Introduction
Pancreatic cancer is a disease with a high mortality
rate that is characterized by the early metastasis to local and
distant organs; the majority of patients present with unresectable
disease at the time of initial diagnosis (1). Therefore, patients with pancreatic
cancer have a poor prognosis, with an overall 5-year survival rate
of <5% in the United States between 1975 and 2008 (2). No significant advances in the treatment
of pancreatic cancer have been made in >10 years, largely due to
of the resistance of the disease to conventional chemotherapy and
radiation therapy (3–6). The involvement of cancer stem cells
(CSC; also termed tumor-initiating cells) in the development of
chemotherapy resistance has been reported in a number of types of
malignancy (7–9). CSCs are a phenotypically distinct
population of cells that are functionally defined by their ability
to form tumors, self-renew and differentiate, as well as their
resistance to chemotherapy (10,11).
Therefore, the development of novel chemotherapeutic agents that
are effective against CSCs is urgently required.
Thalidomide is a non-barbiturate sedative and
hypnotic drug with anti-angiogenic and immunomodulatory properties
(12). Thalidomide is currently used
in the treatment of various types of malignant tumor, including
prostate cancer (13), glioblastoma
(14), glioma (15), renal cell carcinoma (16) and advanced breast cancer (17). The mechanism of action of thalidomide
includes the inhibition of vascular endothelial growth factor,
basic fibroblast growth factor and tumor necrosis factor-α, the
inhibition of angiogenesis, and the stimulation of natural killer
cells (12). However, to the best of
our knowledge, the use of thalidomide in the treatment of
pancreatic cancer has not been assessed and the potential mechanism
of thalidomide-mediated inhibition of tumor cell viability remains
to be elucidated. In the present study, the effect of thalidomide
on pancreatic cancer cells was examined, alongside its mechanism of
action.
Materials and methods
Cell lines and reagents
The human pancreatic cancer SW1990, Capan-2, Panc-1,
Patu8988 and Aspc-1 cell lines were purchased from the American
Type Culture Collection (Manassas, VA, USA) and maintained in
high-glucose Dulbecco's modified Eagle medium (DMEM) supplemented
with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and 1% penicillin-streptomycin (Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a humidified incubator
with 5% CO2. Thalidomide was obtained from Selleck
Chemicals (Houston, TX, USA). The MTT reagent and anti-cluster of
differentiation 133 (CD133) antibody for western blotting were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany; cat.
no. SAB2107606). The phycoerythrin-conjugated CD133 antibody for
flow cytometry was purchased from MiltenyiBiotec, Inc., (Auburn,
CA, USA; cat. no. 130-080-801). Transwell filter inserts were
obtained from Corning (Corning In corporated, Corning, NY, USA).
Rabbit anti-E-cadherin (cat. no. sc-7870), rabbit anti-N-cadherin
(cat. no. sc-7939), and mouse anti-β-actin (cat. no. sc-47778) were
purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX, USA).
Thalidomide was dissolved in dimethyl sulfoxide (DMSO) to a storage
concentration of 50 mmol/l. An equal amount (0.1% v/v) of DMSO was
present in all the treatment groups, including the control.
Cell proliferation assay
Cell viability was measured using the MTT assay,
according to the manufacturer's protocol. Cells were seeded at a
density of 5×104 cells/well in 96-well flat-bottom
microtiter plates. Cells were treated with different concentrations
(0, 6.25, 12.5, 25, 50 or 100 µmol/l) of thalidomide in 200 µl
complete medium for 24, 48 or 72 h at 37°C in an incubator. Cells
were then incubated with MTT (0.25 mg/ml) for 2–4 h at 37°C. The
formazan crystals in the cells were solubilized with a solution
containing 50% dimethylformamide and 20% SDS (pH 4.7). Finally, the
level of MTT/formazan was determined by measuring absorbance at a
wavelength of 570 nm using a microplate reader (SPECTRA; Tecan
Group, Ltd., Mannedorf, Switzerland).
Cell migration assays
SW1990 and Capan-2 cells were plated in 6-well
plates with various concentrations of thalidomide (0, 50, 100
µmol/l). At 48 h, the cells were harvested, washed once in PBS and
resuspended in serum-free high-glucose DMEM. A 200-µl aliquot of
cell suspension was added to Transwell filter inserts, resulting in
a density of 2×104 cells per insert. High-glucose DMEM
containing 10% FBS was added to the lower wells. Migration was
allowed to proceed for 48 h at 37°C. Cells that did not migrate
through the filters were removed using cotton swabs, and cells that
migrated through the inserts were fixed and stained with 1% crystal
violetat room temperature. The number of migrating cells was
observed with phase contrast microscopy (magnification, ×200) and
images were captured.
Flow cytometry
SW1990 and Capan-2 cells were plated in 6-well
plates for 48 h with various concentrations of thalidomide (0, 50,
100 µmol/l), then were harvested, washed twice in cold PBS and
incubated with a phycoerythrin-conjugated CD133 antibody (1:10) for
30 min at 4°C. Mouse IgG1-phycoerythrin (1:10; cat. no. IC002P;
R&D Systems, Inc.) was used as an isotype control antibody.
Non-viable cells were eliminated with 7-aminoactinomycin D (Nanjing
KeyGen Biotech Co., Ltd., Nanjing, China; cat. no. KGA219)
staining. The labeled cells were analyzed by a BD FASCCanto II flow
cytometer 338960 (BD Biosciences, Franklin Lakes, NJ, USA) in
accordance with the manufacturer's protocol and data was analyzed
using FlowJo v10 software (Tree Star, Inc., Ashland, OR, USA).
Gating was implemented on the basis of negative-control staining
profiles.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
A density of 10×104 SW1990 and Capan-2
cells were respectively treated with 100 µmol/l thalidomide for 24
h and harvested, and total RNA was extracted using TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and subjected to reverse transcription
using a PrimeScript RT reagent kit (Takara Bio, Inc., Otsu, Japan).
The mRNA expression was detected by qPCR with an ABI
PRISM® 7900HT Sequence Detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using SYBR-Green Dye
(Takara Bio, Inc.). Primers used for qPCR are listed in Table I. cDNA was amplified with 40 PCR
cycles (0.5 sec at 95°C; 10 sec at 60°C; 10 sec at 72°C). The
relative expression levels were calculated using the
2−ΔΔCq method (18). All
experiments were repeated at ≥3 times.
 | Table I.Polymerase chain reaction primer
sequences. |
Table I.
Polymerase chain reaction primer
sequences.
| Gene | Primer sequence,
5′-3′ |
|---|
| CD133 |
|
|
Forward |
TTCTTGACCGACTGAGACCCA |
|
Reverse |
TCATGTTCTCCAACGCCTCTT |
| E-cadherin |
|
|
Forward |
TCGACACCCGATTCAAAGTGG |
|
Reverse |
GTGGGTTATGAAACCGTAGAGG |
| N-cadherin |
|
|
Forward |
AGCCAACCTTAACTGAGGAGT |
|
Reverse |
GGCAAGTTGATTGGAGGGATG |
| β-actin |
|
|
Forward |
CTGGAACGGTGAAGGTGACA |
|
Reverse |
AAGGGACTTCCTGTAACAATGCA |
Western blotting
SW1990 and Capan-2 cells were treated with 100
µmol/l thalidomide for 24 h and rinsed twice in PBS. The cells were
harvestedand centrifuged at 2,000 × g for 5 min at 4°C, then lysed
for 2 h in radioimmunoprecipitation analysis lysis buffer (Beyotime
Institute of Biotechnology, Haimen, China; cat. no. P0013B) on ice,
and centrifuged at 12,000 × g for 10 min at 4°C. The protein
concentration was determined by using a bicinchoninic acid protein
assay (BCA™ protein assay kit; Pierce; Thermo Fisher
Scientific, Inc.). Aliquots of 40 µg protein were separated by 6%
SDS-PAGE and transferred onto polyvinylidene fluoride membranes.
Non-specific binding was blocked with 5% low-fat milk at room
temperature for 1 h in a covered container. The membranes were
incubated with the relevant primary antibodies overnight at 4°C
(dilutions: E-cadherin, 1:200; N-cadherin, 1:400, CD133, 1:800;
β-actin, 1:1,000). The membranes were washed in PBS with 0.1%
Tween-20 and incubated with the appropriate HRP-conjugated
secondary antibodies (goat anti-rabbit IgG-HRP; 1:2,000; cat. no.
sc-2004; goat anti-mouse IgG-HRP; 1:2,000; cat. no. sc-2005; Santa
Cruz Biotechnology, Inc.) for 1 h at 37°C. The blots were developed
using an enhanced chemiluminescence (ECL)-detection system (Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), dried, and exposed to
ECL film. All experiments were repeated ≥3 times, with similar
results.
Statistical analysis
SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA)
was used for statistical analysis. Data were obtained from three
independent experiments and expressed as the mean ± standard
deviation. The statistical analyses performed included
χ2 test and one-way analysis of variance (ANOVA), which
was followed by Student-Newman-Keuls (SNK) as a post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Thalidomide inhibits the proliferation
of pancreatic cancer cells
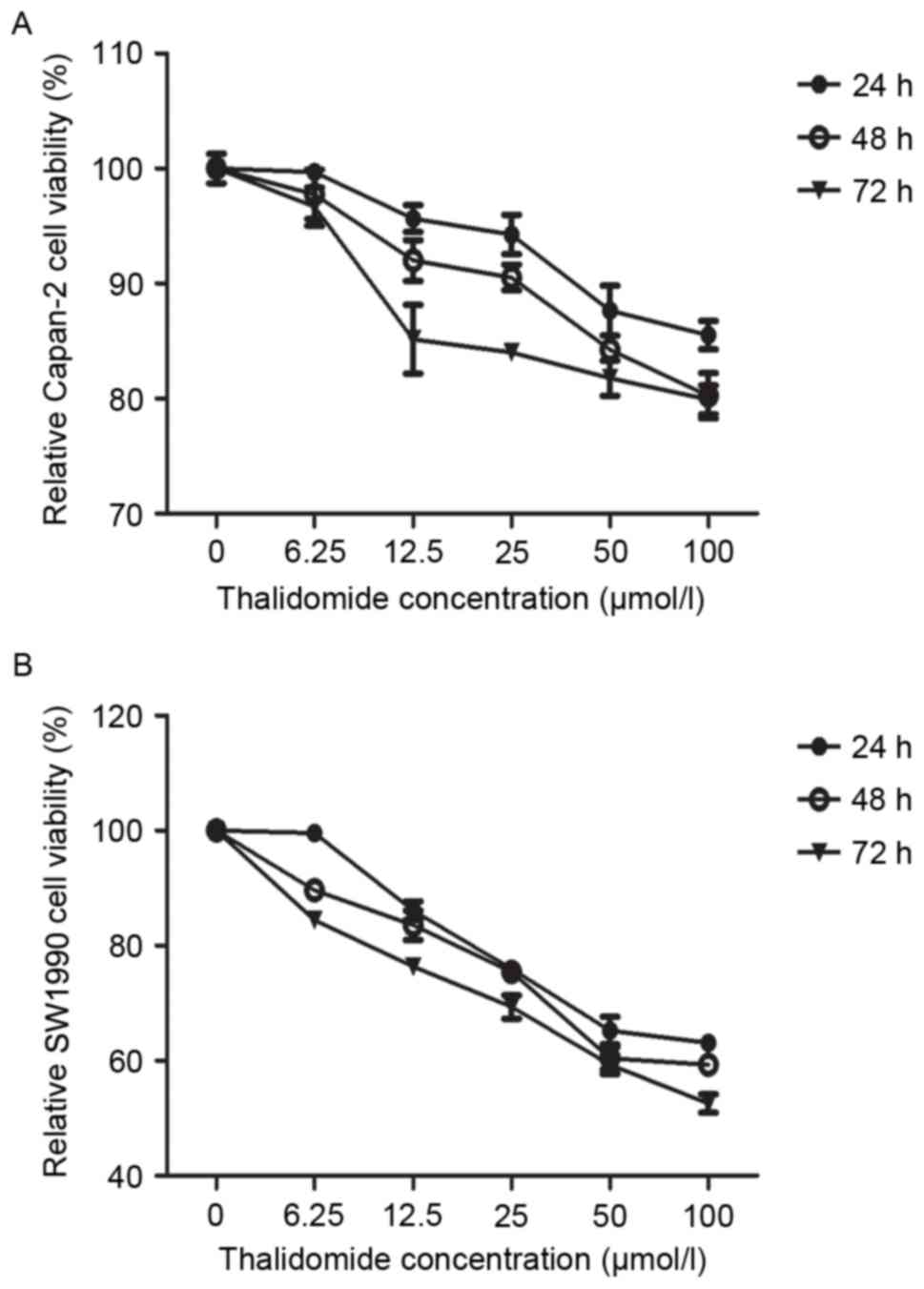
The inhibitory effect of thalidomide on the growth
of human pancreatic cancer cell lines was determined by an MTT
assay. In Capan-2 cells treated with thalidomide, a dose-dependent
decrease in cell number was observed, with a 4–14, 7–19 and
14.8–20% inhibition in cell growth, respectively (Fig. 1A). Thalidomide caused acomparatively
higher dose- and time-dependent inhibition of cell growth in SW1990
cells, with 13–36, 16–40, and 23–47% of growth at 24, 48 and 72 h
at 6.25–100 µmol/l, respectively (Fig.
1B). These data indicated that the mechanism for growth
inhibition by thalidomide may differ between the two cell
lines.
The proliferation of three other pancreatic cancer
cell lines (Panc-1, Patu8988 and Aspc-1) was assessed in
preliminary experiments using the MTT assay (data not shown).
Results obtained from this analysis demonstrated that the
proliferation of these three pancreatic cancer cell lines did not
differ significantly following administration of thalidomide,
whereas the proliferation of the Capan-2 and SW1990 cell lines were
inhibited following treatment. Therefore, Capan-2 and SW1990 cells
were selected for the present study.
Effects of thalidomide on migration in
pancreatic cancer cells
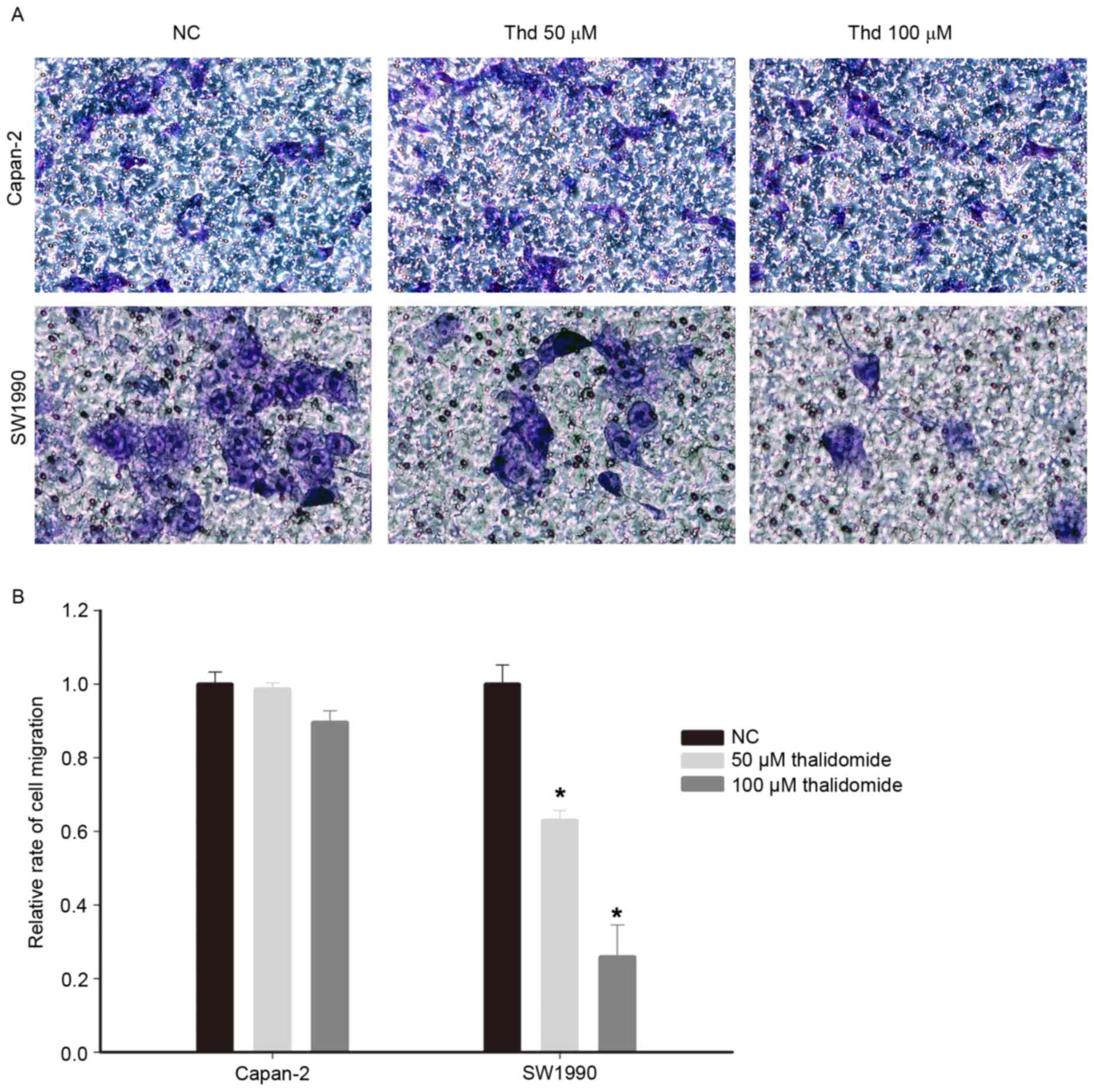
To examine the effect of thalidomide on the motility
of pancreatic cancer cells, a cell migration assay was conducted
with Transwell filter inserts. The motility of SW1990 cells treated
with thalidomide for 48 h was reduced compared withthe control
cells (Fig. 2A and B; P<0.05).
However, the Capan-2 cells exposed to thalidomide treatment
exhibited no significant decrease in motility (Fig. 2A and B), which was consistent with the
pattern of growth inhibition in the two cell lines.
Thalidomide reduces the proportion of
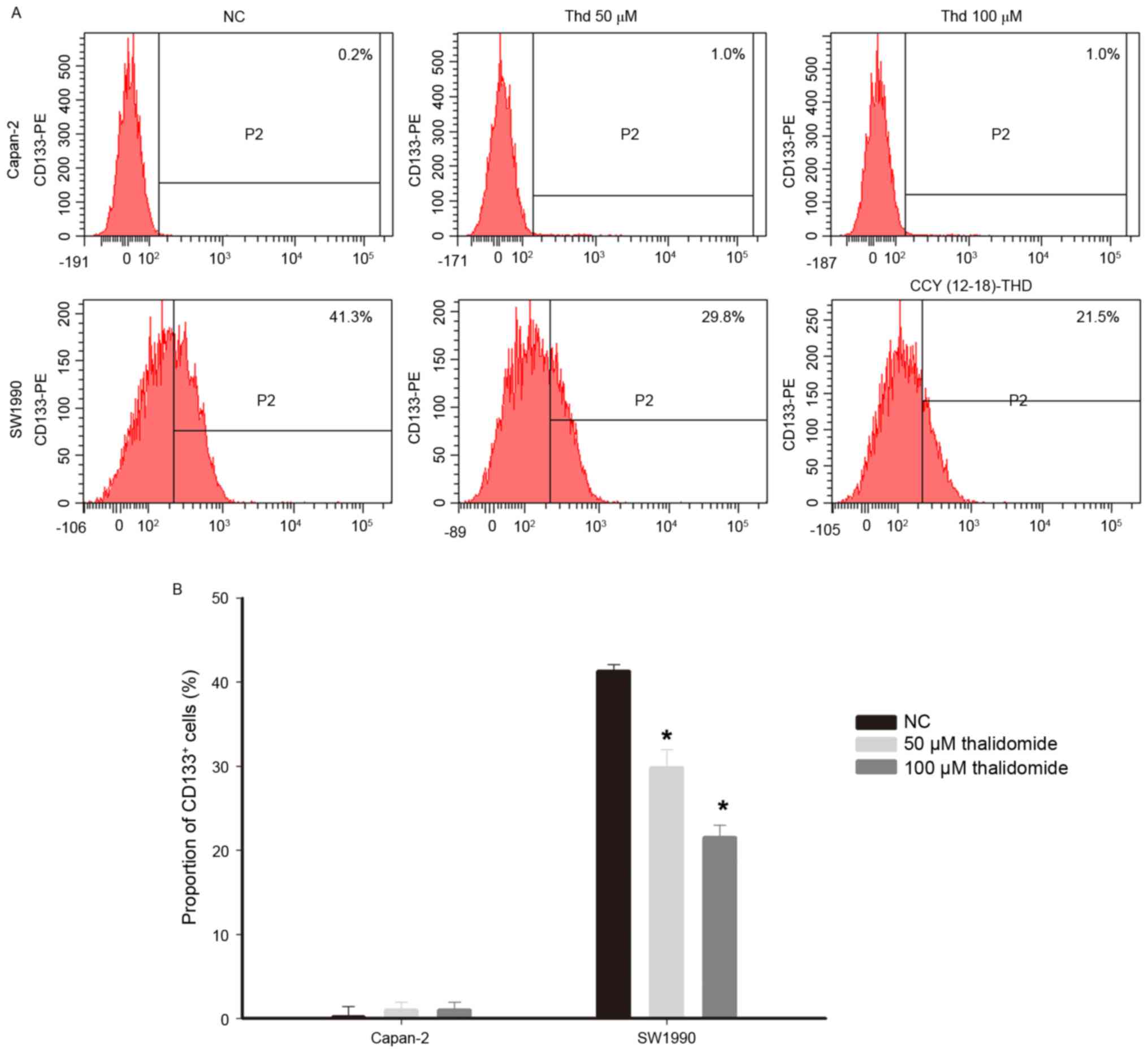
CD133+ cell subpopulations in vitro
CD133 is a cell surface marker of CSCs in pancreatic
cancer (19,20). The effect of thalidomide on the
proportion of pancreatic cancer cells with the CD133+
antigenic phenotype was analyzed by flow cytometry. The proportion
of CD133+ cells in the Capan-2 and SW1990 lines was
different, with <1% of cells exhibiting CD133-positivity in the
Capan-2 cell line compared with >40% in SW1990. This indicated
that the SW1990 line has a higher proportion of CSCs than the
Capan-2 line. The proportion of CD133+ cells in the
Capan-2 cell line were not significantly altered in response to
thalidomide treatment (negative control, 0.2% vs. thalidomide,
1.0%), whereas thalidomide treatment significantly decreased the
proportion of CD133+ cells in the SW1990 cell line
(negative control, 41.3% vs. thalidomide 50 µmol/l, 29.8% and 100
µmol/l, 21.5%; Fig. 3;
P<0.05).
Thalidomide inhibits EMT in pancreatic
cancer cells
The dysregulation of the EMT program contributes to
tumor initiation, invasion and metastatic spread, and is associated
with an increase in tumor cell stemness (21). The teratogenic effect of thalidomide
is well-documented; EMT governs morphogenesis and is activated
during embryogenesis (22).
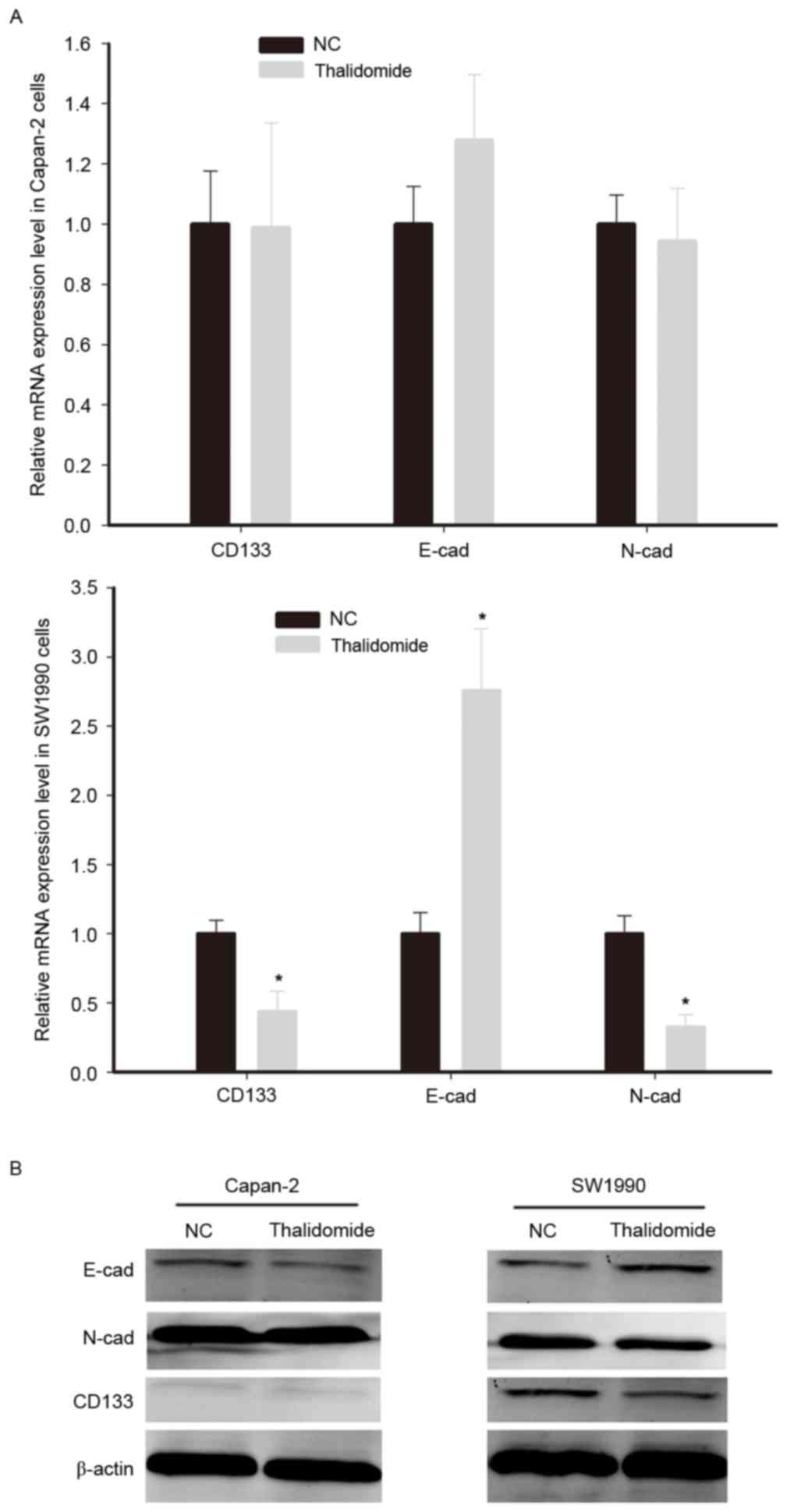
Therefore, the EMT status was assessed in cells exposed to
thalidomide. The primary molecular feature of cancer cells
undergoing EMT is the upregulation of characteristic mesenchymal
genes, including N-cadherin and E-cadherin (23). On the basis of the previous results
that thalidomide could reduce the proportion of CD133+
cells, the expression of N-cadherin, E-cadherin and CD133 were
detected by RT-qPCR and western blotting.
The results of western blotting indicated that the
protein expression of CD133 in Capan-2 cells was markedly lower
than in SW1990 cells. Thalidomide treatment downregulated CD133 and
N-cadherin, and upregulated E-cadherin mRNA and protein expression
in SW1990 cells (Fig. 4). However,
thalidomide did not significantly affect the expression of
E-cadherin, N-cadherin and CD133 mRNA in Capan-2 cells. Therefore,
it was concluded that thalidomide inhibited the EMT program in
CD133+ cells, whereas it exhibited no significant effect
in cells with low CD133 expression.
Discussion
Pancreatic cancer is the fourth-leading cause of
cancer-associated mortality in the United States (24). Unlike other types of cancer, the
survival rate of patients with this disease has not improved
substantially in ~40 years, largely due to its aggressiveness
(25). Emerging evidence indicates
that the aggressiveness of pancreatic cancer may be partly driven
by CSCs, phenotypically distinct cell populations (10,26,27). CD133
is the most commonly expressed CSC marker in several cancer types,
including pancreatic cancer (18,19,28–30).
In the present study, it was demonstrated that
thalidomide inhibits the growth of human pancreatic cancer cells
in vitro. However, the sensitivity to thalidomide differed
between SW1990 and Capan-2 cells, which exhibit different levels of
CD133 expression. The SW1990 cell line, in which the proportion of
CD133+ cells was >40%, was more sensitive to
thalidomide than the Capan-2 cell line, of which <1% were
CD133+ cells, as demonstrated by the increased
inhibition of cell growth and migration. These results indicated
that the sensitivity of pancreatic cancer cells to thalidomide may
dependent on the expression of CD133, and therefore, on the
proportion of CSCs. Thalidomide reduced the CD133+ cell
subpopulation in the SW1990 cell line and downregulated the mRNA
and protein expression of CD133. Taken together, the results
indicated that thalidomide may inhibit the proliferation and
metastasis of pancreatic cancer cells partly by modulating the
expression of CD133, which suggests that the antitumor effects of
thalidomide are partially mediated by inhibiting CSCs.
The epithelial-mesenchymal transition (EMT) is a
process implicated in drug resistance, metastasis and the
generation of CSCs (21,31,32). The
present study analyzed the mRNA and protein expression of the EMT
markers E-cadherin and N-cadherin in pancreatic cancer cells
treated with thalidomide. The results of this analysis revealed
that thalidomide upregulated E-cadherin expression and
downregulated N-cadherin expression in SW1990 cells, whereas there
was no significant effect on these markers in Capan-2 cells. These
results suggested that thalidomide inhibited EMT in SW1990 cells
and not in Capan-2 cells. Previous studies have demonstrated that
CD133 is part of the regulatory network that facilitates EMT; CD133
is a transmembrane protein associated with the signaling pathways
regulating the EMT program, particularly the cadherin switch
(28,33). In liver cancer cells,
CD133+ cells exhibited upregulated N-cadherin expression
and downregulated E-cadherin expression compared with
CD133− cells, which indicates that EMT occurs more
frequently in CD133+ cells than in CD133−
cells (34). Ding et al
(29) reported that CD133 serves an
important role in the regulation of N-cadherin and Slug expression.
However, other molecules are involved in the regulation of EMT, in
addition to CD133. Further study is required to characterize this
regulatory loop, which implies that thalidomide may inhibit EMT at
least partly through the modulation of CD133.
In summary, the data of the present study indicate
that the in vitro efficacy of thalidomide at inhibiting
pancreatic cancer cell growth and migration is mediated by the
downregulation of CD133 and the inhibition of EMT. The efficacy of
thalidomide was dependent on the expression of CD133, as
demonstrated by the different sensitivity to thalidomide of the
SW1990 and Capan-2 pancreatic cancer cell lines, which express
different levels of CD133. The results indicated that thalidomide
may partly target CSCs. However, the differential response of
SW1990 and Capan-2 cells to thalidomide warrants further
investigation at the molecular level in order to further
characterize the mechanisms underlying their sensitivity to
thalidomide. Furthermore, assessing the effect of a combination of
CSC-targeting and non-CSC-targeting therapies could be of value in
optimizing the tumor response, enhancing long-term disease control
and ultimately improving patient survival rates.
Acknowledgements
The present study was supported in part by grants
from the National Natural Science Foundation of China (grant nos.
81372643 and 81270543) and Foundation for Shanghai Science and
Technology Committee (grant no. XBR2013082). The funders had no
role in study design, data collection and analysis, the decision to
publish, or preparation of the study.
References
|
1
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Von Hoff DD, Ramanathan RK, Borad MJ,
Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias
JL, et al: Gemcitabine plus nab-paclitaxel is an active regimen in
patients with advanced pancreatic cancer: A phase I/II trial. J
Clin Oncol. 29:4548–4554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Forssell H, Pröh K, Wester M and Krona H:
Tumor size as measured at initial X-ray examination, not length of
bile duct stricture, predicts survival in patients with
unresectable pancreatic cancer. BMC Cancer. 12:4292012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirono S, Kawai M, Okada KI, Miyazawa M,
Shimizu A, Kitahata Y, Ueno M and Yamaue H: Treatment strategy for
borderline resectable pancreatic cancer with radiographic artery
involvement. Pancreas. 45:1438–1446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma VP, Anderson NT and Geusz ME:
Circadian properties of cancer stem cells in glioma cell cultures
and tumorspheres. Cancer Lett. 345:65–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hale JS, Sinyuk M, Rich JN and Lathia JD:
Decoding the cancer stem cell hypothesis in glioblastoma. CNS
Oncol. 2:319–330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang Y, Ju B, Tian J, Liu F, Yu H, Xiao
H, Liu X, Liu W, Yao Z and Hao Q: Ovarian cancer stem cell-specific
gene expression profiling and targeted drug prescreening. Oncol
Rep. 31:1235–1248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gilbertson RJ and Graham TA: Cancer:
Resolving the stem-cell debate. Nature. 488:462–463. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosen JM and Jordan CT: The increasing
complexity of the cancer stem cell paradigm. Science.
324:1670–1673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stewart AK: Medicine. How thalidomide
works against cancer. Science. 343:256–257. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Song L, Zhou X and Li X: Phase II trial of
granulocyte-macrophage colony-stimulating factor plus thalidomide
in older patients with castration-resistant prostate cancer. Mol
Clin Oncol. 3:865–868. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Milanovic D, Sticht C, Röhrich M, Maier P,
Grosu AL and Herskind C: Inhibition of 13-cis retinoic acid-induced
gene expression of reactive-resistance genes by thalidomide in
glioblastoma tumours in vivo. Oncotarget. 6:28938–28948. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ruiz J, Case D, Enevold G, Rosdhal R,
Tatter SB, Ellis TL, McQuellon RP, McMullen KP, Stieber VW, Shaw EG
and Lesser GJ: A phase II trial of thalidomide and procarbazine in
adult patients with recurrent or progressive malignant gliomas. J
Neurooncol. 106:611–617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tunio MA, Hashmi A, Qayyum A, Naimatullah
N and Masood R: Low-dose thalidomide in patients with metastatic
renal cell carcinoma. J Pak Med Assoc. 62:876–879. 2012.PubMed/NCBI
|
|
17
|
Burris HA III, Jones SF, Shipley D, Meluch
AA, Greco FA, Barton JH, Yardley DA and Hainsworth JD: Phase II
study of capecitabine in combination with thalidomide in patients
with metastatic breast cancer. Cancer Invest. 28:408–412. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hermann PC, Huber SL, Herrler T, Aicher A,
Ellwart JW, Guba M, Bruns CJ and Heeschen C: Distinct populations
of cancer stem cells determine tumor growth and metastatic activity
in human pancreatic cancer. Cell Stem Cell. 1:313–323. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee HJ, You DD, Choi DW, Choi YS, Kim SJ,
Won YS and Moon HJ: Significance of CD133 as a cancer stem cell
markers focusing on the tumorigenicity of pancreatic cancer cell
lines. J Korean Surg Soc. 81:263–270. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lyons JG, Lobo E, Martorana AM and
Myerscough MR: Clonal diversity in carcinomas: Its implications for
tumour progression and the contribution made to it by
epithelial-mesenchymal transitions. Clin Exp Metastasis.
25:665–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wiseman DA, Werner SR and Crowell PL: Cell
cycle arrest by the isoprenoids perillyl alcohol, geraniol, and
farnesol is mediated by p21(Cip1) and p27(Kip1) in human pancreatic
adenocarcinoma cells. J Pharmacol Exp Ther. 320:1163–1170. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guillaumond F, Iovanna JL and Vasseur S:
Pancreatic tumor cell metabolism: Focus on glycolysis and its
connected metabolic pathways. Arch Biochem Biophys. 545:69–73.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Antoniou A, Hébrant A, Dom G, Dumont JE
and Maenhaut C: Cancer stem cells, a fuzzy evolving concept: A cell
population or a cell property? Cell Cycle. 12:3743–3748. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kumar R, Dholakia A and Rasheed Z: Stem
cell-directed therapies in pancreatic cancer. Curr Probl Cancer.
37:280–286. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding Q, Yoshimitsu M, Kuwahata T, Maeda K,
Hayashi T, Obara T, Miyazaki Y, Matsubara S, Natsugoe S and Takao
S: Establishment of a highly migratory subclone reveals that CD133
contributes to migration and invasion through
epithelial-mesenchymal transition in pancreatic cancer. Hum Cell.
25:1–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding Q, Miyazaki Y, Tsukasa K, Matsubara
S, Yoshimitsu M and Takao S: CD133 facilitates
epithelial-mesenchymal transition through interaction with the ERK
pathway in pancreatic cancer metastasis. Mol Cancer. 13:152014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Weng CC, Kuo KK, Su HT, Hsiao PJ, Chen YW,
Wu DC, Hung WC and Cheng KH: Pancreatic tumor progression
associated with CD133 overexpression: Involvement of increased TERT
expression and epidermal growth factor receptor-dependent Akt
activation. Pancreas. 45:443–457. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fan YL, Zheng M, Tang YL and Liang XH: A
new perspective of vasculogenic mimicry: EMT and cancer stem cells
(Review). Oncol Lett. 6:1174–1180. 2013.PubMed/NCBI
|
|
32
|
Hao J, Zhang Y, Deng M, Ye R, Zhao S, Wang
Y, Li J and Zhao Z: MicroRNA control of epithelial-mesenchymal
transition in cancer stem cells. Int J Cancer. 135:1019–1027. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cai C, Yu JW, Wu JG, Lu RQ, Ni XC, Wang SL
and Jiang BJ: CD133 promotes the invasion and metastasis of gastric
cancer via epithelial-mesenchymal transition. Zhonghua Wei Chang
Wai Ke Za Zhi. 16:662–667. 2013.(In Chinese). PubMed/NCBI
|
|
34
|
Na DC, Lee JE, Yoo JE, Oh BK, Choi GH and
Park YN: Invasion and EMT-associated genes are up-regulated in B
viral hepatocellular carcinoma with high expression of CD133-human
and cell culture study. Exp Mol Pathol. 90:66–73. 2011. View Article : Google Scholar : PubMed/NCBI
|