizing EI9 or EI8
splicing site were used for analyzing enrichment of indicated
protein. Data are presented as the mean ± standard deviation.
*P<0.05 vs. siRNA non-silencing control. S6K2, S6 kinase 2; PKM,
pyruvate kinase; PKM1, pyruvate kinase M1 isoform; PKM2, pyruvate
kinase M2 isoform; siRNA, short interfering RNA; qPCR, quantitative
polymerase chain reaction; hnRNPA1, heterogeneous nuclear
ribonucleoprotein A1; hnRNPA2, heterogeneous nuclear
ribonucleoprotein A2; -NS, non-silencing; PTB, RNA binding motif
containing. |
Serine 6 phosphorylation of hnRNPA1 is
required for S6K2 regulating the levels of PKM isoforms
hnRNPA1 has been identified as a substrate of S6K2
kinase (19). Consistent with a
previous study (19), mutation of
either Ser4 or Ser6 residue from serine to
alanine decreased serine phosphorylation of hnRNPA1 (Fig. 2A). Subsequently, the present study
aimed to determine whether this phosphorylation event was involved
in alternative splicing of the PKM gene. hnRNPA1 may induce the
expression of PKM2 and inhibit the expression of PKM1 (5). The results of the present study
demonstrated that the PKM2/PKM1 mRNA ratio was increased by hnRNPA1
in HCT116 cells compared with the vector control (Fig. 2B). In contrast with the wild-type,
hnRNPA1-S6A mutant was unable to increase the mRNA ratio of
PKM2/PKM1 (Fig. 2B). In addition, the
mRNA ratio of PKM2/PKM1 did not significantly differ between the
wild-type and hnRNPA1-S4A mutant (Fig.
2B). Furthermore, mutation at residue Ser6 inhibited
the decrease of the PKM2/PKM1 mRNA ratio induced by S6K2 knockdown
(Fig. 2C). Only Ser6
phosphorylation of hnRNPA1 is required for alternative splicing of
the PKM gene, in spite of Ser4 and Ser6 being
phosphorylating sites of S6K2 (16).
Subsequently, the present study aimed at identifying whether
Ser6 phosphorylation of hnRNPA1 by S6K2 affected the
mRNA ratio of PKM2/PKM1. ChIP analysis indicated that the abundance
of hRNPA1 on the flanking regions of exon 9 was decreased in HCT116
cells transfected with hnRNPA1-S6A mutant, compared with that
transfected with either wild-type or hnRNPA1-S4A mutant (Fig. 2D). Therefore, the results of the
present study indicated that S6K2 selectively phosphorylates
Ser6 residue of hnRNPA1 to regulate alternative splicing
of PKM gene.
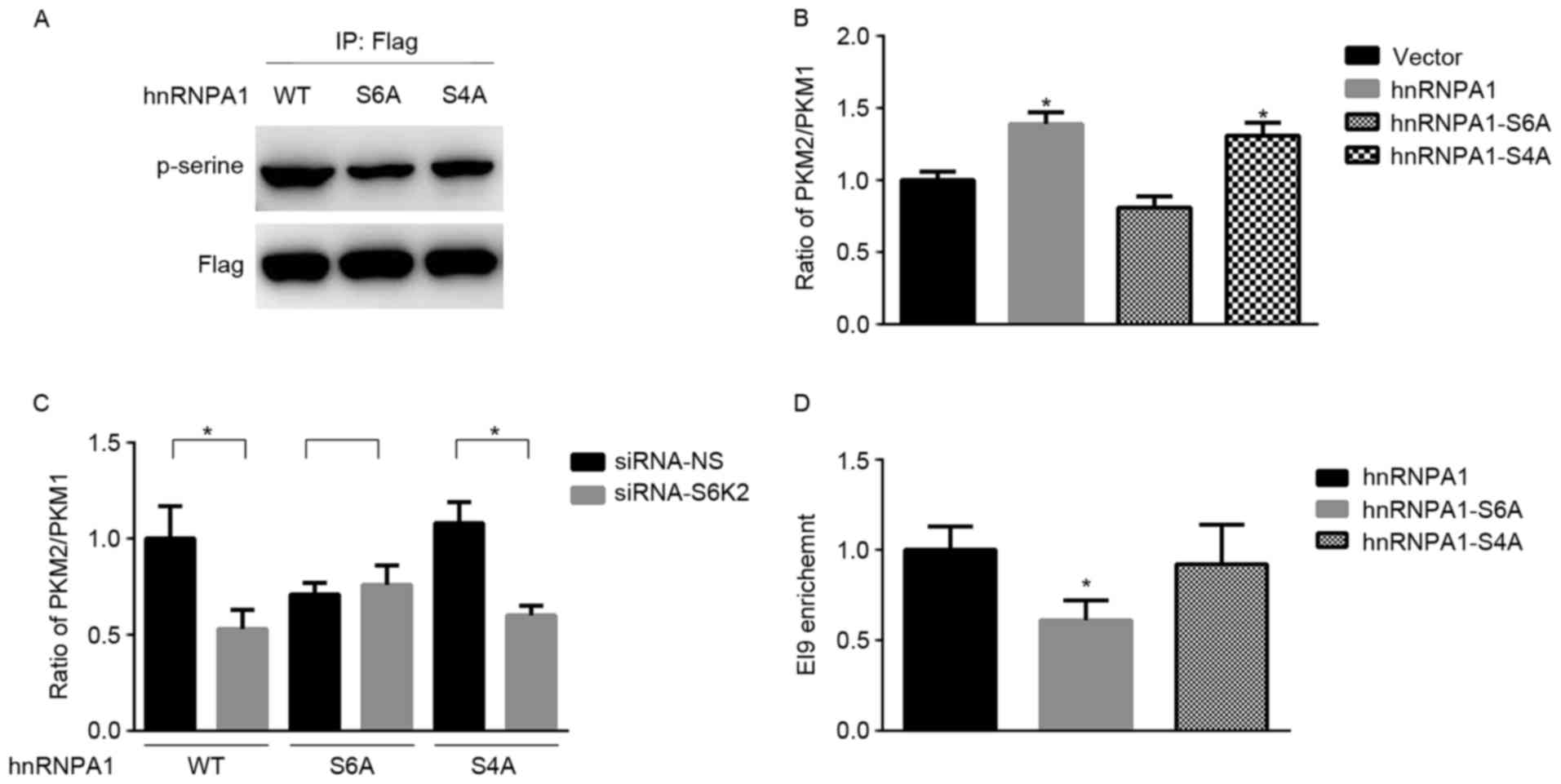 | Figure 2.Serine 6 phosphorylation of hnRNPA1
by S6K2 regulates PKM expression. (A) HCT116 cells were transfected
with wild-type Flag-hnRNPA1, S6A or S4A mutant of Flag-hnRNPA1.
Flag antibodies were used for immunoprecipitation. The
phosphor-serine antibody was applied for determining the serine
phosphorylation in hnRNPA1. (B) HCT116 cells were transfected with
empty vector, wild-type Flag-hnRNPA1, and S6A or S4A mutant of
Flag-hnRNPA1. The ratio of PKM2/PKM1 was calculated according to
their mRNA levels, as analyzed using qPCR. *P<0.05 vs. vector
control. (C) HCT116 cells were co-transfected with siRNA against
S6K2 (NS siRNA as the control) and empty vector, wild-type
Flag-hnRNPA1, S6A or S4A mutant of Flag-hnRNPA1. The ratio of
PKM2/PKM1 was calculated according to their mRNA levels, as
analyzed using qPCR. *P<0.05 vs. siRNA non-silencing control.
(D) HCT116 cells were transfected with wild-type Flag-hnRNPA1 and
S6A or S4A mutant of Flag-hnRNPA1. Flag antibodies were used for
chromatin immunoprecipitation analysis. The primers recognizing EI9
splicing site were used for analyzing enrichment of indicated
protein. *P<0.05 vs. hnRNPA1 group. Data are presented as the
mean ± standard deviation. hnRNPA1, heterogeneous nuclear
ribonucleoprotein A1; PKM, pyruvate kinase; S6A, serine 6 A; S4A,
serine 4 A; PKM2, pyruvate kinase M2 isoform; PKM1, pyruvate kinase
M1 isoform; qPCR, quantitative polymerase chain reaction; siRNA,
short interfering RNA; WT, wild-type; -NS, non-silencing. |
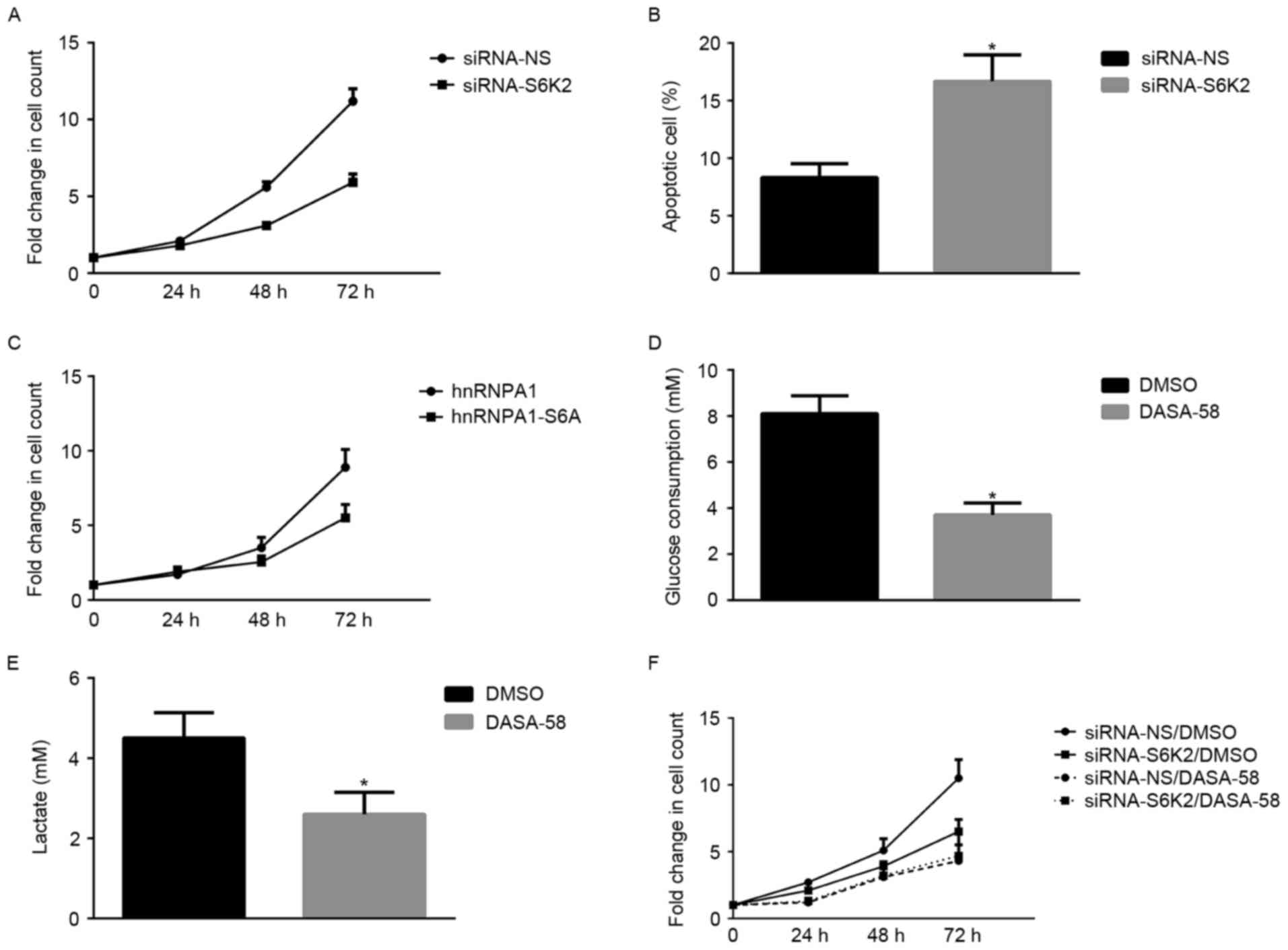
S6K2 promotes glycolysis of CRC cells
via hnRNPA1-mediated alterative splicing of PKM gene
On the basis of the fundamental function of PK in
glycolysis, the present study aimed at determining whether S6K2 may
regulate glycolysis of CRC cell lines. S6K2 knockdown decreased
glycolysis, as indicated by glucose consumption (Fig. 3A) or lactate production (Fig. 3B). S6K2-mediated Ser6
phosphorylation was inhibited by expression of the hnRNPA1-S6A
mutant, but S6K2 did not exhibit a regulatory effect on glycolysis
of HCT116 cells (Fig. 3C and D). To
identify the function of hnRNPA1-mediated splicing in glucose
metabolism, an ASO against exon 10 for PKM gene was used.
Consistent with a previous study (25), this specific ASO against exon 10
decreased the mRNA ratio of PKM2/PKM1 (Fig. 3E). Furthermore, the decrease of
PKM2/PKM1 induced by S6K2 knockdown was inhibited in the presence
of the ASO against exon 10 (Fig. 3E).
In the presence of ASO against exon10, the S6K2-mediated glycolysis
was inhibited (Fig. 3F and G). The
results of the present study suggested that S6K2 modulated
glycolysis of CRC cells by phosphorylating the Ser6
residue of hnRNPA1.
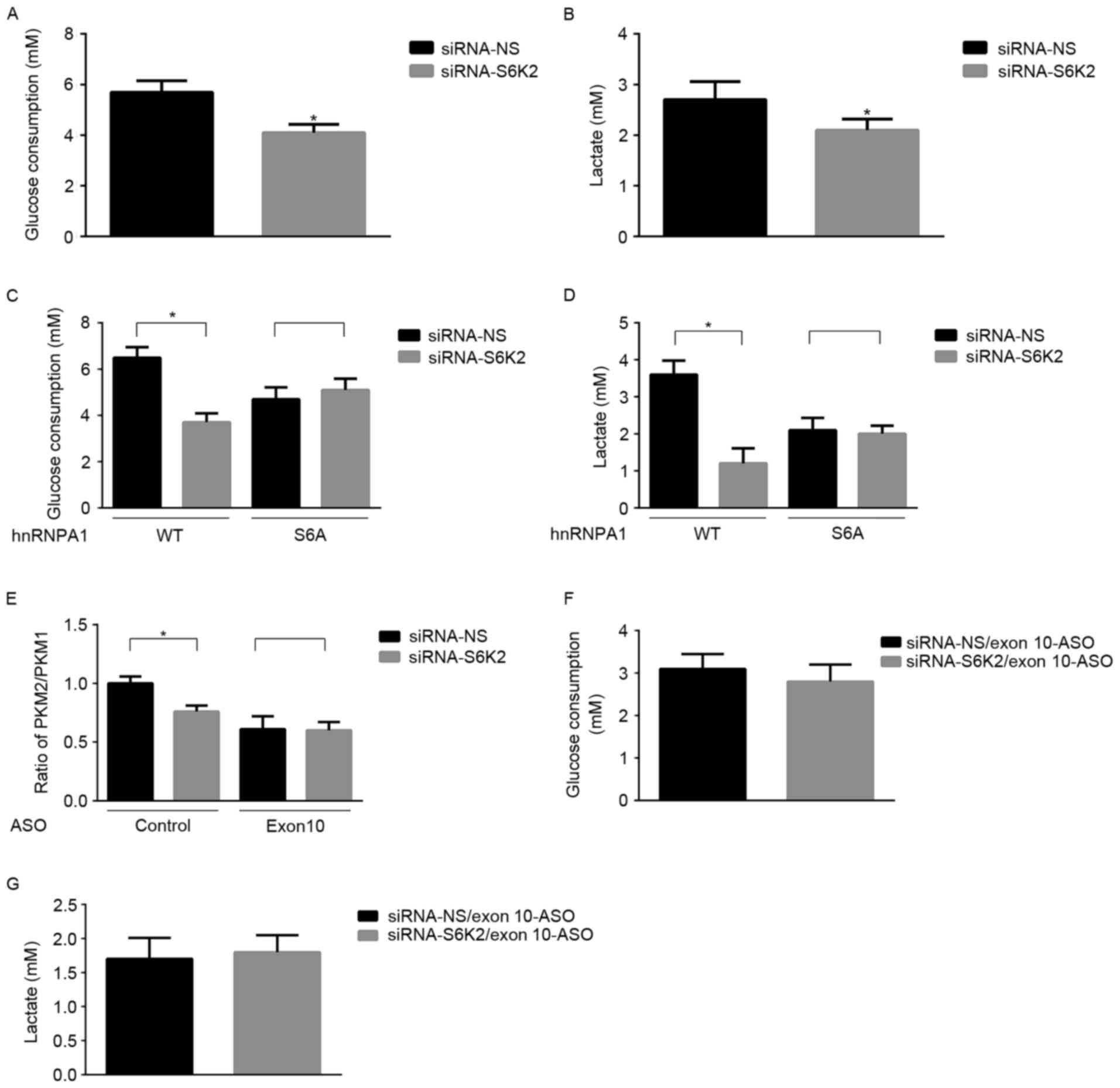 | Figure 3.S6K2 promotes glycolysis of CRC
cells. HCT116 cells were transfected with siRNA against S6K2 or NS
control. (A) Glucose consumption and (B) lactate production were
analyzed in these cells. HCT116 cells were co-transfected with
siRNA against S6K2 (NS siRNA as the control) and wild-type
Flag-hnRNPA1, and S6A or S4A mutant of Flag-hnRNPA1. (C) Glucose
consumption and (D) lactate production were analyzed in these
cells. (E) HCT116 cells were co-transfected with siRNA against S6K2
(NS siRNA as the control) and EI9-ASO against EI9 splicing site or
control ASO. The ratio of PKM2/PKM1 was calculated according to
their mRNA level as analyzed by qPCR. HCT116 cells were
co-transfected with siRNA against S6K2 (NS siRNA as the control)
and EI9-ASO against EI9 splicing site or control ASO. (F) Glucose
consumption and (G) lactate production were analyzed in these
cells. Data are presented as the mean ± standard deviation.
*P<0.05 vs. siRNA non-silencing control. S6K2, S6 kinase 2; CRC,
colorectal cancer; siRNA, short interfering RNA, -NS,
non-silencing; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1;
S6A, serine 6 A; S4A, serine 4 A; ASO, antisense oligonucleotide;
qPCR, quantitative polymerase chain reaction. |
S6K2 promotes proliferation of CRC
cells through activating glycolysis
Subsequently, the present study investigated whether
the metabolic advantages induced by S6K2 regulated the
proliferation of CRC cells. S6K2 knockdown significantly decreased
the growth of HCT116 cells (Fig. 4A).
However, the proportion of apoptotic cells increased following
silencing of S6K2 in HCT116 cells (Fig.
4B). HCT116 cells transfected with hnRNPA1 mutant devoid of
S6K2-mediated Ser6 phosphorylation exhibited a decreased
fold-change in the cell count, compared with those transfected with
wild-type hnRNPA1 (Fig. 4C). To
understand the function of glucose metabolism in S6K2-mediated
proliferation of cancer cells, a PKM2 activator, DASA-58, was used
to modulate glycolysis. DASA-58 inhibits proliferation and
glycolysis of cancer cells by activating PKM2 enzyme activity
(26). Glucose consumption (Fig. 4D) and lactate production (Fig. 4E) of HCT116 cells were inhibited by
DASA-58. In the presence of DASA-58, the effect of S6K2 knockdown
on cell growth was inhibited (Fig.
4F). Therefore, the results of the present study demonstrated
that serine 6 phosphorylation of hnRNPA1 regulated cancer cell
growth, which is associated with PKM2-mediated glycolysis.
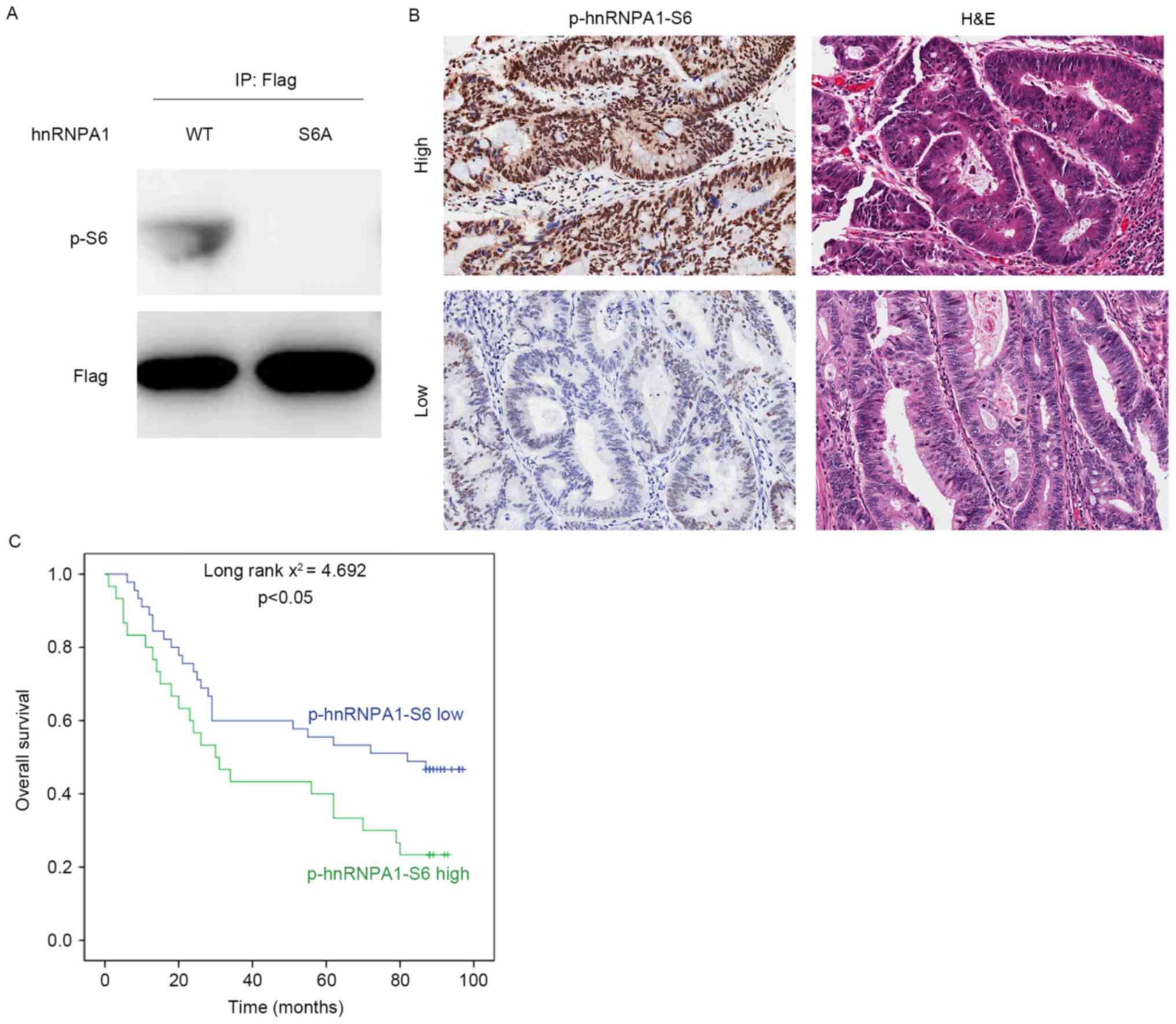
hnRNPA1 phosphorylation by S6K2
predicts poor prognosis of CRC
To analyze the clinical significance of serine 6
phosphorylation of hnRNPA1, a specific antibody against this
modification was generated (Fig. 5A).
Subsequently, IHC analysis was performed in 75 colorectal cancer
tissues. Representative staining images of phosphorylation of
hnRNPA1-S6 are presented in Fig. 5B.
According to semi-quantitative analysis of the level of serine 6
phosphorylation of hnRNPA1, patients were divided into two groups:
High (n=30) and low p-hnRNPA1-S6 level (n=45) groups. The median
survival duration was 44.633 months [95% confidence interval (CI),
32.328–56.938] and 61.822 months (95% CI, 50.984–72.660) for the
high and low p-hnRNPA1-S6 groups, respectively. The Kaplan-Meier
estimator survival analysis indicated that patients belonging to
the high p-hnRNPA1-S6 group exhibited a significantly decreased
overall survival time, compared with those classified as the low
p-hnRNPA1-S6 group (Fig. 5C).
Therefore, the results of the present study suggested that serine 6
phosphorylation of hnRNPA1 may be a useful prognostic marker for
patients with CRC.
Discussion
CRC is one of the most common types of cancer with
high morbidity and mortality rates worldwide (27). In addition to genetic and epigenetic
alterations in oncogenes and tumor suppressors, metabolic
disturbance has an effect on the initiation and progression of CRC
(28). The reprogramming of glucose
metabolism is observed in a number of types of cancer, including
CRC (4). The Warburg effect,
characterized as increased glucose consumption and lactate
production, is the metabolic phenotype observed in CRC (29). An increase in glucose metabolism by
glycolysis, rather than by oxidative phosphorylation, promotes
tumor survival and chemoresistance in CRC (30). For this reason, understanding the
underlying molecular mechanisms and potential targets involved in
glucose metabolic reprogramming may enable the identification of
novel therapeutic strategies for CRC. The results of the present
study demonstrated that the phosphorylation of Ser6 in
hnRNPA1 by S6K2 is required for glycolysis and the proliferation of
CRC cells. In addition, the accumulation of Ser6
phosphorylation was associated with poor survival of patients with
CRC.
Dysregulated glucose metabolism in CRC is regulated
by a group of enzymes or proteins including glucose transporter 1,
hexokinase 2, PKM2 and lactate dehydrogenase A. There is
accumulating evidence demonstrating that PKM2 functions in glucose
metabolic reprogramming in cancer cells (31,32). PKM2
levels are increased in CRC and associated with the tumor stage and
lymph metastasis (33). Expressing
PKM2 isoform, rather than the PKM1 isoform, is a typical
characteristic observed during cancer development (34). In glioblastoma, the splicing factors
(PTB, hnRNPA1 and hnRNPA2) and PKM2 are highly expressed (7). Therefore, inhibition of PKM2 expression
by modulating alternative splicing of the PKM gene may be a useful
strategy to treat cancer. The results of the present study revealed
that S6K2 is an upstream regulator of the alternative splicing of
the PKM gene, by phosphorylating hnRNPA1. On the basis of the
results of the present study, the pan-S6 K inhibitor or specific
S6K2 inhibitor may be exploited for CRC treatment.
hnRNPA1, as a RNA binding protein, participates in
various steps of maturation of nascent transcripts (35). hnRNPA1 participates in alternative
splicing of multiple genes. The binding of hnRNPA1 to a transcript
of BRCA1 DNA repair associated results in exon 18 skipping
(36) and hnRNPA1 has been identified
to repress exon 3 inclusion during transcription of human
immunodeficiency virus type 1 (1). In
regard to alternative splicing of the PKM gene, hnRNPA1 binds to
the flanking of exon 9, which causes exon 10 inclusion and exon 9
exclusion (7). Oncogenic
transcription factor c-MYC promotes hnRNPA1 expression and
subsequent alternative splicing of PKM gene (7). Ser6/4 in hnRNPA1 are phosphorylation
sites of S6K2 kinase and the phosphorylation on these two sites is
involved in the translation of BCL-xL and XIAP (19). Notably, the results of the present
study indicated that only Ser6 phosphorylation is
required for promoting hnRNPA1 to bind to the splicing site of the
PKM gene. Ser6, rather than Ser4,
phosphorylation may cause structural alterations to hnRNPA1, which
facilitated the binding of Ser6 to the splicing site of
the PKM gene. In addition, this suggested that the phosphorylation
pattern of hnRNPA1 by S6K2 may be distinct in regard to the
specific target gene of hnRNPA1.
The results of the present study provided an insight
into how CRC cells may reprogramme glucose metabolism to maintain
its malignant phenotype. It was demonstrated that the
phosphorylation of hnRNPA1 by S6K2 is required for glycolysis and
growth of CRC cells. Inhibiting S6K2-meidated modification of
hnRNPA1 may be a future strategy for CRC treatment.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81201532, 81372195
and 81572719), the Science and Technology Commission of Shanghai
Municipality (grant no. 134119a5600), the Program for Professor of
Special Appointment (Eastern Scholar) at Shanghai Institutions of
Higher Learning (grant no. 1410000157) and the Shanghai Municipal
Commission of Health and Family Planning (grant no.
XYQ2013109).
References
|
1
|
Tange TO, Damgaard CK, Guth S, Valcárcel J
and Kjems J: The hnRNP A1 protein regulates HIV-1 tat splicing via
a novel intron silencer element. EMBO J. 20:5748–5758. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, et al: Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koppenol WH, Bounds PL and Dang CV: Otto
Warburg's contributions to current concepts of cancer metabolism.
Nat Rev Cancer. 11:325–337. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: Meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mazurek S, Boschek CB, Hugo F and
Eigenbrodt E: Pyruvate kinase type M2 and its role in tumor growth
and spreading. Semin Cancer Biol. 15:300–308. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
David CJ, Chen M, Assanah M, Canoll P and
Manley JL: HnRNP proteins controlled by c-Myc deregulate pyruvate
kinase mRNA splicing in cancer. Nature. 463:364–368. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang W, Xia Y, Hawke D, Li X, Liang J,
Xing D, Aldape K, Hunter T, Yung Alfred WK and Lu Z: PKM2
phosphorylates histone h3 and promotes gene transcription and
tumorigenesis. Cell. 150:685–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Christofk HR, Vander Heiden MG, Harris MH,
Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL and
Cantley LC: The M2 splice isoform of pyruvate kinase is important
for cancer metabolism and tumour growth. Nature. 452:230–233. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Calabretta S, Bielli P, Passacantilli I,
Pilozzi E, Fendrich V, Capurso G, Fave GD and Sette C: Modulation
of PKM alternative splicing by PTBP1 promotes gemcitabine
resistance in pancreatic cancer cells. Oncogene. 35:2031–2039.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun Y, Zhao X, Zhou Y and Hu Y: miR-124,
miR-137 and miR-340 regulate colorectal cancer growth via
inhibition of the Warburg effect. Oncol Rep. 28:1346–1352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pearce LR, Komander D and Alessi DR: The
nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol.
11:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pardo OE and Seckl MJ: S6K2: The neglected
s6 kinase family member. Front Oncol. 3:1912013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Savinska LO, Lyzogubov VV, Usenko VS,
Ovcharenko GV, Gorbenko ON, Rodnin MV, Vudmaska MI, Pogribniy PV,
Kyyamova RG, Panasyuk GG, et al: Immunohistochemical analysis of
S6K1 and S6K2 expression in human breast tumors. Eksp Onkol.
26:24–30. 2004.PubMed/NCBI
|
|
17
|
Lyzogubov VV, Lytvyn DI, Dudchenko TM,
Lubchenko NV, Pogrybniy PV, Nespryadko SV, Vinnitska AB, Usenko VS,
Gout IT and Filonenko VV: Immunohistochemical analysis of S6K1 and
S6K2 expression in endometrial adenocarcinomas. Exp Oncol.
26:287–293. 2004.PubMed/NCBI
|
|
18
|
Pardo OE, Wellbrock C, Khanzada UK, Aubert
M, Arozarena I, Davidson S, Bowen F, Parker PJ, Filonenko VV, Gout
IT, et al: FGF-2 protects small cell lung cancer cells from
apoptosis through a complex involving PKCepsilon, B-Raf and S6K2.
EMBO J. 25:3078–3088. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roy R, Durie D, Li H, Liu BQ, Skehel JM,
Mauri F, Cuorvo LV, Barbareschi M, Guo L, Holcik M, et al: hnRNPA1
couples nuclear export and translation of specific mRNAs downstream
of FGF-2/S6K2 signalling. Nucleic Acids Res. 42:12483–12497. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Datta SR, Dudek H, Tao X, Masters S, Fu H,
Gotoh Y and Greenberg ME: Akt phosphorylation of BAD couples
survival signals to the cell-intrinsic death machinery. Cell.
91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Y, He W, Luo M, Zhou Y, Chang G, Ren
W, Wu K, Li X, Shen J, Zhao X and Hu Y: SREBP1 regulates
tumorigenesis and prognosis of pancreatic cancer through targeting
lipid metabolism. Tumour Biol. 36:4133–4141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun Y, Zhao X, Yao Y, Qi X, Yuan Y and Hu
Y: Connexin 43 interacts with Bax to regulate apoptosis of
pancreatic cancer through a gap junction-independent pathway. Int J
Oncol. 41:941–948. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun Y, Zhao X, Luo M, Zhou Y, Ren W, Wu K,
Li X, Shen J and Hu Y: The pro-apoptotic role of the regulatory
feedback loop between miR-124 and PKM1/HNF4alpha in colorectal
cancer cells. Int J Mol Sci. 15:4318–4332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Z, Jeon HY, Rigo F, Bennett CF and
Krainer AR: Manipulation of PK-M mutually exclusive alternative
splicing by antisense oligonucleotides. Open Biol. 2:1201332012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Anastasiou D, Yu Y, Israelsen WJ, Jiang
JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A, et al:
Pyruvate kinase M2 activators promote tetramer formation and
suppress tumorigenesis. Nat Chem Biol. 8:839–847. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Global Burden of Disease Cancer
Collaboration, . Fitzmaurice C, Dicker D, Pain A, Hamavid H,
Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, et
al: The Global Burden of Cancer 2013. JAMA Oncol. 1:505–527. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
DeBerardinis RJ, Lum JJ, Hatzivassiliou G
and Thompson CB: The biology of cancer: Metabolic reprogramming
fuels cell growth and proliferation. Cell Metab. 7:11–20. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang S and Fang X: Advances in glucose
metabolism research in colorectal cancer. Biomed Rep. 5:289–295.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vellinga TT, Borovski T, de Boer VC,
Fatrai S, van Schelven S, Trumpi K, Verheem A, Snoeren N, Emmink
BL, Koster J, et al: SIRT1/PGC1α-dependent increase in oxidative
phosphorylation supports chemotherapy resistance of colon cancer.
Clin Cancer Res. 21:2870–2879. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even Warburg did not anticipate.
Cancer cell. 21:297–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hirschey MD, DeBerardinis RJ, Diehl AME,
Drew JE, Frezza C, Green MF, Jones LW, Ko YH, Le A, Lea MA, et al:
Dysregulated metabolism contributes to oncogenesis. Semin Cancer
Biol. 35 Suppl:S129–S150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou CF, Li XB, Sun H, Zhang B, Han YS,
Jiang Y, Zhuang QL, Fang J and Wu GH: Pyruvate kinase type M2 is
upregulated in colorectal cancer and promotes proliferation and
migration of colon cancer cells. IUBMB Life. 64:775–782. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wong N, De Melo J and Tang D: PKM2, a
central point of regulation in cancer metabolism. Int J Cell Biol.
2013:2425132013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jean-Philippe J, Paz S and Caputi M: hnRNP
A1: The Swiss army knife of gene expression. Int J Mol Sci.
14:18999–19024. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goina E, Skoko N and Pagani F: Binding of
DAZAP1 and hnRNPA1/A2 to an exonic splicing silencer in a natural
BRCA1 exon 18 mutant. Mol Cell Biol. 28:3850–3860. 2008. View Article : Google Scholar : PubMed/NCBI
|