Introduction
Gastric cancer (GC) is one of the most common
malignant tumors worldwide, particularly in China and other Asian
countries (1). Chemotherapy is the
standard strategy used to manage GC; however, the majority of
patients fail to achieve the ideal initial response and/or develop
resistance to chemotherapy. Multidrug resistance (MDR) is one of
the primary mechanisms for failure of GC treatment. A previous
study demonstrated that aberrant drug transportation and cell
apoptosis contributed to chemotherapeutic resistance as drug
transporters were demonstrated to be an essential component in
intracellular drug metabolism (2).
P-glycoprotein (P-gp) is one of the most studied transporters in
drug resistance. A large population of chemotherapeutic drugs are
substrates for P-gp, and thus expression or function of P-gp was
associated with MDR in several types of cancer (2,3). Certain
types of cancer cells, including breast cancer, head and neck
squamous cell carcinoma and colorectal cancer cells are able to
activate several signaling transduction pathways in response to
drug stimuli, and also alter the expression and protein activity of
apoptosis-associated molecules in order to resist the action of
therapy and allow cancer cells to survive (2,4).
Currently, multiple molecules, including glutathione S-transferase,
breast cancer resistance protein, PI3K/Akt, Bcl-2 and p53 have been
proved to be associated with MDR in chemotherapy (2–4). However,
the underlying molecular mechanism and associated molecular
interactions remain unclear.
Transcription factors (TFs) are able to target
promoter regions of different genes, leading to the regulation of a
large group of target genes. Previous studies have identified that
TFs serve multiple critical roles in cancer MDR. The inactivation
or mutation of p53, a well-known master tumor suppressor, was
reported to induce drug resistance through the modulation of
apoptosis-associated proteins (5–7). Nuclear
factor-κB (NF-κB) was also revealed to promote MDR by targeting
apoptosis-associated molecules or microRNAs (8–10).
Furthermore, NF-κB may also possess a compensatory function with
p53 through mutual interaction and thereby regulate the response to
5-fluorouracil treatment (8). As well
as the classical association with drug response, there is also a
series of MDR-associated TFs that have been identified. In
drug-resistant GC cells, the expression of zinc ribbon
domain-containing 1 (ZNRD1) is increased, and therefore inhibition
of ZNRD1may be able to improve drug sensitivity to various
chemotherapeutics (11). A previous
study also reported that a significant decrease in cut-like
homeobox 1 (CUTL1) transcriptional activity may participate in
doxorubicin resistance (12).
Therefore, targeting MDR-associated TFs may be an efficient
strategy in MDR reversion.
In the present study, a high-throughput TF
activation-profiling assay was utilized to analyze activities of
TFs between drug-resistant and drug-sensitive GC cells. A total of
15 TFs with aberrant activities were detected. Among these TFs, few
previous reports have commented on the association between
hepatocyte nuclear factor (HNF)-4α and MDR. Furthermore, ectopic
expression of HNF-4α promoting drug resistance in GC was exhibited,
whereas the loss of HNF-4α increased drug sensitivity in
vitro. In addition, it was revealed that HNF-4α was able to
regulate MDR by targeting B-cell lymphoma 2 (Bcl-2), without marked
effects on intracellular chemotherapeutic drug transportation.
Materials and methods
Patient characteristics of tissue
specimens
In total, 126 GC tissue samples and 69 chronic
gastritis (CG) tissue samples were obtained from the Pathology
Department of Xijing Hospital (Shaanxi, China). GC tissue samples
were from 60 males and 66 females, and were categorized on the
basis of age (above or below 56 years), tumor differentiation
(well, moderate and poor), tumor stage (T1-4) and lymph node
metastasis (N0-N3). Corresponding clinical data were obtained from
medical records and all patients with GC were followed-up for 70
months. The present study was approved by the Institutional Review
Board of Xijing Hospital, Xi'an, China.
Cell culture and TF Activation
Profiling Plate Array
Human GC cell lines including KATO III, AGS and
immortalized human gastric epithelial cell line GES-1 were obtained
from the Shanghai Institute of Biochemistry and Cell (Chinese
Academy of Sciences, Shanghai, China) and maintained within the
Department of Hepatobiliary Surgery, the First Affiliated Hospital
(Chongqing Medical University, Chongqing, China). Human GC cell
lines GC9811, SGC7901 and MDR variant SGC7901/VCR (derived from
SGC7901 by stepwise selection with vincristine) were obtained from
the Department of Digestive Diseases, Xijing Hospital (Shaanxi,
China). All cells were cultured in RPMI-1640 medium (Hyclone; GE
Healthcare, Logan, UT, USA) containing 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1%
penicillin/streptomycin and maintained at 37°C under an atmosphere
of 5% CO2. The nucleoprotein extracts of SGC7901 and
SGC7901/VCR cells were prepared and subjected to TF Activation
Profiling Plate Array (Signosis, Inc., Santa Clara, CA, USA),
according to the manufacturer's protocol. The TF Activation
Profiling Plate Array was used to determine the activities of 96
TFs in one plate. The activity of each TF was automatically
recorded and 1.5 was set as the threshold value for screening
over-activated TFs.
Lentiviral infection and stable cell
variants
Lentiviruses expressing HNF-4α, specific small
interfering RNA (siRNA) or the corresponding controls were products
from Shanghai Genechem Co., Ltd. (Shanghai, China). Target cells
were infected with lentiviruses according to the manufacturer's
protocol and mixed stable clones were isolated and subjected to
in vitro drug sensitivity assay, apoptosis assay,
intracellular adriamycin concentration analysis and western
blotting.
In vitro drug sensitivity assay
Drug sensitivity was evaluated in vitro using
an MTT assay (Merck KGaA, Darmstadt, Germany) as described
previously (11). Briefly, cells
(5×103) were seeded into 96-well plates and incubated at
37°C with Adriamycin, 5-fluorouracil, cisplatin, vincristine and
mitomycin for 48 h at 0.01-, 0.1-, 1- and 10-fold peak
concentration in human sera. Peak concentrations for Adriamycin,
5-fluorouracil, cisplatin, vincristine and mitomycin were 0.4,
10.0, 3.0, 0.5 and 3.0 µg/ml respectively. MTT was added to the
wells and the optical density at wave length 570 nm was measured 4
h later. The inhibition rates and half-maximal inhibitory
concentration (IC50) values were then calculated.
Apoptosis assay
GC SGC7901 cells and variants overexpressing HNF-4α
were treated with 0.25 µg/ml vincristine. SGC7901/VCR cells and
their variants with knockdown of HNF-4α were treated with 2.5 µg/ml
vincristine. Following incubation at 37°C for 24 h with
vincristine, the apoptotic cells were analyzed using flow cytometry
using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis
detection kit (BD Biosciences, Franklin Lakes, NJ, USA) as
described previously (11). Briefly,
cell samples were sequentially incubated with Annexin V-fluorescein
isothiocyanate and propidium iodide (PI) following the
manufacturer's protocol and then analyzed with a flow cytometer
(FACSCalibur; BD Biosciences, San Jose, CA, USA) using a 530/30 nm
signal detector for Annexin V-FITC and a 582/42 nm signal detector
for PI. The data were subsequently analyzed by Flow J software
(version 7.6.5; Tree Star, Inc., San Carlos, CA, USA). The upper
left and lower left quadrants represented late and early apoptosis,
respectively. The total apoptosis ratio was calculated by adding
the late and early apoptosis proportions.
Intracellular Adriamycin concentration
analysis
The intracellular accumulation and retention of
Adriamycin was determined using flow cytometry. GC cells and their
variants were inoculated into 6-well plates and allowed to adhere
overnight at 37°C. Adriamycin (5 mg/ml) was added and cells were
incubated at 37°C in Adriamycin-containing RPMI-1640 medium with
10% fetal bovine serum for 1 h. To detect Adriamycin retention,
cells were transferred to Adriamycin-free RPMI-1640 medium with 10%
fetal bovine serum for another 1 h and then trypsinized, washed,
resuspended in phosphate buffered saline (PBS) and subjected to
flow cytometry. A flow cytometer (FACSCalibur; BD Biosciences, San
Jose, CA, USA) was used with a 582/42 nm signal detector for
intracellular Adriamycin. The data were subsequently analyzed by
Flow J software (version 7.6.5; Tree Star, Inc.). Mean fluorescence
intensity of Adriamycin was obtained and expressed as the mean ±
standard error of the mean. The Adriamycin-releasing index was
calculated as 100% × (mean fluorescence intensity of
accumulation-mean fluorescence intensity of retention)/(mean
fluorescence intensity of accumulation). Experiments were performed
in triplicate.
Western blotting
Cells were lysed in radioimmunoprecipitation buffer
(Beyotime Institute of Biotechnology, Haimen, China) supplemented
with 1 mM phenylmethylsulfonyl fluoride and 10 µg/ml each of
pepstatin A, leupeptin, chymostatin and aprotinin (Roche
Diagnostics, Basel, Switzerland). Protein concentration was
measured with a Bicinchoninic acid Protein Assay kit according to
the manufacturer's protocol (Thermo Scientific Pierce, Rockford,
IL, USA). Western blots were performed according to standard
methods as described previously (8).
Equal amounts of protein (50 µg) were loaded onto a SDS-PAGE gel
(8–12% polyacrylamide) and subjected to electrophoresis at 200 V
for 50 min, transferred to nitrocellulose and blocked overnight at
4°C in blocking buffer (NaCl 250 mmol/l, 0.02% Tween 20, 5% goat
serum and 3% bovine serum albumin). Primary antibodies were added
for 3 h at room temperature. Blots were washed, and species-matched
peroxidase-conjugated secondary antibody was added (1:2,000).
Labeled bands from washed blots were detected using an enhanced
chemiluminescence kit (Amersham, Louisville, CO, USA). Primary
antibodies against HNF-4α (1:1,000; cat. no. 3113; Cell Signaling
Technology, Inc., Danvers, MA, USA), Bcl-2-associated X protein
(Bax; 1:500; cat. no. sc-6236; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), Bcl-2 homologous antagonist killer (Bak; 1:500;
cat. no. sc-832, Santa Cruz Biotechnology, Inc.), B-cell lymphoma
extra-large (Bcl-xL; 1:500; cat. no. sc-7195, Santa Cruz
Biotechnology, Inc.), caspase-3 (1:1,000; cat. no. 9662, Cell
Signaling Technology, Inc.), cleaved caspase-3 (1:1,000; cat. no.
9661, Cell Signaling Technology, Inc.), Bcl-2 (1:500; cat. no.
04-436, Merck KGaA, Darmstadt, Germany) and β-actin (1:2,000; cat.
no. MABT825, Merck KGaA) were used. The secondary antibodies
included horseradish peroxidase (HRP)-conjugated anti-rabbit
immunoglobulin (Ig)G (1:2,000; cat. no. 7074, Cell Signaling
Technology, Inc.) and HRP-conjugated anti-mouse IgG (1:3,000; cat.
no. 7076, Cell Signaling Technology, Inc.).
Tissue specimens and
immunohistochemistry
Immunohistochemical examination was performed using
the streptavidin-biotin complex method. Fresh gastric tissues were
fixed in 4% formalin overnight at room temperature and embedded in
paraffin. The tissue blocks were cut into sections (4 µm thick).
Prior to staining, the sections were treated with 0.3% hydrogen
peroxide in 100% methanol for 30 min at room temperature and then
washed in PBS. Following incubation with normal goat serum for 10
min, the sections were incubated with anti-HNF-4α (1:200; cat. no.
3113, Cell Signaling Technology, Inc.) overnight at 4°C. After
incubation, the sections were incubated at room temperature for 1
h. They were washed twice in PBS and treated with biotinylated goat
anti-rabbit IgG (1:100; cat. no. SA2002, Boster, Wuhan, China) and
peroxidase-conjugated streptavidin (1:100; cat. no. SA2002, Boster)
for 30 min at room temperature. They were then reacted with 0.02%
diaminobenzidine tetrahydrochloride containing 0.005% hydrogen
peroxide for 4 min and counterstained with hematoxylin. Rabbit
normal serum was used to replace the primary antibody as a blank
control. The stained sections were observed using a light
microscope (magnification, ×200). The expression of HNF-4α was
evaluated according to the ratio of positive cells per specimen (R)
and staining intensity (I) as described previously (13). A total score (RxI) of 0 to 12 was
calculated and graded as follows: negative (−, 0 to 2), weak
positive (+, 3 to 5), moderate positive (++, 6 to 9) and strong
positive (+++, 10 to 12).
Statistical analysis
SPSS software (version 17.0; SPSS, Inc., Chicago,
IL, USA) was used to perform statistical analysis. One-way analysis
of variance or two-tailed unpaired Student's t-test was used to
analyze the data of TF activity, IC50 values, cell
apoptosis and intracellular Adriamycin. A χ2 test was
applied to detect the significance of the difference in HNF-4α
expression frequency in human gastric tissues and its
clinicopathological association in GC. Kaplan-Meier estimator
survival curves were created to analyze the association of HNF-4α
with patient survival, and the log-rank test was used to compare
the difference of survival curves among groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Identification of MDR-associated TFs
by TF Activation Profiling Plate Array
To identify TFs involved in MDR in GC,
nucleoproteins were extracted from chemo-sensitive (SGC7901) and
chemo-resistant GC cells (SGC7901/VCR) for TF Activation Profiling
Plate Array. Results presented in Table
I identify the increased activity of 15 TFs in chemo-resistant
GC cells SGC7901/VCR compared with SGC7901. Among the top three
aberrantly activated TFs, NF-κB and hypoxia-inducible factor
(HIF)-1 were frequently reported to regulate drug resistance in GC,
followed by HNF-4α, a well-known TF in hepatocyte differentiation
and whose potential roles in MDR were not previously completely
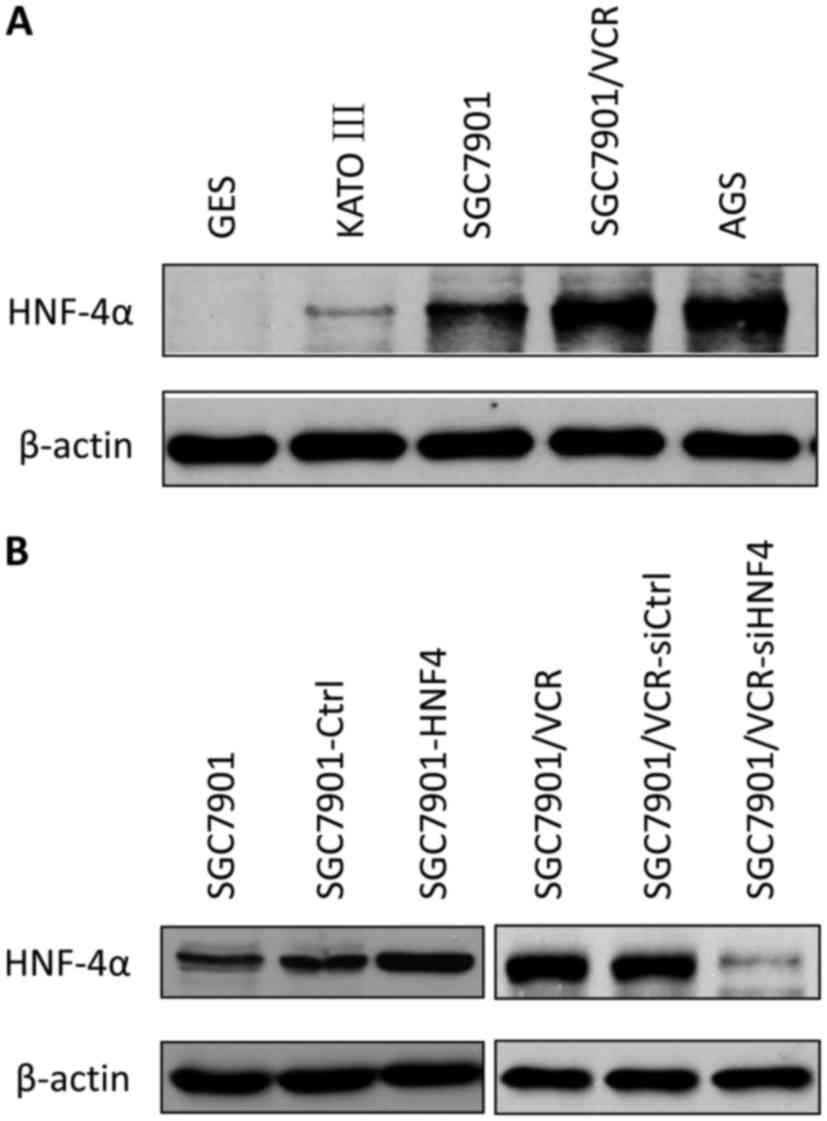
investigated. The western blot analysis determined the expression
levels of HNF-4α in multiple GC cell lines. HNF-4α was increased in
GC cell lines compared with immortalized gastric epithelial cells,
and the expression of HNF-4α was also upregulated in
chemo-resistant cells (SGC7901/VCR) compared with chemo-sensitive
cells (SGC7901; Fig. 1A).
 | Table I.Screening for aberrantly activated TFs
in chemo-resistant GC cells. |
Table I.
Screening for aberrantly activated TFs
in chemo-resistant GC cells.
| Transcription
factor | Activity ratio
(VCR/SGC7901) | P-value |
|---|
| NF-κB | 2.17 | 0.006 |
| HIF-1 | 1.95 | 0.026 |
| HNF-4α | 1.92 | 0.019 |
| RXR | 1.89 | 0.025 |
| GATA | 1.87 | 0.021 |
| STAT1 | 1.74 | 0.031 |
| STAT3 | 1.69 | 0.024 |
| 4-Oct | 1.68 | 0.017 |
| C/EBP | 1.64 | 0.004 |
| KLF4 | 1.58 | 0.011 |
| Snail | 1.56 | 0.028 |
| NFAT | 1.55 | 0.035 |
| TCF/LEF | 1.54 | 0.007 |
| AP1 | 1.52 | 0.009 |
| ATF2 | 1.5 | 0.016 |
HNF-4α regulates MDR of GC cells in
vitro
Cell models with stable lentiviral transfection for
HNF-4α or its siRNA were established (Fig. 1B) and drug sensitivities were measured
using MTT assays (Table II). The
overexpression of HNF-4α markedly promoted resistance to
chemotherapeutic drugs, which demonstrated an increase in the
IC50 values of Adriamycin, vincristine, 5-fluorouracil,
cisplatin and mitomycin. Furthermore, inhibition of HNF-4α resulted
in a decrease in the IC50 values of these drugs
(Table II).
| Table II.Effects of HNF-4α on drug
sensitivities of GC cells. |
Table II.
Effects of HNF-4α on drug
sensitivities of GC cells.
|
| IC50
values, µg/ml |
|---|
|
|
|
|---|
| Cell line | ADR | 5-Fu | CDDP | VCR | MMC |
|---|
| SGC7901 | 0.52±0.05 | 3.11±0.22 | 1.63±0.21 | 0.25±0.01 | 1.39±0.16 |
| SGC7901-Ctrl | 0.61±0.11 | 2.89±0.34 | 1.93±0.28 | 0.29±0.08 | 1.56±0.23 |
| SGC7901-HNF4α |
1.58±0.29a |
6.98±0.73a |
4.16±0.52a |
1.73±0.31a |
4.55±0.67a |
| SGC7901/VCR | 5.44±0.47 | 9.17±1.15 | 7.65±0.94 | 5.86±0.69 | 8.22±1.03 |
|
SGC7901/VCR-siCtrl | 6.21±0.79 | 10.83±1.57 | 7.02±0.86 | 6.29±0.88 | 9.03±1.27 |
|
SGC7901/VCR-siHNF4α |
2.95±0.38b |
8.21±0.93b |
5.14±0.78b |
3.46±0.52b |
6.11±0.94b |
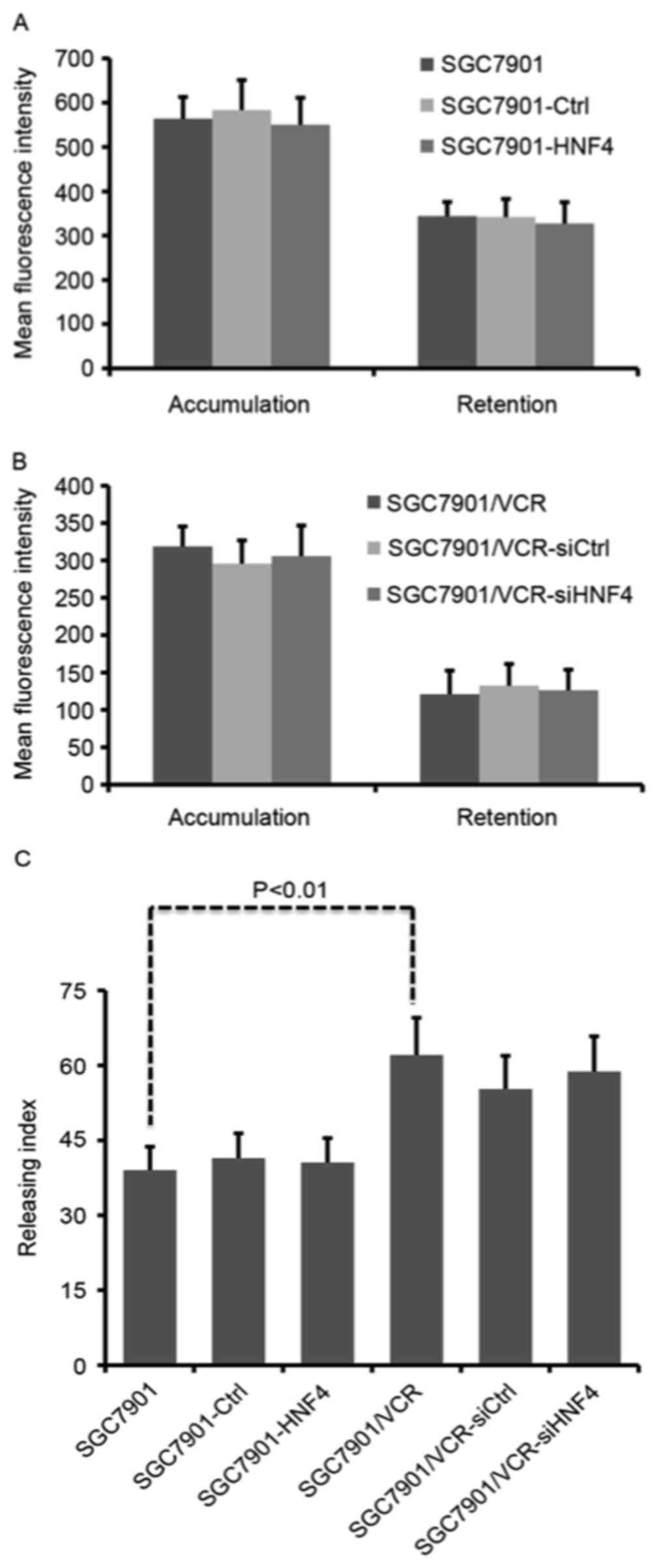
HNF-4α has no effect on drug transport
in GC cells
Enhanced drug efflux activity is one of the primary
causes of drug resistance. To test whether HNF-4α was able to
affect drug transportation, intracellular Adriamycin assays were
performed using flow cytometry. Fluorescence intensity of
accumulated and retained Adriamycin was markedly decreased in
SGC7901/VCR cells compared with SGC7901 cells (Fig. 2A and B), indicating that drug
transportation was active in chemo-resistant GC cells. However,
neither ectopic expression nor knockdown of HNF-4α was able to lead
to any change in Adriamycin accumulation and retention (Fig. 2A and B). Although SGC7901/VCR cells
exhibited a significantly increased Adriamycin releasing index
compared with SGC7901 cells (P<0.01), modulation of HNF-4α
expression displayed no influence on Adriamycin release from GC
cells (Fig. 2C).
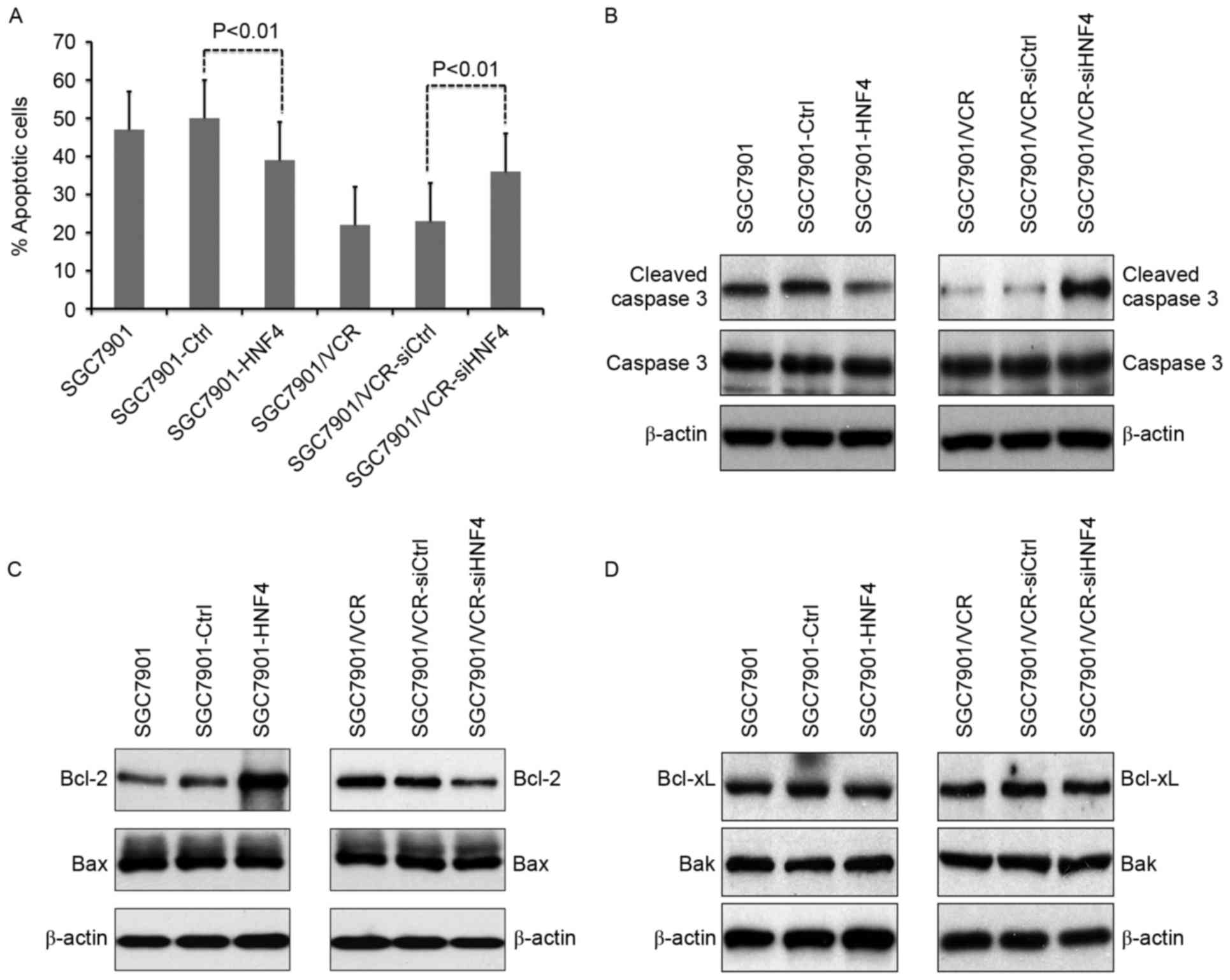
HNF-4α modulates cell apoptosis
through targeting Bcl-2 expression
The effects of HNF-4α on vincristine-triggered cell
apoptosis were evaluated in vitro. It was revealed that cell
apoptosis was suppressed in SGC7901 cells with HNF-4α
overexpression (Fig. 3A). Conversely,
increasing apoptosis in SGC7901/VCR cells was observed following
HNF-4α knockdown (Fig. 3A). Cleaved
caspase-3 was also analyzed in vincristine-treated cells (Fig. 3B). As indicated in Fig. 3B, ectopic expression of HNF-4α
resulted in decreased cleaved caspase-3, whereas knockdown of
HNF-4α led to enhanced cleaved caspase-3. Detection of expression
levels of the apoptosis-associated molecules Bcl-2, Bax, Bcl-xL and
Bak demonstrated that the expression of Bcl-2 was upregulated in
HNF-4α-overexpressed cells, as well as downregulated in cells with
knock-down of HNF-4α (Fig. 3B).
Furthermore, the expression of Bax, Bcl-xL and Bak were not
influenced by HNF-4α (Fig. 3C and
D).
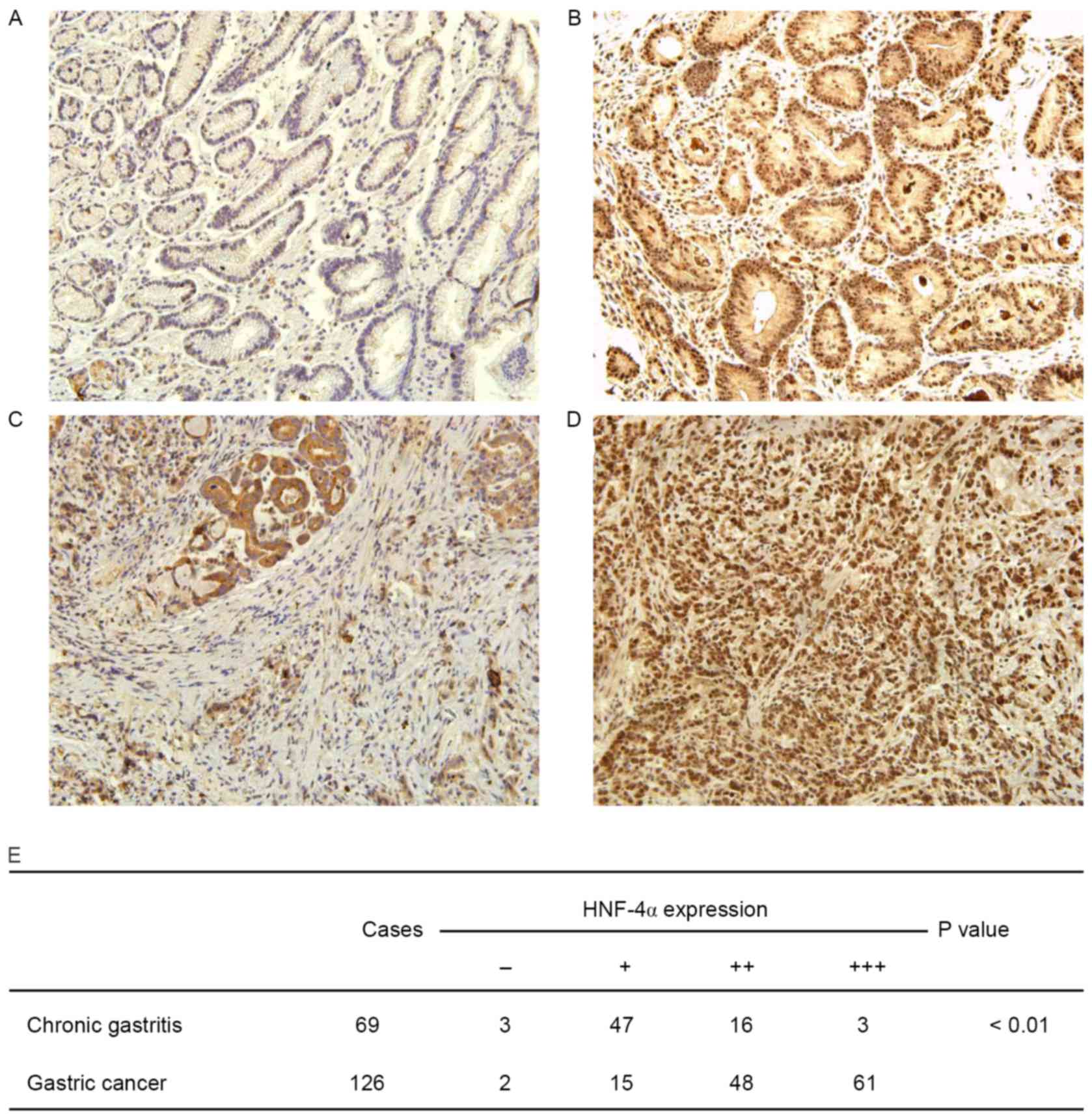
HNF-4α is overexpressed in human GC
tissues
To elucidate the clinical relevance of HNF-4α in GC,
an immunohistochemical assay to determine HNF-4α expression in
human GC tissues was performed. ACG tissue specimen was used as a
control. It was demonstrated that HNF-4α was extensively expressed
in GES cells, localized in both nuclei and cytoplasm (Fig. 4A-D). Staining of HNF-4α was weak in
CG, although it was much stronger in GC tissues. As presented in
Fig. 4E, the difference in HNF-4α
staining in GC and CG was statistically significant (P<0.01).
The clinical and pathological association of HNF-4α expression was
further analyzed. It was demonstrated that HNF-4α expression in GC
tissues was associated with tumor stage and lymph node metastasis;
however, it was not associated with age, sex or tumor
differentiation (Table III). All
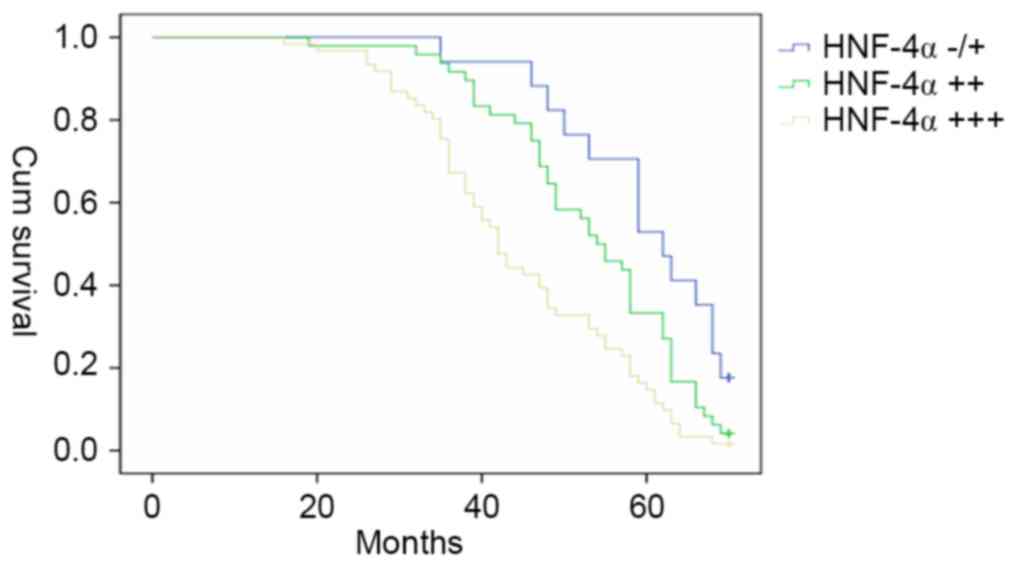
patients with GC were followed-up for 70 months (n=126). The
Kaplan-Meier estimator survival curves were plotted according to
the HNF-4α level in gastric cancer tissues, and the patients with
strong positive (+++) HNF-4α expression exhibited the poorest
survival rate among those three groups (Fig. 5).
| Table III.Clinicopathological association of
HNF-4α in human GC tissues. |
Table III.
Clinicopathological association of
HNF-4α in human GC tissues.
|
| HNF-4α
expression |
|
|---|
|
|
|
|
|---|
|
Characteristics | – | + | ++ | +++ | P-value |
|---|
| n | 2 | 15 | 48 | 61 |
|
| Age (years) |
|
|
|
| 0.682 |
|
<56 | 0 | 8 | 23 | 32 |
|
|
≥56 | 2 | 7 | 25 | 29 |
|
| Gender |
|
|
|
| 0.563 |
|
Male | 1 | 6 | 26 | 27 |
|
|
Female | 1 | 9 | 22 | 34 |
|
|
Differentiation |
|
|
|
| 0.499 |
|
Well | 1 | 5 | 15 | 18 |
|
|
Moderate | 1 | 3 | 20 | 21 |
|
|
Poor | 0 | 7 | 13 | 22 |
|
| Tumor stage |
|
|
|
| 0.015 |
| T1 | 0 | 6 | 8 | 7 |
|
| T2 | 1 | 4 | 13 | 14 |
|
| T3 | 1 | 2 | 15 | 18 |
|
| T4 | 0 | 3 | 12 | 22 |
|
| Lymph node
metastasis |
|
|
|
| 0.007 |
| N0 | 0 | 4 | 10 | 6 |
|
| N1 | 1 | 5 | 12 | 9 |
|
| N2 | 0 | 3 | 12 | 20 |
|
| N3 | 1 | 3 | 14 | 26 |
|
Discussion
On the basis of the high-throughput profiling
analysis, several subsets of TFs were suggested to serve a role in
drug resistance in cancer. In the present study, TF Activation
Profiling Plate Arrays were performed using GC cell models with
distinct chemo-sensitivities. Previous studies have identified
several TFs associated with drug resistance in GC. For example, it
was revealed that HIF-1-dependent pathways were activated in
chemo-resistant GC cells, and MGr1-antigen (MGr1-Ag)/37 kDa laminin
receptor precursor (37LRP), mitogen-activated protein
kinases/extracellular-signal-related kinases and phosphatase and
tensin homologue (PTEN)/protein kinase B (Akt) were frequently
identified to be synergistically altered with HIF-1 (14–16).
NF-κB, a key TF in inflammation was also identified to be
associated with MDR in GC. Inhibition of NF-κB activation was able
to reverse drug resistance, and pathways including p53 and PTEN/Akt
were also involved in NF-κB activation (8,17,18). In addition, the NF-κB-Snail-Bcl-2 axis
was also identified to be associated with mitochondrial antioxidant
manganese superoxide dismutase-induced cisplatin resistance in lung
cancer (19). These previous studies
assist in confirming the reliability of the results of the present
study.
HNF-4α is a member of the orphan steroid hormone
nuclear receptor superfamily, and it activates a diverse set of
liver genes, including transthyretin and α1-antitrypsin
in early liver development (20).
However, the roles of HNF-4α in cancer development are not yet
understood. According to a recent study, HNF-4α was demonstrated to
be downregulated by hepatitis B viral protein, leading to
cytochrome P450 2E1gene inhibition (21), which suggested the activation of this
signaling pathway may contribute to hepatocarcinogenesis (21). Intrahepatic cholangiocarcinoma (IHCC)
was associated with the genetic alterations in isocitrate
dehydrogenase 1 (IDH1) and IDH2. A recent study identified that IDH
mutation was able to block the hepatocyte differentiation of liver
progenitor cells by suppressing HNF-4α, indicating a functional
role in IDH-driven IHCC pathogenesis (22). In a study by Schwartz et al
(23), knockdown of HNF-4α in
colorectal cancer led to a decreased proliferation rate, and
inhibited the proliferation in HT22 and Caco2 cells.
The present study identified that HNF-4α was also
significantly over-activated in chemo-resistant GC cells; however,
its exact function in GC was unclear. In the present study, the
effects of HNF-4α on MDR of GC were investigated, and it was
demonstrated that the chemo-sensitivities of GC cells maybe
significantly affected by HNF-4α. Chemo-resistance was promoted by
HNF-4α through the inhibition of cell apoptosis; however, this
phenomenon was not associated with drug transportation. The results
of the present study suggest that HNF-4α may serve an important
role in malignant phenotypes of GC. Multiple studies are in
agreement with these results (24,25).
Kojima et al (24) performed
immunohistochemical studies in 35 cases of gastric adenocarcinomas
and corresponding non-neoplastic gastric tissues GC. It was
demonstrated that in non-neoplastic and neoplastic gastric
glandular cells, the expression of HNF-4α was associated with the
intestinal phenotype, which suggested that HNF-4α may participate
in the development and maintenance of the intestinal phenotype of
the gastric mucosa and adenocarcinomas. Hepatoid carcinomas,
including α-fetoprotein-producing gastric carcinoma cells also
demonstrated an upregulation in HNF-4α (25). This phenomenon may be attributed to
its function in liver development and its transactivation of
liver-associated genes. In the present study, HNF-4α was markedly
expressed in CG, significantly upregulated in GC tissues, and
associated with GC tumor stage and differentiation, indicating that
HNF-4α serves a role in gastric carcinogenesis. Furthermore, the
HNF-4α expression level in GC tissues is inversely associated with
patient survival. Therefore, future studies evaluating the
prognostic value of HNF-4α for patients with GC are
recommended.
The results of the present study indicate the
potential for HNF-4α to regulate MDR of GC; however, its underlying
molecular mechanism remains unclear. An important mechanism of
HNF-4α regulation may be epigenetic modulation. The effects of
HNF-4α on cell proliferation have been observed in various
colorectal cancer cells, and histone deacetylase inhibitor
targeting HNF-4α were able to suppress proliferation of colon
cancer cells (26). Notably, NF-κB,
HIF-1 and HNF-4α were the most over-activated TFs in the present
study, and their mutual interaction may also contribute to the
function of HNF-4α. According to a previous study in hepatic cells,
tumor necrosis factor-α or other factors (latent membrane protein 1
of the Epstein-Barr virus and wild-type forms of NF-κB signalling
mediators) was able to suppress the transcriptional activity of
HNF-4-dependent promoters by triggering the NF-κB response
(27). Furthermore, it was shown that
this inhibition could be accounted for by a decrease in DNA binding
and the downregulation of the transactivation potential of the
activation functions 1 and 2 (AF-1 and AF-2) domains of HNF-4α
(27). In primary rat hepatocytes,
glucokinase gene expression was associated with HIF-1 and HNF-4 in
a PI3K/Akt-dependent signaling pathway (28). Another study demonstrated that the
transitional change occurred from the interaction of HIF-1 and
HNF-4 under hypoxic conditions (29).
Considering the fact that HIF-1 serves important roles in MDR in
GC, the cross-talk of HIF-1 and HNF-4 may be the potential
mechanism underlying the regulatory function of HNF-4 in MDR. The
present study demonstrated that ectopic expression of HNF-4α was
able to upregulate Bcl-2 in GC cells. This effect was specific for
Bcl-2 owing to several other apoptosis-related molecules including
Bax, Bak and Bcl-xL not being influenced by HNF-4α. It is
reasonable to hypothesize that HNF-4α promoted MDR of GC cells, at
least through regulating Bcl-2 expression.
To conclude, HNF-4α is a TF that regulates MDR in
GC. It inhibits drug-induced apoptosis and promotes GC-related MDR
in vitro; however, further studies are required to further
elucidate the underlying molecular mechanisms. The results of the
present study suggest that HNF-4α may serve as an important target
for MDR management.
Glossary
Abbreviations
Abbreviations:
|
GC
|
gastric cancer
|
|
HIF
|
hypoxia-inducible factor
|
|
HNF
|
hepatocyte nuclear factor
|
|
IDH
|
isocitrate dehydrogenase
|
|
IHCC
|
intrahepatic cholangiocarcinoma
|
|
MDR
|
multidrug resistance
|
|
NF-κB
|
nuclear factor-κB
|
|
P-gp
|
P-glycoprotein
|
|
TF
|
transcription factor
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fodale V, Pierobon M, Liotta L and
Petricoin E: Mechanism of cell adaptation: When and how do cancer
cells develop chemoresistance? Cancer J. 17:89–95. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Noguchi K, Katayama K and Sugimoto Y:
Human ABC transporter ABCG2/BCRP expression in chemoresistance:
Basic and clinical perspectives for molecular cancer therapeutics.
Pharmgenomics Pers Med. 7:53–64. 2014.PubMed/NCBI
|
|
4
|
Elkholi R, Renault TT, Serasinghe MN and
Chipuk JE: Putting the pieces together: How is the mitochondrial
pathway of apoptosis regulated in cancer and chemotherapy? Cancer
Metab. 2:162014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pflaum J, Schlosser S and Müller M: p53
family and cellular stress responses in cancer. Front Oncol.
4:2852014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amelio I and Melino G: The p53 family and
the hypoxia-inducible factors (HIFs): Determinants of cancer
progression. Trends Biochem Sci. 40:425–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ozaki T, Nakamura M and Shimozato O: Novel
implications of DNA damage response in drug resistance of malignant
cancers obtained from the functional interaction between p53 family
and RUNX2. Biomolecules. 5:2854–2876. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Endo F, Nishizuka SS, Kume K, Ishida K,
Katagiri H, Ishida K, Sato K, Iwaya T, Koeda K and Wakabayashi G: A
compensatory role of NF-κB to p53 in response to 5-FU-based
chemotherapy for gastric cancer cell lines. PLoS One. 9:e901552014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prabhu L, Mundade R, Korc M, Loehrer PJ
and Lu T: Critical role of NF-κB in pancreatic cancer. Oncotarget.
5:10969–10975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li F, Zhang J, Arfuso F, Chinnathambi A,
Zayed ME, Alharbi SA, Kumar AP, Ahn KS and Sethi G: NF-κB in cancer
therapy. Arch Toxicol. 89:711–731. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong L, Qiao T, Han Y, Han S, Zhang X, Lin
T, Gao J, Zhao P, Chen Z and Fan D: ZNRD1 mediates resistance of
gastric cancer cells to methotrexate by regulation of IMPDH2 and
Bcl-2. Biochem Cell Biol. 84:199–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao L, Pan Y, Gang Y, Wang H, Jin H, Tie
J, Xia L, Zhang Y, He L, Yao L, et al: Identification of GAS1 as an
epirubicin resistance-related gene in human gastric cancer cells
with a partially randomized small interfering RNA library. J Biol
Chem. 284:26273–26285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Su L, Liu X, Chai N, Lv L, Wang R, Li X,
Nie Y, Shi Y and Fan D: The transcription factor FOXO4 is
down-regulated and inhibits tumor proliferation and metastasis in
gastric cancer. BMC Cancer. 14:3782014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu L, Sun L, Zhang H, Li Z, Ning X, Shi
Y, Guo C, Han S, Wu K and Fan D: Hypoxia-mediated up-regulation of
MGr1-Ag/37LRP in gastric cancers occurs via
hypoxia-inducible-factor 1-dependent mechanism and contributes to
drug resistance. Int J Cancer. 124:1707–1715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu L, Zhang H, Sun L, Gao Y, Jin H, Liang
S, Wang Y, Dong M, Shi Y, Li Z, et al: ERK/MAPK activation involves
hypoxia-induced MGr1-Ag/37LRP expression and contributes to
apoptosis resistance in gastric cancer. Int J Cancer. 127:820–829.
2010.PubMed/NCBI
|
|
16
|
Chen F, Zhuang M, Zhong C, Peng J, Wang X,
Li J, Chen Z and Huang Y: Baicalein reverses hypoxia-induced 5-FU
resistance in gastric cancer AGS cells through suppression of
glycolysis and the PTEN/Akt/HIF-1α signaling pathway. Oncol Rep.
33:457–463. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou W, Fu XQ, Zhang LL, Zhang J, Huang X,
Lu XH, Shen L, Liu BN, Liu J, Luo HS, et al: The
AKT1/NF-kappaB/Notch1/PTEN axis has an important role in
chemoresistance of gastric cancer cells. Cell Death Dis.
4:e8472013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhi X, Tao J, Xiang G, Cao H, Liu Z, Yang
K, Lv C and Ni S: APRIL induces cisplatin resistance in gastric
cancer cells via activation of the NF-κB pathway. Cell Physiol
Biochem. 35:571–585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen PM, Cheng YW, Wu TC, Chen CY and Lee
H: MnSOD overexpression confers cisplatin resistance in lung
adenocarcinoma via the NF-κB/Snail/Bcl-2 pathway. Free Radic Biol
Med. 79:127–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hayashi Y, Wang W, Ninomiya T, Nagano H,
Ohta K and Itoh H: Liver enriched transcription factors and
differentiation of hepatocellular carcinoma. Mol Pathol. 52:19–24.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Lou G, Li C, Wang X, Cederbaum AI,
Gan L and Xie B: HBx inhibits CYP2E1 gene expression via
downregulating HNF4α in human hepatoma cells. PLoS One.
9:e1079132014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Saha SK, Parachoniak CA, Ghanta KS,
Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D,
Cornella H, et al: Mutant IDH inhibits HNF-4α to block hepatocyte
differentiation and promote biliary cancer. Nature. 513:110–114.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schwartz B, Algamas-Dimantov A, Hertz R,
Nataf J, Kerman A, Peri I and Bar-Tana J: Inhibition of colorectal
cancer by targeting hepatocyte nuclear factor-4alpha. Int J Cancer.
124:1081–1089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kojima K, Kishimoto T, Nagai Y, Tanizawa
T, Nakatani Y, Miyazaki M and Ishikura H: The expression of
hepatocyte nuclear factor-4alpha, a developmental regulator of
visceral endoderm, correlates with the intestinal phenotype of
gastric adenocarcinomas. Pathology. 38:548–554. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Supriatna Y, Kishimoto T, Furuya M,
Tochigi N, Ishiguro H, Tosh D and Ishikura H: Expression of
liver-enriched nuclear factors and their isoforms in
alpha-fetoprotein-producing gastric carcinoma cells. Exp Mol
Pathol. 82:316–321. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Algamas-Dimantov A, Yehuda-Shnaidman E,
Peri I and Schwartz B: Epigenetic control of HNF-4α in colon
carcinoma cells affects MUC4 expression and malignancy. Cell Oncol
(Dordr). 36:155–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nikolaidou-Neokosmidou V, Zannis VI and
Kardassis D: Inhibition of hepatocyte nuclear factor 4
transcriptional activity by the nuclear factor kappaB pathway.
Biochem J. 398:439–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roth U, Curth K, Unterman TG and Kietzmann
T: The transcription factors HIF-1 and HNF-4 and the coactivator
p300 are involved in insulin-regulated glucokinase gene expression
via the phosphatidylinositol 3-kinase/protein kinase B pathway. J
Biol Chem. 279:2623–2631. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang W, Tsuchiya T and Yasukochi Y:
Transitional change in interaction between HIF-1 and HNF-4 in
response to hypoxia. J Hum Genet. 44:293–299. 1999. View Article : Google Scholar : PubMed/NCBI
|