Introduction
Tuberous sclerosis complex (TSC) is an autosomal
dominant disease, which typically causes benign tumors to develop
in the brain and other vital organs, including the kidney, skin and
liver (1,2). Global statistics from previous studies,
performed in 2000 and 2011, revealed that 1/6,000-10,000 people are
born with TSC and there are 2,000,000 people with TSC (3,4) TSC is a
genetic disease, with 33% of patients with TSC having inherited the
disease, and 66% of patients with TSC having developed the disease
due to a gene mutation (5). A
definitive diagnosis of TSC often depends on clinical
manifestations and genetic testing, both of which can make
definitively diagnosing the patient challenging. The majority of
previous studies have described individual cases, but family
heredity cases are limited (6).
Identifying and studying cases of familial hereditary TSC may
improve early genetic diagnosis, and thus enable early
intervention, which may increase the quality of life for patients
with TSC. The present case report described different clinical
manifestations and therapeutic prognoses of four patients with TSC
from one family. Written informed consent was obtained from all
patients.
Case report
A 52-year-old female, with a 20-year history of TSC,
presented at the First Affiliated Hospital of Dalian Medical
University (Dalian, China) with abdominal distention, abdominal
pain and anorexia for the past 2 months. The patient, hereafter
known as patient 1, had epilepsy for 50 years with a mean seizure
frequency of 2–3 times/month, and schizophrenia for 12 years with
intelligence degeneration. The patient had been taken antiepileptic
and antipsychotic drugs but the symptoms were not well controlled.
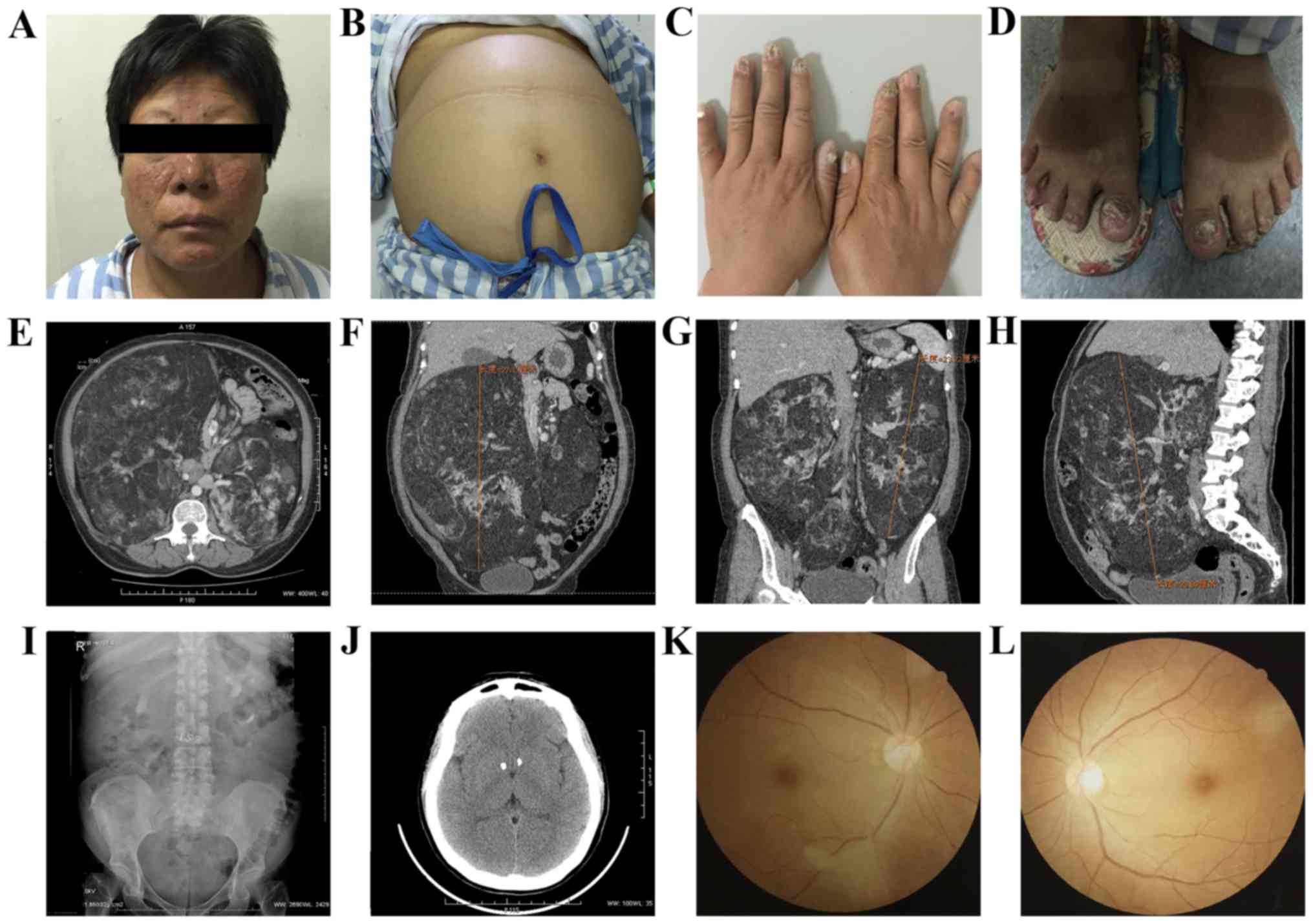
Physical examination (Fig. 1)
revealed facial angiofibromas beside the nasal alar (Fig. 1A), an enlarged abdomen (Fig. 1B), where a large smooth bilateral mass
was observed with a nonclear boundary, and ungual fibromas
(Fig. 1C and D). Biochemical tests
demonstrated that the blood creatinine level was 91 µmol/l, red
blood cell count was 2.98×109 cells/l and hemoglobin
level was 90 g/l. Other lab tests, including the testing of cruor,
liver function and electrolyte, revealed normal results.
Fundus examination results revealed suspicious
retinal pigment spots (Fig. 1K and
L). An abdominal color Doppler ultrasound indicated that there
were intrahepatic calcifications and the abdominal cavity exhibited
hyperechoic light mass shadows on either side, which were
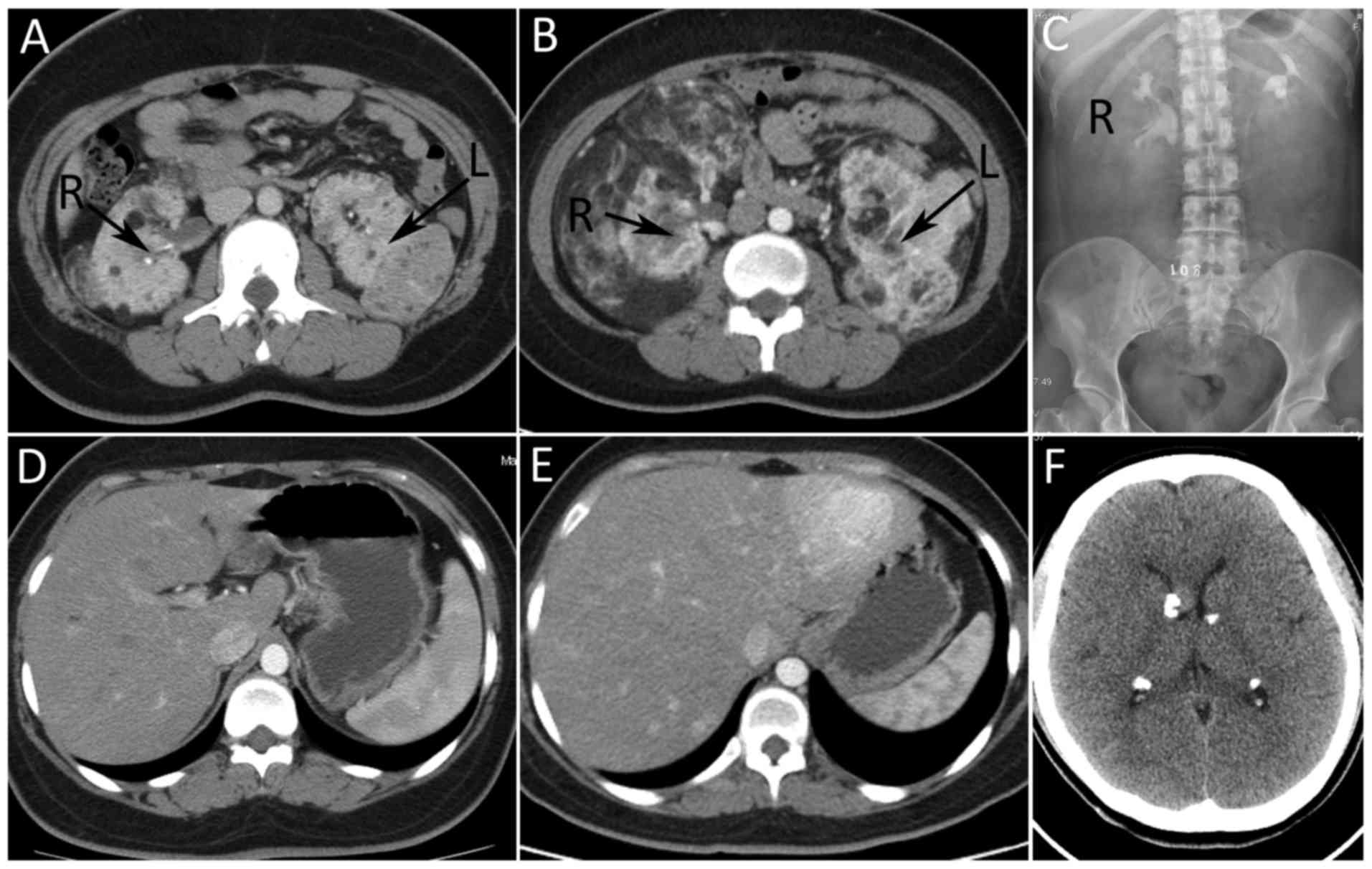
considered renal angiomyolipoma (RAML). Abdominal computed
tomography (CT) (Fig. 1E-H) revealed
that large bilateral mass shadows replaced the normal renal
morphology. The masses were determined to be 28×25×26 and 23×15×11
cm on the right, and left sides, respectively. The CT values were
between-61 and −20HU. Intravenous urography (IVU; Fig. 1I) demonstrated bilateral partial
hydronephrosis and multiple calcification lesions were identified
in the head CT (Fig. 1J). The present
case was diagnosed as TSC, according to its clinical diagnostic
criteria (7). Treatment advice was
administered to the patient where surgery was recommended and
preoperative alternative arterial embolization could be chosen to
decrease the intraoperative bleeding risk. Renal transplantation or
long-term replacement therapy of hemodialysis could be applied
following surgery. Mammalian target of rapamycin (mTOR) inhibitors,
including sirolimus and everolimus, were selected as suitable
treatment options. Furthermore, radiofrequency ablation with a
small dose of mTOR inhibitors was suggested. However, due to the
high risk of the surgery and the high drug costs, the patient
refused surgical, and drug treatments.
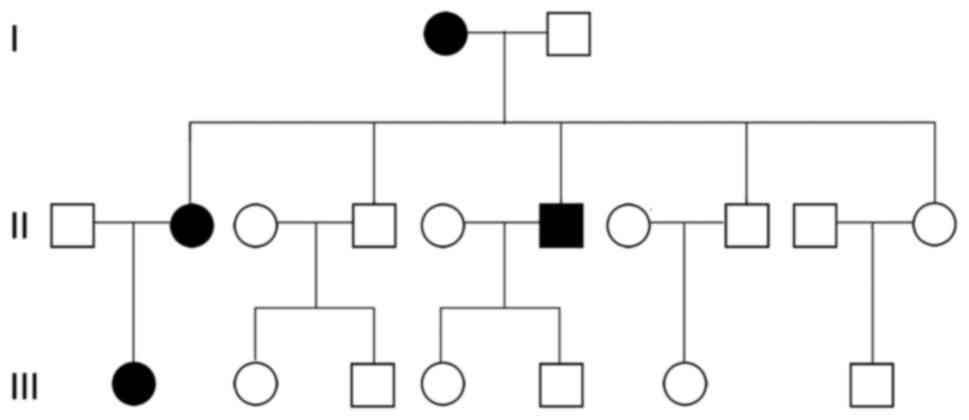
Tracing the disease in the family of the patients
revealed that four family members experienced the disease
successively, which included the patient (patient 1), the patient's
mother (patient 2), the patient's second elder brother (patient 3)
and the patient's daughter (patient 4; Fig. 2). However, the clinical manifestations
were not exactly the same (Table I).
Patient 2 exhibited facial angiofibromas, ungula fibromas and
intelligence degeneration. In addition, bilateral RAML was observed
and two surgeries were performed (left nephrectomy and right
nephron-sparing surgery); however, the patient's mother succumbed
to other diseases.
 | Table I.Common and distinct clinical
manifestations observed in four patients, from the same family,
with tuberous sclerosis complex. |
Table I.
Common and distinct clinical
manifestations observed in four patients, from the same family,
with tuberous sclerosis complex.
| Clinical
manifestation and imaging examination | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|
| Facial
angiofibromas | + | + | + | + |
| Ungual
fibromas | + | + | + | Fingernail -
toenails + |
| Retinal pigment
spots | +/− | − | − | − |
| Shagreen patch | − | − | − | + |
| Intelligent
degeneration | + | + | − | + |
| Seizure
history | + | − | + | − |
| Head CT | Multiple
calcification | Not done | Multiple
calcification | Subependymal giant
cell astrocytoma and calcification |
| Bilateral renal
AML | + | + | + | + |
| Liver CT | Calcification | − | − | Angiofibromas |
Patient 3, 55 years of age at present, was admitted
to the First Affiliated Hospital of Dalian Medical University
(Dalian, China) in June 2002, presenting with a 3-month history of
bilateral waist discomfort. Patient 3 had epilepsy for >12 years
without intelligence degeneration, and did not take medicine
regularly. Physical examination revealed facial angiofibromas and
ungual fibromas. An abdominal CT identified a soft tissue mass in
the left kidney (size, 6×7 cm) and multiple uniform density masses
in the right kidney (size, between 0.5 and 2 cm). Furthermore, a
head CT demonstrated calcification beside the two cerebral
ventricles. Biochemistry test results were all normal, with the
exception of blood creatinine, which was 84 µmol/l. According to
the examination results, a left nephrectomy was performed. The
surgery and recovery were successful; however, image documents were
lost and patient 3 moved to a different city. The patient was
followed up via the telephone and the general condition of the
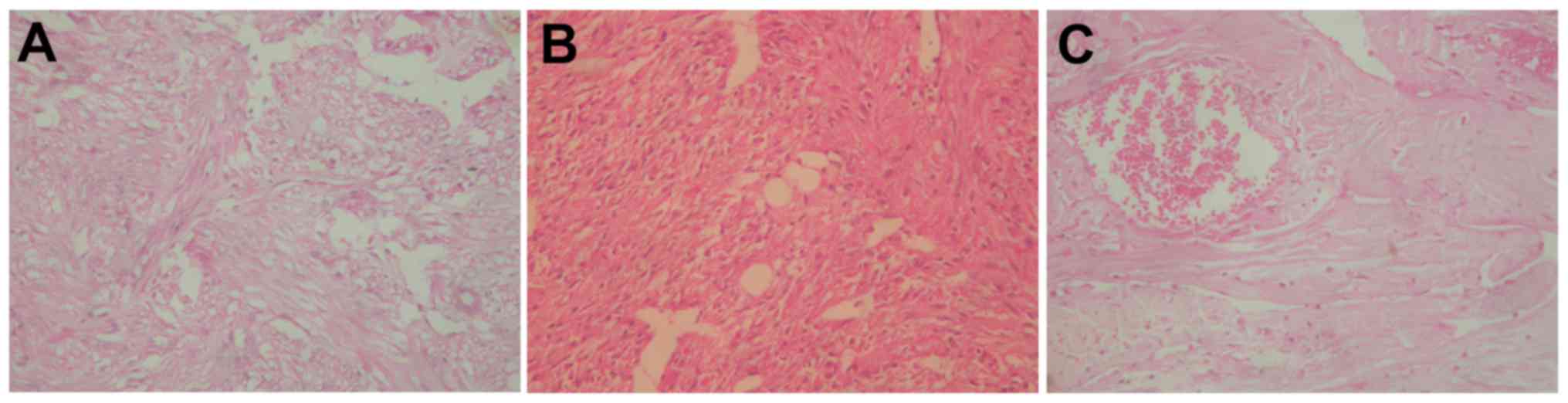
patient improved. Fig. 3 presents a
hematoxylin and eosin stain, which revealed a classical RAML
structure with a majority of smooth muscle and an absence of fat
composition. The procedure for hematoxylin and eosin staining
included dewaxing, with xylene, staining (hematoxylin, 2 min; and
eosin, 3 min; both at room temperature), dehydrating,
transparentizing and mounting. Sections were 5 µm. The procedure
for immunohistochemistry included dewaxing, hydration, antigen
retrieval, primary antibody incubation, secondary antibody
incubation, streptaridin-peroxidase reaction, color development,
redyeing, dehydrating, transparentizing and mounting. Tissues were
paraffin embedded, the fixative used was 10% formalin, at room
temperature for between 30 and 50 min. Sections were placed in an
oven at 60°C for 20 min and subsequently in xylene solution for 10
min two times. Sections were placed in 100% absolute ethanol for 5
min two times, 95% ethanol for 2 min, 80% ethanol for 2 min, 70%
ethanol for 2 min, distilled water for 5 min, and washed with PBS
for 5 mins three times. Following this, sections were blocked with
5% normal goat serum (Shanghai Jianglai Biotech Co., Ltd.,
Shanghai, China) at room temperature for 20 min, incubated with 3%
H2O2 for 10 min at room temperature, to block
the activity of endogenous peroxidase, and washed with PBS (three
times, 5 min each). Sections were subsequently incubated with
primary antibodies overnight at 4°C. Primary antibody details are
listed in Table II. Following this,
the MaxvisionTM Kit (rabbit) was used (cat. no. KIT-5030; Fuzhou
Maixin Biotech Co., Ltd., Fuzhou, China) for secondary antibody
incubation (37°C for 30 min). Subsequently, sections were washed
with PBS five times for 5 min each. Chromogen was detected using
3,3′-diaminobenzidine and sections were stained with haematoxylin
at room temperature for 5 sec, for nuclear protein, and 50 sec, for
endochylema (or envelope protein). A light microscope was used at
magnification, ×100.
| Table II.Primary antibody details. |
Table II.
Primary antibody details.
| Antibody | Cat. no. |
|---|
| Melanoma | MAB-0098 |
| β-actin | Kit-0032 |
| CD34 | Kit-0004 |
| CD68 | MAB-0041 |
| Desmin | Kit-0023 |
| Actin (smooth
muscle) | Kit-0006 |
| Ki-67 antigen | Kit-0005 |
| Vimentin | Kit-0019 |
| CD10/calla | MAB-0338 |
|
Pan-cytokeratin | Kit-0009 |
| EMA | Kit-0011 |
| S-100 Protein | Kit-0007 |
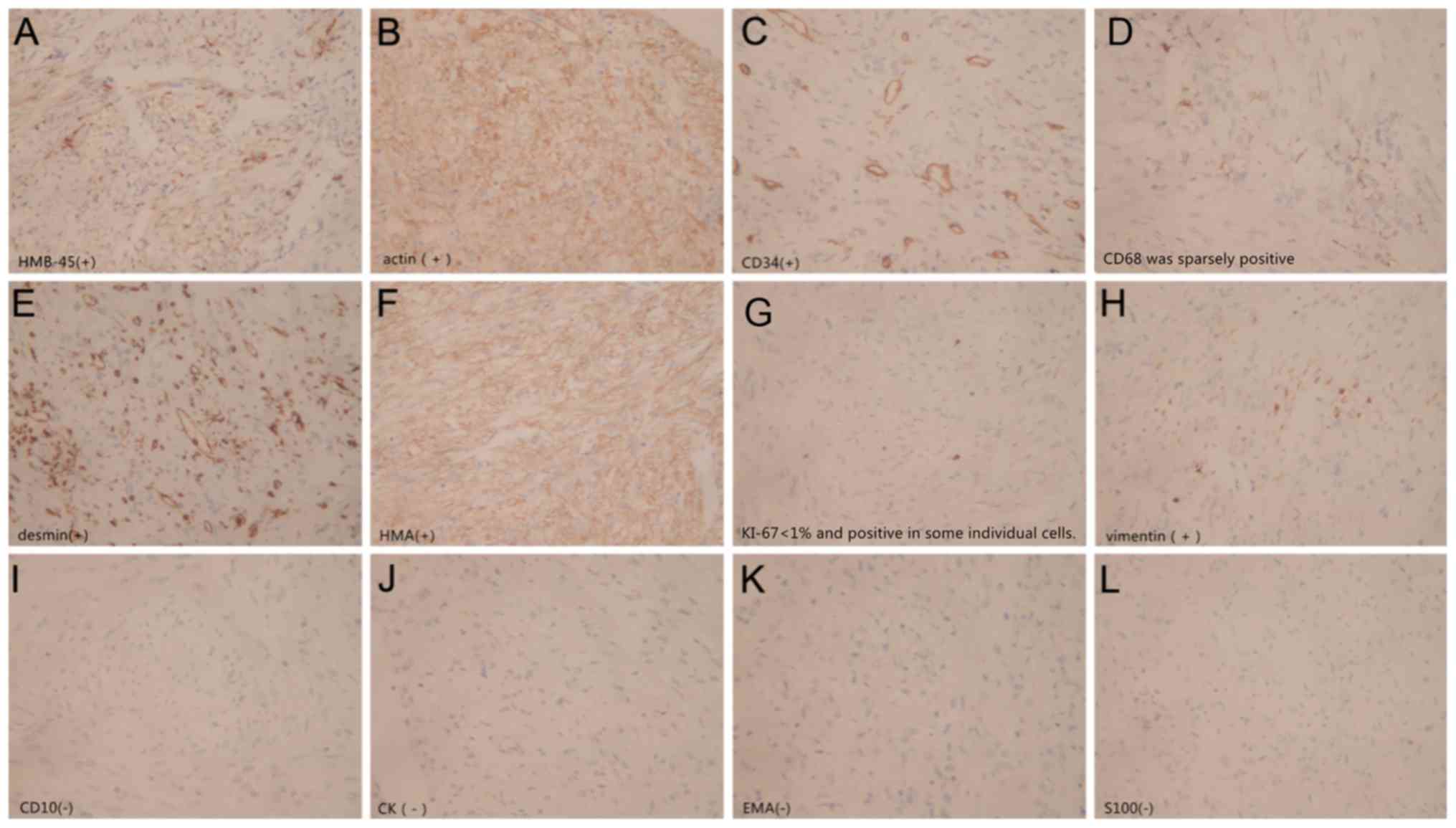
Immunohistochemistry results identified the
following: Homatropinemethylbromide45-positive; 5-(N,
N-Hexamethylene) amiloride (HMA)-positive; actin-positive;
desmin-positive; vimentin-positive; cluster of differentiation
(CD)34-positive; CD68-sparsely positive; S-100 protein-negative;
CD10-negative; epithelial membrane antigen-negative;
cytokeratin-negative; Ki-67 protein <1%-positive (Fig. 4). The pathological diagnosis was
classical RAML.
Patient 4, 24 years of age at present, was diagnosed
with TSC in November 2009. At the latest return visit (June 2015),
intelligence decreased without record of epilepsy. Patient 4
exhibited facial angiofibromas; however, patient 4 also exhibited a
shagreen patch on the back (size, 5×3 cm), which the other members
of this family did not have. Although fibroma was developed under
the toenails of patient 4, the fingernails were observed to be
normal. Biochemical tests revealed the following results, which
were all in the normal range: Red blood cell count,
3.84×109 cells/l; and hemoglobin, 113 g/l. The renal CT,
performed 6 years prior, revealed multiple angiomyolipoma (AML) in
the two kidneys (diameter, between 0.5 and 4 cm), but the renal
morphology was normal. Compared with the previous CT, at present,
the number and the volume of AML increased significantly. The
largest AMLs in the left and right kidneys were determined to be
15×11×8 and 17×10×9 cm, respectively. The CT value was between-80
and 23HU (Fig. 5A and B). The
bilateral renal morphology was vague and the renal parenchyma were
eroded. No lesion was identified in the liver 6 years prior, but
multiple hemangiomas were now observed (Fig. 5D and E). IVU (Fig. 5C) revealed that the right upper
calyceal and the left inferior calyceal were not developed.
Furthermore, a head CT (Fig. 5F)
identified a subependymal giant cell astrocytoma in the septum
pellucidum near the interventricular foramen (diameter, 1.2 cm),
and multiple calcification lesions were observed. A comparison
between head CT scans was not conducted as patient 4 did not have a
head CT performed 6 years prior. A combination of surgical with
medical treatments, including everolimus and sirolimus, was
suggested, and the necessity of treatments was introduced, but the
patient refused therapy.
Discussion
TSC is an autosomal dominant disease, which
typically develops in the kidneys, brain, heart, skin, liver and a
number of other organs, and exhibits individual differences, in
spite of patients exhibiting the same type of genetic mutation
(8). It has been identified that the
genes associated with TSC are TSC1 and TSC2, which are two tumor
suppressor genes (9) TSC1 is located
at region q34 of chromosome 9 and encodes the protein Hamartin, and
TSC2 is located at region p13 of chromosome 16 and encodes the
protein Tuberin (9,10). The Hamartin-Tuberin protein complex
may suppress the transduction of mTOR pathway (11). Once TSC1 and TSC2 are mutated, the
Hamartin-Tuberin protein complex cannot form, thus activating the
mTOR signaling pathway and resulting in benign tumors involving a
number of organs all over the body (12). The lesions typically occur in the
brain, kidney, heart, liver, pulmonary and skin, causing epilepsy,
mental retardation, autism, dysfunctions of liver, lung, and
kidney.
A definite diagnosis of TSC includes clinical
manifestations (Table III)
(7) and genetic testing. The
identification of a mutation (in either TSC1 or TSC2) is sufficient
to diagnose TSC, without clinical manifestation. A pathogenic
mutation is defined as a mutation that inactivates the function of
the TSC1 or TSC2 proteins (out-of-frame indel or non-sense
mutation), prevents protein synthesis (large genomic deletion),.
Other TSC1 or TSC2 variants which exhibit a less certain effect on
protein function do not meet these criteria and are not sufficient
to make a definite diagnosis of TSC. Between 10 and 25% of patients
with TSC exhibit no mutation identified by conventional genetic
testing; therefore, a normal genetic result does not exclude TSC
(7).
| Table III.Clinical diagnostic criteria of
tuberous sclerosis complex. |
Table III.
Clinical diagnostic criteria of
tuberous sclerosis complex.
| A, Major
features |
|---|
|
|---|
| 1. Hypomelanotic
macules (≥3; diameter, ≤5 mm) |
| 2. Angiofibromas
(≥3) or fibrous cephalic plaque |
| 3. Ungual fibromas
(≥2) |
| 4. Shagreen
patch |
| 5. Multiple retinal
hamartomas |
| 6. Cortical
dysplasiasa |
| 7. Subependymal
nodules |
| 8. Subependymal
giant cell astrocytoma |
| 9. Cardiac
rhabdomyoma |
| 10.
Lymphangioleiomyomatosisb |
| 11. Angiomyolipomas
(≥2)b |
|
| B, Minor
features |
|
| 1. ‘Confetti’ skin
lesions |
| 2. Dental enamel
pits (>3) |
| 3. Intraoral
fibromas (≥2) |
| 4. Retinal achromic
patch |
| 5. Multiple renal
cysts |
| 6.
Nonrenalhamartomas |
Genetic testing maybe used for a definitive
diagnosis of a patient with suspected TSC and for early diagnosis
(compared with clinical manifestation), in particular for the early
detection of the offspring of patients, which enables early
intervention. The clinical manifestations often depend on the
changes in the mutational gene. Although there have been a number
of studies on TSC gene detection, this is not typically used for
diagnosis in China due to its expensive costs. Furthermore,
patients in the familial case could not afford genetic testing and
did not wish to undergo it. The patients with TSC discussed in the
present case report are from the same family, but there is no
genetic diagnostic evidence for whether TSC is hereditary. It was
hypothesized that all or some of the patients in this family
developed TSC due to gene mutation.
The development of TSC RAML is slow, but due to the
lack of specific treatment, the prognosis is unfavorable (13). Classical treatments include partial
nephrectomy, selective renal arterial embolization, radiofrequency
and cryotherapy (14). Previous
studies on mTOR inhibitors, including everolimus and sirolimus,
indicate that these effectively decreased tumor size of RAML,
subependymal giant cell astrocytoma and lymphangioleiomyomatosis
(15–17). It has been identified that using the
mTOR inhibitor combined with the classical treatments may aid
controlling the disease and decrease the adverse reactions of drugs
(14,16,18).
It has been demonstrated that the incidence rate of
neurological disease associated with TSC are as follows: Epilepsy,
between 66 and 93%; subependymal nodules, between 90 and 100%;
subependymal giant cell astrocytoma, between 5 and 20%; intelligent
degradation, between 44 and 64%; and infantile spasms, 45%
(19). A previous study identified
that 90% of cases of TSC-associated epilepsy were caused by brain
tumors (20). Patients 1 and 3 had a
history of epilepsy, but head CTs of the two patients revealed only
partial calcification. The head CT of patient 4 suggested
subependymal giant cell astrocytoma but there was no history of
epilepsy; this phenomenon rarely happens and is difficult to
explain, and the traditional anti-epilepsy therapies have limited
effect. It has been revealed that mTOR inhibitors may decrease
seizure frequency, improve learning deficits and reduce the size of
tumor in order to relieve compression of tumor on its surrounding
brain tissue (15).
The incidence of TSC has been identified to exhibit
no significant association with the age or sex of patients
(21) and the majority of patients
develop clinical symptoms after 3 years of age (22). Clinical manifestations are delayed,
whereas a genetic diagnosis may reveal TSC earlier. Therefore,
clinical symptoms are of limited use in the early diagnosis of the
disease. Patient 1 in the present study had suffered from epilepsy
since the age of 2, which is rare. It has been demonstrated that
RAML expresses receptors of estrogen and lutein, and tumors develop
more rapidly in pregnant women (22–24). In
addition, administration of mTOR inhibitors has led to female
adolescent amenorrhoea (25–27). However, Sauter et al (28) considered that the use of drugs
containing estrogenis not associated with renal angiomyolipoma in
tuberous sclerosis. Additional research is required to understand
if there is any association between the two.
A total of 80% of patients with TSC are complicated
with RAML, with females exhibiting an increased incidence of TSC
RAML compared with males (22);
however, the severity of the illness remains unknown. AML is
divided into classical and epithelioid AML. Patients with TSC may
exhibit classical or epithelioid AML, but the incidence of
epithelioid AML is increased compared with that of the classical
type (29). Pathologically,
epithelioid RAML exhibits histological variances with tumor cells
demonstrating destructive and invasive development, and malignant
tendency. Patient 3 exhibited classical AML with no proliferation
of epithelioid cells, necrotized tumor tissue, nuclear division,
atypia or invasive growth under microscopy. TSC-associated renal
manifestations include cysts, angiomyolipoma, epithelioid tumor,
renal cell carcinoma and eosinophilic cell tumor, and may coexist
or exist independently (30–33). The incidence of TSC-associated RAML is
20% of RAML and typically develops bilaterally (34). Imaging techniques enable TSC RAML to
be distinguished from the classical type. A tumor of the former
type develops destructively in multiple centers and typically
involves two sides, whereas the classical type develops separately
and involves only one side. Patients with bilateral giant renal
hamartoma are rare globally. To the best of our knowledge, the
largest bilateral TSC RAML (right side) identified was 32×16×12 cm
in size and had a mass of 7.7 kg (35). In addition, the largest unilateral
renal AML (left side) identified was 39×25×9 cm in size with a mass
of 7.5 kg (36). TSC RAML is
typically accompanied with an aneurysm and with the developmentof
the RAML, aneurysm increases. If a tumor is >4 cm, or the
diameter of aneurysm is >5 mm, the patient exhibits an increased
risk of tumor rupture hemorrhage, which is the primary cause of TSC
RAML-associated mortality (37,38). For
patients with RAML, close follow-ups maybe advised if the tumor is
<4 cm and there are no clinical symptoms. Patients with tumors
>4 cm, aneurysm with diameter is >5 mm or clinical
manifestations (hemorrhage, fever, abdominal pain, hypertension and
hematuria subsequent to RAML) are administered treatment. A
previous study identified that tumors with a diameter >10 cm
were termed ‘giant AMLs’, which are easily ruptured causing
hemorrhage, and require increased attention (39). For patients with TSC RAML that rupture
to form hemorrhages, selective renal arterial embolization may be
the selected treatment. Although there was no obvious hemorrhage in
the giant RAML of the patient in this case, low hemoglobin
suggested possible slow hemorrhage with small vessel rupture.
The patients in the present case report exhibited no
renal function failure and a previous study revealed that renal
failure rarely occurs in TSC (21).
Thus, it was presumed that possibly residual normal renal tissue,
which is difficult to distinguish with the naked eye, serves an
important role in retaining renal function, but it is difficult to
explain how a limited amount of tissue is able to retain normal
renal function. It is hypothesized that genetic mutation causes
normal renal tissue to be replaced by abnormal components, which
exhibit physiological functions similar to normal renal tissue.
However, further study is required.
In symptomatic TSC, clinical intervention is
administered in the majority of cases to decrease the incidence
rate of rupture hemorrhage and preserve renal function. A previous
study identified that there are >1,300 types of TSC1 and TSC2
mutations (40), which may be
associated with the different clinical manifestations between
patients with TSC. The mutation rate of TSC2 in the patients with
TSC is significantly increased and the associated clinical
manifestations are of increased severity compared with that of TSC1
(33). The clinical application of
mTOR inhibitors in the treatment of TSC has been revealed to
achieve a therapeutic effect (41).
mTOR inhibitors may be used to treat patients with TSC that exhibit
subependymal giant cell astrocytoma, lymphangioleiomyomatosis, RAML
and facial angiofiromasin due to the immunosuppressive actions
exhibited on malignant cells (26).
Previous studies have revealed that regular medication for 1 year
significantly decreased the volume of RAML (42,43). In
larger tumors, the effect was more significant; however, renal
tissue damaged by tumors may not always be recovered, in addition,
there is a chance of tumor relapse following drug withdrawal
(44) and the rate of AML rupture
hemorrhage may not decrease.
The adverse reactions of mTOR inhibitors include
oral cavity ulcers, stomatitis, convulsion, an acne-like rash,
joint pain, fever, respiratory tract infection and skin lesions,
and the severity of adverse reactions depends on the dose of the
drug (16,39). A previous study identified that the
side-effects of drug treatment alone are strong, but the
combination of classical treatment and a small dose of mTOR
inhibitors may achieve improved results (27). In addition, in cases of RAML where
surgery may be required, the administration of an mTOR inhibitor
prior to surgery may decrease the tumor volume and the difficulty
of surgery (16). Small dose
maintenance therapy subsequent to surgery may exhibit improve the
maintenance of the surgery result, control disease progression and
decrease tumor recurrence (16,45).
Percutaneous cryoablation therapy is typically used in the
treatment of renal cell carcinoma, but is rarely used in RAML. To
the best of our knowledge, only one study has demonstrated that
percutaneous cryoablation may be combined with a small dose of mTOR
inhibitors to treat TSC RAML to achieve a curative effect and a
decrease in adverse drug reactions (14).
The bilateral renal morphology of patient 4 was
acceptable, the RAML progressed rapidly 6 years subsequently with
the development of tumors in multiple centers and almost all the
normal kidney morphology was absent. If mTOR inhibitors had been
administered in the early stage, prior to the alterations of the
kidney morphology, normal renal tissue may have been preserved, the
development of a subependymal astrocytoma in the brain may have
been prevented and the facial angiofibroma may have been treated.
Therefore, mTOR inhibitors may have decreased the development of
the disease and improved the quality of life for the patient. A
previous study has indicated that early application of the drug
treatment, particularly in infants and young children, may prevent
tumor development, improve the symptoms of epilepsy and other
TSC-associated symptoms (15). All
patients in the present case report were not administered mTOR
inhibitors due to the increased cost associated with this
treatment, which health insurers in China do not cover. Therefore,
the present case report lacks information on the clinical
application of this class of drug, and there are a limited number
of studies on the association between estrogen, mTOR inhibitors and
disease.
mTOR inhibitors may be used in the treatment of
TSC-associated diseases, but cannot prevent the disease being
genetically passed on to the children of patients. Therefore, it is
recommended that patients with TSC have a fetal gene detection
performed during pregnancy. For the treatment of patients with TSC,
early application of drug treatment is advised as this may prevent
the development of tumors, epilepsy and other diseases associated
with TSC. In addition, regular re-examination is important because
the progression speed of TSC is distinct, depending on the
individual patient. Tumors in certain patients with TSC have been
revealed to develop dominantly in a short term, but there have
additionally been patients with TSC that experience long-term
survival with a tumor (15). Patients
with TSC with rapidly developing tumors may be administered mTOR
inhibitor therapy combined with surgery, arterial embolization,
radiofrequency ablation or cryoablation. Novel combination
treatment methods include small dose mTOR inhibitor with partial
renal nephrectomy and small dose of mTOR inhibitor maintenance
treatment, or arterial embolization with partial renal nephrectomy
and mTOR inhibitor, or mTOR inhibitor with cyroablation, all of
which may exhibit therapeutic prospects in the future. Additional
research is required. The present case report described rare cases
in order to provide more appropriate diagnostic and therapeutic
methods for patients with TSC. These include fetal gene detection
during early pregnancy, early application of drugs and the
combination of multiple treatments.
References
|
1
|
Zhang Y, Gan J, Pu Z, Xu Mm, Wang Lf, Li
Yh and Liu Zg: TSC1 R509X Mutation in a chinese family with
tuberous sclerosis complex. Neuromolecular Med. 17:202–208. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cabrera-López C, Bullich G, Marti T,
Català V, Ballarín J, Bissler JJ, Harris PC, Ars E and Torra R:
Insight into response to mTOR inhibition when PKD1 and TSC2 are
mutated. BMC Med Genet. 16:392015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hallett L, Foster T, Liu Z, Blieden M and
Valentim J: Burden of disease and unmet needs in tuberous sclerosis
complex with neurological manifestations: Systematic review. Curr
Med Res Opin. 27:1571–1583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hyman MH and Whittemore VH: National
Institutes of Health consensus conference: Tuberous sclerosis
complex. Arch Neurol. 57:662–665. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rabito MJ and Kaye AD: Tuberous sclerosis
complex: Perioperative considerations. Ochsner J. 14:229–239.
2014.PubMed/NCBI
|
|
6
|
Wang GX, Wang DW, Yi CY, Qu JS and Wang
YL: Mutational analyses of the TSC1 and TSC2 genes in cases of
tuberous sclerosis complex in Chinese Han children. Genet Mol Res.
12:1168–1175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Northrup H and Krueger DA: International
Tuberous Sclerosis Complex Consensus Group: Tuberous sclerosis
complex diagnostic criteria update: Recommendations of the 2012
iinternational tuberous sclerosis complex consensus conference.
Pediatr Neurol. 49:243–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sancak O, Nellist M, Goedbloed M,
Elfferich P, Wouters C, Maat-Kievit A, Zonnenberg B, Verhoef S,
Halley D and van den Ouweland A: Mutational analysis of the TSC1
and TSC2 genes in a diagnostic setting: Genotype-phenotype
correlations and comparison of diagnostic DNA techniques in
tuberous sclerosis complex. Eur J Hum Genet. 13:731–741. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Slegtenhorst M, de Hoogt R, Hermans C,
Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A,
Halley D, Young J, et al: Identification of the tuberous sclerosis
gene TSC1 on chromosome 9q34. Science. 277:805–808. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Habib SL: Tuberous sclerosis complex and
DNA repair. Adv Exp Med Biol. 685:84–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Inoki K, Corradetti MN and Guan K L:
Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet.
37:19–24. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Henske EP, Rasooly R, Siroky B and Bissler
J: Tuberous sclerosis complex, mTOR, and the kidney: Report of an
NIDDK-sponsored workshop. Am J Physiol Renal Physiol.
306:F279–F283. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yi L, Guo F and Zhang YB: Diagnosis and
analysis of cases of tuberous sclerosis. J Clin Exp Med.
10:819–822. 2015.(In Chinese).
|
|
14
|
Krummel T, Garnon J, Lang H, Gangi A and
Hannedouche T: Percutaneous cryoablation for tuberous
sclerosis-associated renal angiomyolipoma with neoadjuvant mTOR
inhibition. BMC Urol. 14:772014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Curatolo P and Moavero R: mTOR inhibitors
in tuberous sclerosis complex. Curr Neuropharmacol. 10:404–415.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Staehler M, Sauter M, Helck A, Linsenmaier
U, Weber L, Mayer K and Fischereder M: Nephron-sparing resection of
angiomyolipoma after sirolimus pretreatment in patients with
tuberous sclerosis. Int Urol Nephrol. 44:1657–1661. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sasongko TH, Ismail NF and Zabidi-Hussin
Z: Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane
Database Syst Rev. 7:CD0112722016.PubMed/NCBI
|
|
18
|
Peces R, Cuestalópez E, Peces C and Selgas
R: Giant bilateral renal angiomyolipomas and
lymphangioleiomyomatosis presenting after two successive
pregnancies successfully treated with surgery and rapamycin.
ScientificWorldJournal. 11:2115–2123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hallett L, Foster T, Liu Z, Blieden M and
Valentim J: Burden of disease and unmet needs in tuberous sclerosis
complex with neurological manifestations: Systematic review. Curr
Med Res Opin. 27:1571–1583. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Crino PB, Nathanson KL and Henske EP: The
tuberous sclerosis complex. N Engl J Med. 355:1345–1356. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
O'Callaghan FJ, Noakes MJ, Martyn CN and
Osborne JP: An epidemiological study of renal pathology in tuberous
sclerosis complex. BJU Int. 94:853–857. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mascarenhas R and McLaughlin P:
Haemorrhage from angiomyolipoma of kidney during pregnancy-a
diagnostic dilemma. Ir Med J. 94:83–84. 2001.PubMed/NCBI
|
|
23
|
Raft J, Lalot JM, Meistelman C and
Longrois D: Influence of pregnancy on renal angiomyolipoma. Gynecol
Obstet Fertil. 33:898–906. 2005.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boorjian SA, Sheinin Y, Crispen PL, Lohse
CM, Kwon ED and Leibovich BC: Hormone receptor expression in renal
angiomyolipoma: Clinicopathologic correlation. Urology. 72:927–932.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bissler JJ, Kingswood JC, Radzikowska E,
Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N,
Brakemeier S, de Vries PJ, et al: Everolimus for angiomyolipoma
associated with tuberous sclerosis complex or sporadic
lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised,
double-blind, placebo-controlled trial. Lancet. 381:817–824. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Franz DN: Everolimus in the treatment of
subependymal giant cell astrocytomas, angiomyolipomas, and
pulmonary and skin lesions associated with tuberous sclerosis
complex. Biologics. 7:211–221. 2013.PubMed/NCBI
|
|
27
|
Roa J, Garcia-Galiano D, Castellano JM,
Gaytan F, Pinilla L and Tena-Sempere M: Metabolic control of
puberty onset: New players, new mechanisms. Mol Cell Endocrinol.
324:87–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sauter M, Sigl J, Schotten KJ, Kreuzer L,
Günthner-Biller M and Fischereder M: Use of oestrogen-containing
medication is not associated with renal angiomyolipoma in tuberous
sclerosis: Findings from a survey. Int Urol Nephrol. 47:707–708.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang KH, Huang CY, Chung SD, Pu YS, Shun
CT and Chen J: Malignant epithelioid angiomyolipoma of the kidney.
J Formos Med Assoc. 106 2 Suppl:S51–S54. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kavaney PB and Fielding I: Angiomyolipoma
and renal cell carcinoma in same kidney. Urology. 6:643–646. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Corsenca A, Aebersold F, Moch H, Bird P,
Weber M, Hofbauer G, Wüthrich RP and Serra AL: Combined nephrectomy
and pre-emptive renal transplantation in a tuberous sclerosis
patient with angiomyolipoma, renal carcinoma and life-threatening
abdominal haemorrhages. Nephrol Dial Transplant. 22:3330–3333.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eble JN: Angiomyolipoma of kidney. Semin
Diagn Pathol. 15:21–40. 1998.PubMed/NCBI
|
|
34
|
Cichocki M, Sosnowski M and Jablonowski Z:
A giant renal angiomyolipoma (AML) in a patient with septo-optic
dysplasia (SOD). Eur J Med Res. 19:462014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hussain M, Mubarak M, Sultan G, Sultan G,
Ahmed E, Yunus M, Salehi MA, Asif M, Naqvi Anwer SA and Rizvi SA:
Renal transplant in a tuberous sclerosis patient with bilateral
giant renal angiomyolipomas and concurrent renal carcinoma. Saudi J
Kidney Dis Transpl. 24:318–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Taneja R and Singh DV: Giant renal
angiomyolipoma: Unusual cause of huge abdominal mass. J Clin
Imaging Sci. 3:562013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miyoshi Y, Iwao K, Nawa G, Yoshikawa H,
Ochi T and Nakamura Y: Frequent mutations in the beta-catenin gene
in desmoid tumors from patients without familial adenomatous
polyposis. Oncol Res. 10:591–594. 1998.PubMed/NCBI
|
|
38
|
Yamakado K, Tanaka N, Nakagawa T,
Kobayashi S, Yanagawa M and Takeda K: Renal angiomyolipoma:
Relationships between tumor size, aneurysm formation, and rupture.
Radiology. 225:78–82. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mitra A, Jain Kumar S, Gupta D and Chandra
Murthy Kaza R: Giant angiomyolipoma of the kidney presenting as
anaemia-a rare presentation. Open J Urol. 2:75–77. 2012. View Article : Google Scholar
|
|
40
|
Curatolo P, Bombardieri R and Jozwiak S:
Tuberous sclerosis. Lancet. 372:657–668. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang G, Yang L, Yang X, Shi X, Wang J, Liu
Y, Ju J and Zou L: Efficacy and safety of a mammalian target of
rapamycin inhibitor in pediatric patients with tuberous sclerosis
complex: A systematic review and meta-analysis. Exp Ther Med.
9:626–630. 2015.PubMed/NCBI
|
|
42
|
Bissler JJ, McCormack FX, Young LR, Elwing
JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J,
et al: Sirolimus for angiomyolipoma in tuberous sclerosis complex
or lymphangioleiomyomatosis. N Engl J Med. 358:140–151. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cabrera-López C, Martí T, Catalá V, Torres
F, Mateu S, Ballarín J and Torra R: Assessing the effectiveness of
rapamycin on angiomyolipoma in tuberous sclerosis: A two years
trial. Orphanet J Rare Dis. 7:872012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Peng ZF, Yang L, Wang TT, Han P, Liu ZH
and Wei Q: Efficacy and safety of sirolimus for renal
angiomyolipoma in patients with tuberous sclerosis complex or
sporadic lymphangioleiomyomatosis: A systematic review. J Urol.
192:1424–1430. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Paul E and Thiele E: Efficacy of sirolimus
in treating tuberous sclerosis and lymphangioleiomyomatosis. N Engl
J Med. 358:190–192. 2008. View Article : Google Scholar : PubMed/NCBI
|