Introduction
Glioblastomas (GBMs) are brain tumors with extensive
vascularization that are associated with tumor malignancy (1). Tumor vasculature is structurally and
functionally abnormal, and the vessels are highly permeable,
tortuous, dilated, saccular and are poorly covered by pericytes
(2). Excessive blood leakage
associated with vessel immaturity increases the interstitial
pressure in the tumor and facilitates invasive tumor cell
penetration into the circulation, allowing the metastatic spread of
cancer (3). A key feature of GBMs is
the extensive network of abnormal vasculature, characterized by
glomeruloid structures and endothelial hyperplasia (4). Due to the location of GBM in the brain,
the primary clinical problem is the edematous swelling and dramatic
increase of intracerebral pressure caused by the disrupted blood
brain barrier (BBB) and leaky blood vessels (5,6). A current
therapeutic approach against GBMs is targeted against this
vascularization process (7).
Targeting endothelial cells (ECs) is a major focus of anti-vascular
therapy (1). However, targeting
glioma angiogenesis by vascular endothelial growth factor
inhibition has only exhibited a minor effect on patient survival
(8). At present, the biological
characteristics of ECs in GBMs are poorly characterized, and the
cellular and molecular mechanisms underlying the vascularization in
GBMs require additional study.
Endothelial cell-cell junctions are critical for
vascular integrity, EC growth, vascular development and the
maintenance of vascular function (9).
Endothelial junctional proteins also control multiple signaling
events intracellularly via membrane proteins, clustering of
signaling molecules and growth factor receptors (10). Afadin, originally identified as an
actin-binding protein, is localized at adherens junctions and is
expressed in epithelial cells, neurons, fibroblasts and ECs
(11,12). Afadin is an adaptor protein that binds
to numerous scaffolding and actin-binding proteins, and contributes
to the association of immunoglobulin-like cell adhesion molecule
nectins with other cell-cell adhesion molecules and intracellular
signaling systems (11). Afadin has
multiple functions in cell-cell adhesion and in cell movement,
proliferation, survival and polarization (13). Afadin has also been demonstrated to
regulate integrin-mediated cell adhesion and cell migration,
although it appears that the function of Afadin in the positive or
negative regulation of cell motility is context-dependent (14,15).
Afadin also directly interacts with the tight junction-associated
protein junction adhesion molecule-A, thereby affecting tight
junction assembly during progression of the polarity program
(16).
The phosphoinositide 3-kinase (PI3K)/protein kinase
B (AKT) pathway is a complex and central regulator of a number of
fundamental cellular processes (17)
and regulates all phenotypes that contribute to the progression of
human cancer, including GBM (17,18). The
AKT pathway was previously demonstrated to be activated in EC
tumors (19,20). Sustained endothelial AKT activation
mediated increased blood vessel size and generalized edema caused
by chronic vascular permeability (21). AKT signaling in the tumor vasculature
was sensitive to rapamycin, suggesting that rapamycin may affect
tumor growth in part by acting as a vascular AKT inhibitor
(21). A previous study indicated
that Afadin is phosphorylated by all AKT isoforms and that this
phosphorylation elicits a re-localization of Afadin from the
adherens junctions to the nucleus (15). The present study attempted to clarify
the functions of the PI3K/AKT pathway and Afadin in the GBM tumor
vasculature.
Materials and methods
Reagents
The PI3K inhibitor, LY294002 (Calbiochem; Merck
KGaA, Darmstadt, Germany) was used at a final concentration of 10
µM. The antibodies used for immunostaining and western blotting
were anti-cluster of differentiation (CD) 31 (cat. no. 910003;
BioLegend, Inc., San Diego, CA, USA), anti-Afadin (cat. no.
ab11337; Abcam, Cambridge, UK), anti-phosphorylated
(phospho)-Afadin Ser1718 (cat. no. 5485; Cell Signaling Technology,
Inc., Danvers, MA, USA), anti-AKT (cat. no. 9272; Cell Signaling
Technology, Inc.), anti-phospho-AKT Ser473 (cat. no. 4060; Cell
Signaling Technology, Inc.), anti-vascular endothelial
(VE)-cadherin (cat. no. AF938; R&D Systems, Inc., Minneapolis,
MN, USA), anti-p-endothelial nitric oxide synthase (cat. no. 9571;
Cell Signaling Technology, Inc.), anti-p-ERK (cat. no. 9101; Cell
Signaling Technology, Inc.) and anti-ERK (cat. no. 4695; Cell
Signaling Technology, Inc.).
Isolation of primary ECs
GBM or general traumatic surgical specimens were
collected in accordance with a protocol approved by the Ethics
Committee of Clinical Investigation of Chongqing Medical University
(Chongqing, China). These specimens were obtained subsequent to the
procurement of written informed consent by the neurosurgical team
at the Children's Hospital of Chongqing Medical University
(Chongqing, China). Surgical samples from 9 GBM patients (7 males
and 2 females) were included. The median age was 9 years and the
age range was between 2 and 13 years. Inclusion criteria included
confirmation of a histopathological diagnosis of classic GBM by two
independent pathologists (Chongqing Medical University). Pediatric
patients with histopathological diagnosis of other types of tumor
were excluded from the present study. Control human brain vascular
ECs (HBVECs) were obtained from 5 pediatric brain traumatic cases
that had not previously been diagnosed with cancer. All tissues
were collected between January and November 2014. ECs were derived
from GBM tissues (GBM-ECs) or general traumatic surgical specimens
(HBVECs) and characterized as previously described (22). Briefly, GBM tumors or general
traumatic brain tissues were disaggregated using 1-mg/ml
collagenase IV (Sigma-Aldrich; Merck KGaA) for 60 min at 37°C.
Isolated cells and vessel fragments were passed through a 70-µm
mesh, collected by centrifugation at 800 × g for 10 min at 4°C, and
seeded into 100-mm dishes with modified M-199 medium (9.87 g/l
medium M-199; cat. no. 11150059; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 10 ml/l 100X basal medium Eagle vitamin solution
(cat. no. 21010046; Thermo Fisher Scientific, Inc.), 1 g/l glucose
and 2.2 g/l NaHCO3 containing 20% heat-inactivated
low-endotoxin fetal bovine serum (Thermo Fisher Scientific, Inc.),
100 µg/ml neomycin, 20 U/ml nystatin and 2 mmol/l L-glutamine
(complete medium), and cultured for a week at 37°C with 5%
CO2. Culture medium was changed twice a week. To purify
the ECs, the cultured cells were subjected to
fluorescence-activated cell sorting for CD31-positive cells using
fluorescein isothiocyanate (FITC)-conjugated anti-CD31 (1:100; cat.
no. 303103, BioLegend, Inc.). Cells were incubated with anti-CD31
antibody at 400 µg/ml for 30 min at 4°C at a volume of 2 µl of
antibody per 105 cells. The sorted cells were then
collected in EC growth medium (EGM2; Lonza Group Ltd., Basel,
Switzerland), plated in 24-well tissue culture plates and incubated
at 37°C with 5% CO2.
Immunocytofluorescence analysis
Cells were grown on Lab-Tek II chamber slide systems
(Thermo Fisher Scientific, Inc.), fixed with 4% paraformaldehyde in
phosphate buffered saline (PBS; pH 7.4) overnight at 4°C, rinsed
with PBS and blocked with PBS containing 5% BSA (cat. no. A2058;
Sigma-Aldrich; Merck KGaA) for 30 min at room temperature. Primary
antibodies against VE-cadherin [cat. no. sc-9989; 1:100 dilution in
PBS containing 0.1% (v/v) Tween-20 (T-PBS); Santa Cruz
Biotechnology, Inc., Dallas, TX, USA]; cluster of differentiation
(CD) 31 (cat. no. 550389; 1:200; BD Biosciences, Franklin Lakes,
NJ, USA); Afadin (cat. no. ab11337; 1:100; Abcam) and phospho-AKT
Ser473 (cat. no. 4060; 1:300; Cell Signaling Technology, Inc.) were
applied to the surface of the slide and incubated overnight at 4°C.
The following day, cells were incubated with secondary antibodies
Alexa Fluor 594- or 488-conjugated goat anti-rabbit immunoglobulin
G (IgG) [heavy and light (H+L)] or anti-mouse IgG (H+L) (cat. no.
A-11037 and A-11001; 1:500; Thermo Fisher Scientific, Inc.) for 1 h
at room temperature. Finally, cells were counterstained with DAPI
(1:5,000 dilution in T-PBS; Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature. Negative controls, consisting of secondary
antibodies alone, were performed on each section of tissue studied.
Immunofluorescence photomicrographs were captured using a
fluorescence microscope (Nikon Corporation, Tokyo, Japan).
EC low-density lipoprotein (LDL)
uptake
The ability of isolated ECs to take up acetylated
(Ac) -LDL was examined using Ac-LDL labeled with
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
(Dil). The primary cells were seeded at an initial density of
2×105 cells/cm2. Cultures grown on Lab-Tek II
chamber slide systems (cat. no. 154526, Thermo Fisher Scientific,
Inc.) were incubated with 15 µg/ml Dil-Ac LDL (Thermo Fisher
Scientific, Inc.) in EGM2 for 20 min, washed with PBS (pH 7.4) and
immediately observed by inverted fluorescence microscopy using a
rhodamine filter.
Permeability assay
HBVECs or GBM-ECs (2×105 cells) were
seeded on Transwell filters (0.4-µm pore size, Costar; Corning
Incorporated, Corning, NY, USA) in 24-well dishes and grown in EGM2
until they reached 100% confluence. For the inhibition assay,
GBM-ECs were pre-incubated for 24 h in the presence of LY294002 (10
µM) or with 1 µl of dimethylsulfoxide as a vehicle control. Then,
FITC-dextran (70,000 Da; Sigma-Aldrich; Merck KGaA) at a final
concentration of 1 mg/ml was added to the upper chamber. At 30 min,
50 µl samples were removed from the lower chamber. The fluorescence
of these samples was measured at an excitation wavelength of 485 nm
and an emission wavelength of 530 nm using a fluorescence plate
reader (BioTek Instruments, Inc., Winooski, VT, USA).
Transwell migration assay
Cell migration was evaluated using a Transwell
system (Costar; Corning Incorporated) comprised of 8-µm
polycarbonate filter inserts in 24-well plates. Briefly, HBVECs and
GBM-ECs (passaged 2–3 times) were harvested using trypsin in EGM2.
Next, 600 ml EGM2 medium was added to the lower chambers, while
1×105 HBVECs or GBM-ECs were plated in the upper
chambers in EGM2. After 4 h of incubation, cells on the bottom of
the Transwell membrane were fixed with 4% paraformaldehyde at 37°C
for 20 min and stained with 1% crystal violet at 37°C for 10 min;
the non-migrating cells in the upper chamber were removed with
blunt-end swabs. The membranes were washed three times with PBS and
images were captured under a fluorescence microscope (Olympus
Corporation, Tokyo, Japan). Finally, the level of cell migration
was quantified by counting four fields of view. The mean count of
triplicated chambers is presented.
Western blot analysis
HBVECs and GBM-ECs were cultured in 6-cm dishes.
Proteins were extracted using 2X SDS sample buffer (Sigma-Aldrich;
Merck KGaA) and quantified using a Bradford protein assay kit
(P0006C; Beyotime Institute of Biotechnology). The extracted
proteins (30 µg/lane) were separated using 4–15%
Protean® TGX™ Precast Protein gels (cat. no. 4561083;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins were
transferred onto polyvinylidene difluoride membranes. Following
blocking with 5% bovine serum albumin overnight at 4°C, the
membranes were incubated with the aforementioned antibodies
overnight at 4°C. anti-Afadin (1:1,000; cat. no. ab11337; Abcam,
Cambridge, UK), anti-phosphorylated (phospho)-Afadin Ser1718
(1:1,000; cat. no. 5485; Cell Signaling Technology, Inc., Danvers,
MA, USA), anti-AKT (1:1,000; cat. no. 9272; Cell Signaling
Technology, Inc.), anti-phospho-AKT Ser473 (1:1,000; cat. no. 4060;
Cell Signaling Technology, Inc.), anti-p-endothelial nitric oxide
synthase (1:1,000; cat. no. 9571; Cell Signaling Technology, Inc.)
and anti-p-ERK (1:1,000; cat. no. 9101; Cell Signaling Technology,
Inc.). GAPDH (1:2,500; cat. no. 2118; Cell Signaling Technology,
Inc.) was used as loading control. Membranes were subsequently
incubated with horseradish peroxidase-conjugated anti-mouse and
anti-rabbit secondary antibodies (cat. nos. ab97040 and ab7090;
1:2,500; Abcam) for 1 h at room temperature. Following two membrane
washes, membranes were developed with 3,3′-diaminobenzidine
tetrahydrochloride and imaged/quantified using a Sigma-Aldrich
microDOC gel documentation system (cat. no. Z692557; Merck
KGaA).
Statistical analysis
Independent Student's t-tests were used to determine
statistical significance. Statistical tests were performed in
Microsoft Excel 2013 (Microsoft Corporation, Redmond, WA, USA). All
P-values were derived from at least three independent experiments.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ECs isolated from human GBM tumors
demonstrate increased permeability and motility
To characterize the phenotype of GBM-derived ECs,
primary GBM-ECs and HBVECs were isolated from 9 and 5 patients,
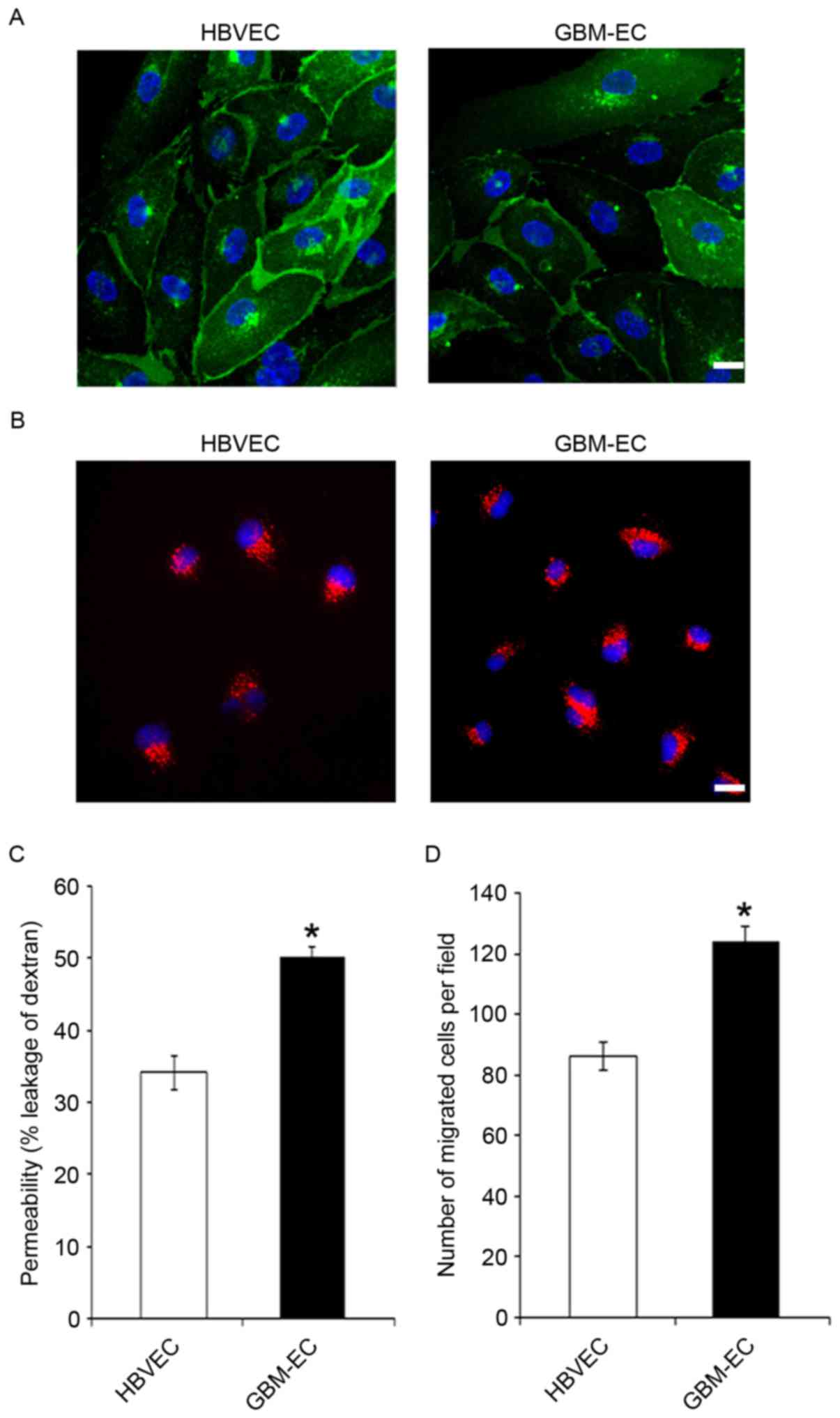
respectively. CD31 immunostaining (Fig.
1A) and uptake of Dil-acetylated LDL (Fig. 1B) demonstrated the purity of primary
isolated brain ECs. The results indicate that these two types of
ECs were positive for CD31, and that GBM-ECs exhibit a similar
morphology and LDL uptake activity to HBVECs (Fig. 1A and B). Permeability of the EC
barrier was assessed by comparing the transport of 70 kDa
fluorescent dextran across the endothelial monolayer. HBVECs or
GBM-ECs were seeded into the upper chamber of 0.4 µm Transwell
filters. The degree of leakage was determined by measuring the
intensity of FITC-dextran fluorescence in the lower chamber. The
results demonstrate that FITC-dextran leakage was significantly
increased in GBM-ECs compared with HBVECs (Fig. 1C), indicating significantly disrupted
endothelial barrier function in GBMs. Cell motility is of
particular interest in the design of anti-cancer therapeutics, as
endothelial migration is required for tumor angiogenesis (23). Next, EC migration was evaluated using
the Transwell migration assay. HBVECs or GBM-ECs were plated onto
the upper chambers of an 8-µm Transwell system. Cells were allowed
to migrate for 4 h through the 8-µm pores of the polycarbonate
filters. As indicated in Fig. 1D,
compared with HBVECs, a significantly increased number of GBM-ECs
migrated to the lower side of the filter. This result demonstrated
that endothelial migration is induced in GBMs.
Adherens junction protein Afadin is
phosphorylated and re-localized in GBM-ECs
The integrity of intercellular junctions is a major
determinant of permeability of the brain endothelium (24). The adherens junction protein, Afadin,
participates in the initial step of junction formation and serves a
fundamental role in the regulation of cell-cell contact (25). The expression and localization of
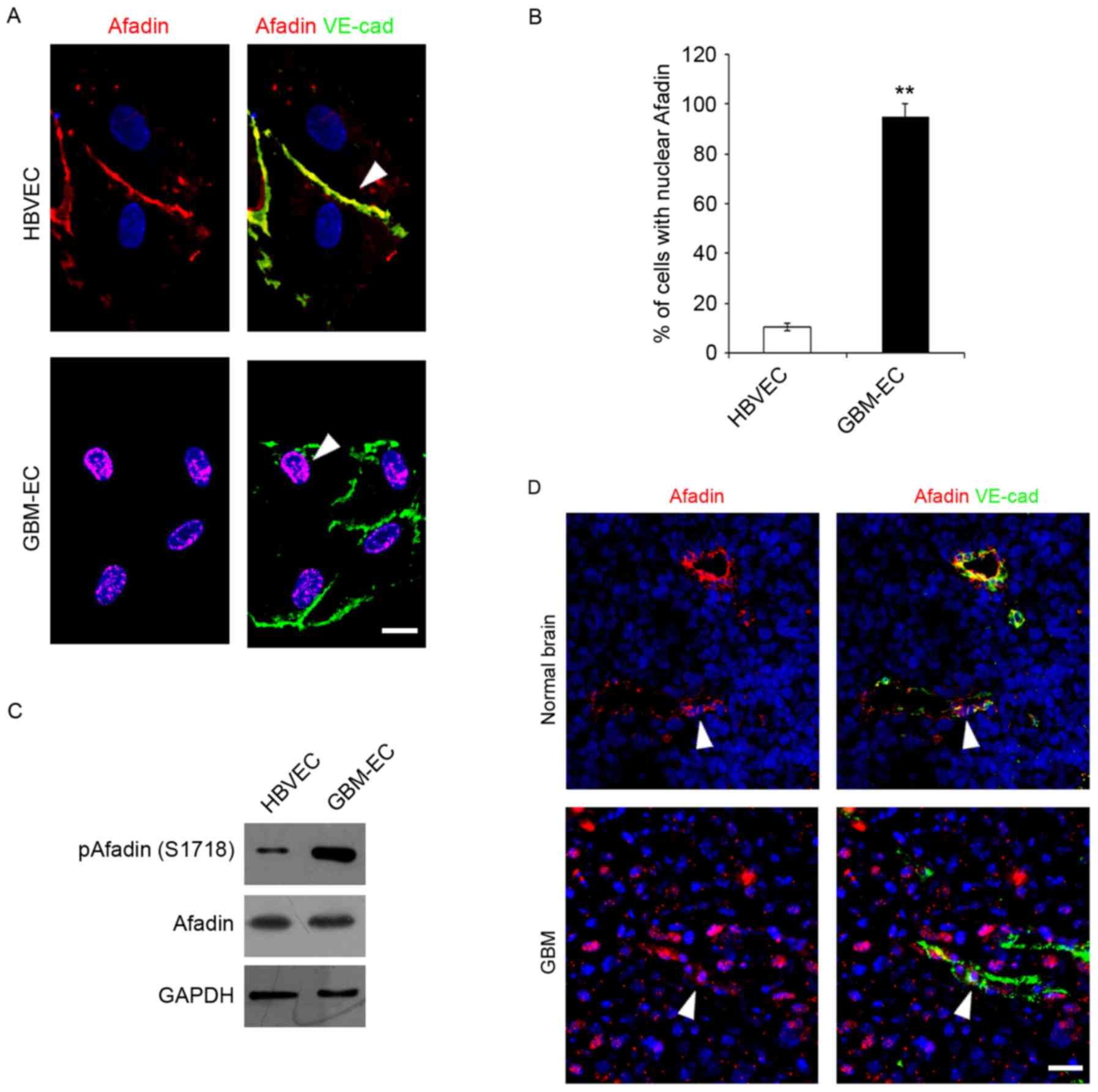
Afadin was detected in ECs Immunofluorescence microscopy indicated
that total Afadin exhibited a predominantly membrane-restricted
localization in HBVECs and was concentrated at the cell-cell
contact sites between adjacent HBVECs, which was co-localized with
a marker of endothelial cell-cell contact, VE-cadherin (Fig. 2A). However, Afadin membrane
localization and its distribution along the cell borders was
markedly decreased, accompanied by the appearance of its nuclear
localization in GBM-ECs (Fig. 2A).
Quantification of the nuclear translocation was evaluated using
co-staining with DAPI and this revealed that a significantly higher
percentage of GBM-ECs stained positive for nuclear Afadin compared
with HBVECs (Fig. 2B). Notably,
western blot analysis revealed that total Afadin protein expression
did not change in GBM-ECs, while phosphorylation of Afadin at
serine 1718 (Ser1718) was markedly induced in GBM-ECs compared with
HBVECs (Fig. 2C). Finally, Afadin
localization in human GBMs was assessed. GBM tumors and normal
brain tissue obtained from archival pathology specimens were used
for localization analysis and evaluated using immunofluorescence.
Afadin expression in ECs was assessed by co-immunostaining for
Afadin and VE-cadherin. A marked increase in the nuclear
localization of Afadin in the ECs of GBM vessels compared with
normal brain tissue was identified. Fig.
2D presents representative images of normal brain tissue and
GBM specimens. Taken together, these results suggest that Afadin is
phosphorylated and re-localized in GBM-ECs.
AKT pathway is activated in
GBM-ECs
A previous study has indicated that the PI3K/AKT
pathway serves a critical function in the pathogenesis of
vasculature and BBB interruption in tumors (26). Whether the PI3K/AKT pathway is
activated in human GBM-ECs was next examined. To investigate the
level of AKT activation, AKT phosphorylation was analyzed using the
immunofluorescence staining of human GBMs. The results demonstrated
that numerous tumor ECs expressed phospho-AKT in GBMs, while
quiescent vessels in the normal brain did not exhibit a high
intensity of phospho-AKT (Fig. 3A).
Western blot analyses of cell lysates confirmed the immunostaining
results by demonstrating elevated levels of phospho-AKT,
standardized to the level of total AKT, in GBM-ECs compared with
HBVECs (Fig. 3B). Additionally, AKT
activation was associated with the activation of downstream
signaling pathways, as evidenced by the increased phosphorylation
of extracellular signal-related kinase and endothelial nitric oxide
synthase (Fig. 3B). These results
demonstrated that the PI3K/AKT pathway is activated in GBM-ECs.
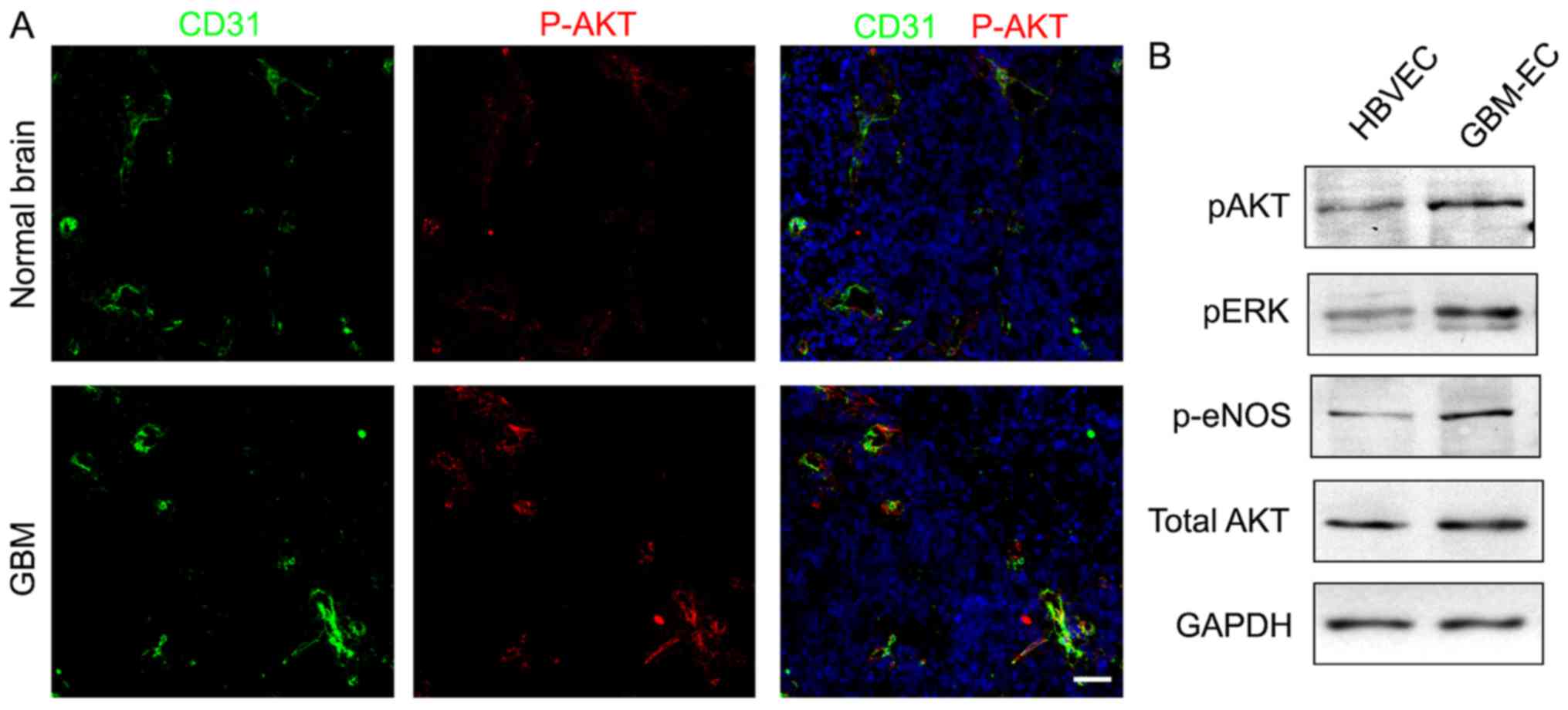 | Figure 3.Immunocytofluorescence analysis and
western blot assay detecting phosphorylation of AKT. (A)
Phosphorylation levels of AKT in normal brain vessels and GBM
vessels. P-AKT (red), CD31 (green) and DAPI (blue). Representative
images are presented. Scale bars, 100 µm. (B) Western blotting
demonstrated phosphorylation levels of AKT, ERK, eNOS, and total
expression level of AKT in HBVECs and GBM-ECs. p, phosphorylated;
AKT, protein kinase B; CD31, cluster of differentiation; ERK,
extracellular signal-related kinase; eNOS, endothelial nitric oxide
synthase; HBVEC, human brain vascular endothelial cells; GBM-ECs,
glioblastoma endothelial cells. |
Phosphorylation and re-localization of
Afadin is regulated by the PI3K/AKT pathway in GBM-ECs
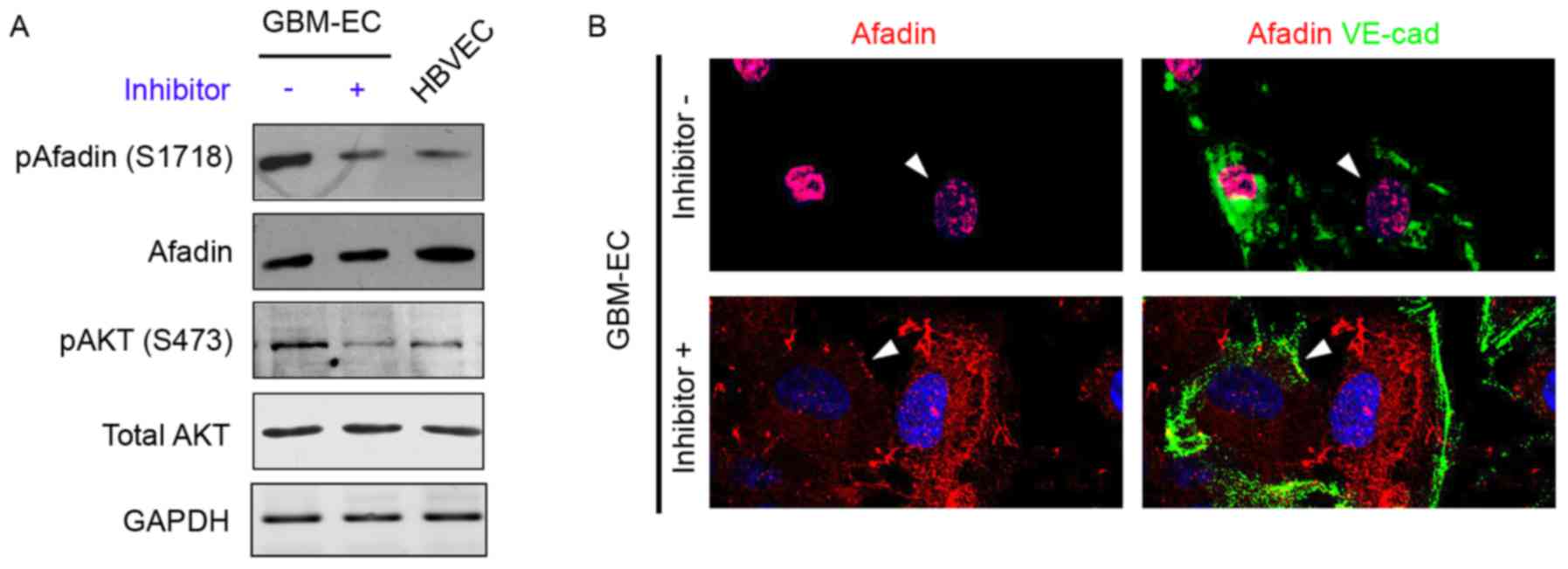
A previous study demonstrated that PI3K/AKT
signaling phosphorylates Afadin at Ser1718 in cancer epithelial
cells (15); therefore, our group
hypothesized that the induction of Afadin phosphorylation and
re-localization is mediated by the PI3K/AKT pathway in GBM-ECs. To
examine this hypothesis, GBM-ECs were treated with the PI3K
inhibitor LY294002 (10 µM) for 4 h, which decreased AKT activity
(Fig. 4A). The inhibition of AKT in
GBM-ECs prevented the Ser1718-phosphorylation of Afadin (Fig. 4A). The effect of AKT on Afadin
localization was investigated. Notably, the nuclear localization of
Afadin was reversed by LY29400 treatment in GBM-ECs. Afadin protein
accumulated at cell-cell junctions in LY29400-treated GBM-ECs. In
contrast, Afadin accumulation was not observed at cell-cell
junctions in untreated cells (Fig.
4B). The cellular localization of Afadin in the LY29400-treated
GBM-ECs was in agreement with the localization observed in
HBVECs.
Effects of the PI3-K/AKT/Afadin
pathway on the permeability and migration of GBM-ECs
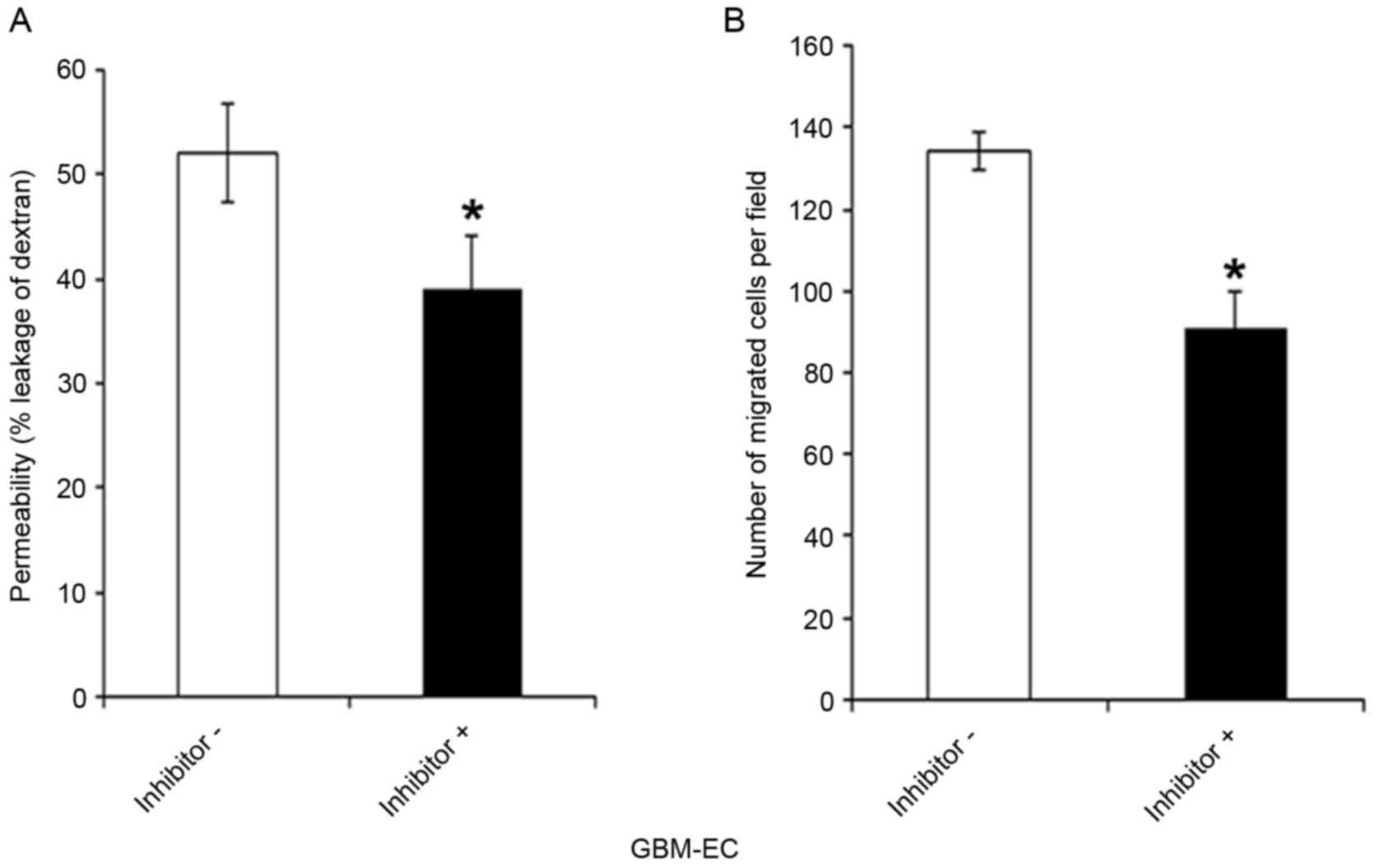
Whether the PI3K/AKT/Afadin pathway regulates brain
endothelial permeability and motility, which are dependent on
intact adherens junctions, was next investigated. GBM-ECs were
pretreated with LY294002 inhibitor or control vehicle for 4 h.
LY294002 exposure significantly decreased the FITC-Dextran leakage
of GBM-ECs compared with those treated with the DMSO control
(Fig. 5A). As indicated in Fig. 5B, pre-treatment of cells with LY294002
inhibitors significantly reduced GBM-EC migration in the Transwell
experiments. Taken together, these results suggest that the nuclear
localization of Afadin and phosphorylation at Ser1718 by the AKT
pathway is relevant for high permeability and pathological
angiogenesis in GBM vessels.
Discussion
To develop novel and effective strategies against
GBMs, which are highly vascularized, it is mandatory to understand
the biological characteristics of ECs in GBMs. Considering the
different vascular biology of different tumors and organs, and that
ECs tend to change their phenotype in response to culture
conditions (27), there are
significant benefits associated with the use of freshly isolated
ECs from GBMs for the investigation of GBM-induced angiogenesis. In
the present study, GBM-ECs were established using 9 patient-derived
GBM EC cultures and HBVECs were isolated from normal brain tissues.
There were no apparent differences in EC morphology between the
HBVECs and GBM-ECs. However, a significantly higher permeability
and a marked increase in migration of GBM-ECs compared with HBVECs
were observed, indicating that ECs are abnormally activated in
GBMs.
The intercellular junctions of ECs serve an
important barrier function that regulates permeability to small
molecules (25). Previous studies
have indicated that Afadin regulates migration and cell-cell
adhesion in other cell types, including fibroblasts and epithelial
cells (28,29). A previous study indicated that Afadin
is associated with angiogenesis through Rap1 signaling (30). The present study has demonstrated that
Afadin is phosphorylated and re-localized in GBM-ECs compared with
HBVECs in human brain tissues and in vitro cultures,
suggesting the potential of phosphorylation and nuclear
localization of Afadin as a novel marker of pathological
angiogenesis in GBMs.
The PI3K/AKT pathway is a frequently unregulated
pathway in cancer; with 86% of GBM clinical samples exhibiting
alterations in the receptor tyrosine kinases/PI3K pathway (31,32).
Furthermore, sustained endothelial activation of AKT1 has been
demonstrated to induce the formation of structurally abnormal blood
vessels that recapitulate the aberrations of tumor vessels
(31). However, downstream mediators
of AKT in brain tumor vasculatures remain poorly understood. AKT
induces phosphorylation of Afadin in epithelial cells (15). However, the involvement of AKT-Afadin
in angiogenesis is unknown. The present study revealed that the
phosphorylation of AKT was significantly induced in GBM-ECs
compared with HBVECs, indicating that the PI3K/AKT pathway is
activated in GBM-ECs. Notably, to the best of our knowledge, these
data demonstrate for the first time that the phosphorylation and
re-localization of Afadin is PI3K/AKT pathway-dependent in human
brain ECs. The inhibition of AKT by a PI3K inhibitor was revealed
to prevent Ser1718-phosphorylation and the nuclear localization of
Afadin in GBM-ECs.
The present study demonstrated that AKT-mediated
phosphorylation and re-localization of Afadin was critically
involved in the modulation of brain endothelial permeability. The
pharmacological inhibition of AKT activity was associated with a
decreased permeability in GBM-ECs, as evidenced by the decreased
passage of 70 kDa dextran. GBM-EC migration was revealed to be
downregulated by inhibition of AKT activity. Therefore, the present
study has identified a function for the PI3K/AKT/Afadin pathway in
the regulation of EC permeability and migration. These data on the
molecular interactions of the PI3K/AKT pathway and Afadin may
assist to explain an important regulatory mechanism of AKT-mediated
function, including the control of EC permeability and
migration.
In conclusion, the data of the present study
demonstrated that AKT-mediated phosphorylation and re-localization
of Afadin is an important mechanism for the regulation of GBM
endothelial permeability and motility. Dysregulation of AKT
activity contributes to Afadin-associated defects in vascular
permeability or angiogenesis associated with GBM metastasis or
chronic edema. Therefore, treatment with drugs targeting the
PI3K/AKT/Afadin pathway may be beneficial in reducing the
angiogenic potential of GBMs. Additional studies are required to
understand how this pathway is integrated into the various
signaling networks that regulate brain EC permeability and
migration.
Acknowledgements
The present study was supported by the special fund
of Chongqing Key Laboratory (grant no. cstc2012gg-yyjs0838), from
Chongqing Science and Technology Commission Scientific and
Technological Projects, and from the Chongqing Natural Science
Foundation (grant no. cstc2015jcyjBX0144).
References
|
1
|
Norden AD, Drappatz J and Wen PY:
Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol.
5:610–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jain RK: Normalization of tumor
vasculature: An emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goel S, Duda DG, Xu L, Munn LL, Boucher Y,
Fukumura D and Jain RK: Normalization of the vasculature for
treatment of cancer and other diseases. Physiol Rev. 91:1071–1121.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang R, Chadalavada K, Wilshire J, Kowalik
U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C and
Tabar V: Glioblastoma stem-like cells give rise to tumor
endothelium. Nature. 468:829–833. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wolburg H, Noell S, Fallier-Becker P, Mack
AF and Wolburg-Buchholz K: The disturbed blood-brain barrier in
human glioblastoma. Mol Aspects Med. 33:579–589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mrugala MM: Advances and challenges in the
treatment of glioblastoma: A clinician's perspective. Discov Med.
15:221–230. 2013.PubMed/NCBI
|
|
7
|
Gerstner ER and Batchelor TT:
Antiangiogenic therapy for glioblastoma. Cancer J. 18:45–50. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pàez-Ribes M, Allen E, Hudock J, Takeda T,
Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D and Casanovas O:
Antiangiogenic therapy elicits malignant progression of tumors to
increased local invasion and distant metastasis. Cancer cell.
15:220–231. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bazzoni G and Dejana E: Endothelial
cell-to-cell junctions: Molecular organization and role in vascular
homeostasis. Physiol Rev. 84:869–901. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takai Y, Ikeda W, Ogita H and Rikitake Y:
The immunoglobulin-like cell adhesion molecule nectin and its
associated protein afadin. Annu Rev Cell Dev Biol. 24:309–342.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mandai K, Nakanishi H, Satoh A, Obaishi H,
Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, et al:
Afadin: A novel actin filament-binding protein with one PDZ domain
localized at cadherin-based cell-to-cell adherens junction. J Cell
Biol. 139:517–528. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ogita H, Rikitake Y, Miyoshi J and Takai
Y: Cell adhesion molecules nectins and associating proteins:
Implications for physiology and pathology. Proc Jpn Acad Ser B Phys
Biol Sci. 86:pp. 621–629. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Su Li, Hattori M, Moriyama M, Murata N,
Harazaki M, Kaibuchi K and Minato N: AF-6 controls
integrin-mediated cell adhesion by regulating Rap1 activation
through the specific recruitment of Rap1GTP and SPA-1. J Biol Chem.
278:15232–15238. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elloul S, Kedrin D, Knoblauch NW, Beck AH
and Toker A: The adherens junction protein afadin is an AKT
substrate that regulates breast cancer cell migration. Mol Cancer
Res. 12:464–476. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takai Y and Nakanishi H: Nectin and
afadin: Novel organizers of intercellular junctions. J Cell Sci.
116:17–27. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arcaro A and Guerreiro AS: The
phosphoinositide 3-kinase pathway in human cancer: Genetic
alterations and therapeutic implications. Curr Genomics. 8:271–306.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McDowell KA, Riggins GJ and Gallia GL:
Targeting the AKT pathway in glioblastoma. Curr Pharm Des.
17:2411–2420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase-AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Keunen O, Johansson M, Oudin A, Sanzey M,
Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, et al:
Anti-VEGF treatment reduces blood supply and increases tumor cell
invasion in glioblastoma. Proc Natl Acad Sci USA. 108:pp.
3749–3754. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Phung TL, Ziv K, Dabydeen D, Eyiah-Mensah
G, Riveros M, Perruzzi C, Sun J, Monahan-Earley RA, Shiojima I,
Nagy JA, et al: Pathological angiogenesis is induced by sustained
Akt signaling and inhibited by rapamycin. Cancer cell. 10:159–170.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhai X, Liang P, Li Y, Li L, Zhou Y, Wu X,
Deng J and Jiang L: Astrocytes regulate angiogenesis through the
Jagged1-mediated notch1 pathway after status epilepticus. Mol
Neurobiol. 53:5893–5901. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Eccles SA: Parallels in invasion and
angiogenesis provide pivotal points for therapeutic intervention.
Int J Dev Biol. 48:583–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dejana E, Tournier-Lasserve E and
Weinstein BM: The control of vascular integrity by endothelial cell
junctions: Molecular basis and pathological implications. Dev Cell.
16:209–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wallez Y and Huber P: Endothelial adherens
and tight junctions in vascular homeostasis, inflammation and
angiogenesis. Biochim Biophys Acta. 1778:794–809. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Posada-Duque RA, Barreto GE and
Cardona-Gomez GP: Protection after stroke: Cellular effectors of
neurovascular unit integrity. Front Cell Neurosci. 8:2312014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dudley AC: Tumor endothelial cells. Cold
Spring Harb Perspect Med. 2:a0065362012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Severson EA, Lee WY, Capaldo CT, Nusrat A
and Parkos CA: Junctional adhesion molecule a interacts with afadin
and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels and
enhance cell migration. Mol Biolo Cell. 20:1916–1925. 2009.
View Article : Google Scholar
|
|
29
|
Fournier G, Cabaud O, Josselin E, Chaix A,
Adélaïde J, Isnardon D, Restouin A, Castellano R, Dubreuil P,
Chaffanet M, et al: Loss of AF6/afadin, a marker of poor outcome in
breast cancer, induces cell migration, invasiveness and tumor
growth. Oncogene. 30:3862–3874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tawa H, Rikitake Y, Takahashi M, Amano H,
Miyata M, Satomi-Kobayashi S, Kinugasa M, Nagamatsu Y, Majima T,
Ogita H, et al: Role of afadin in vascular endothelial growth
factor-and sphingosine 1-phosphate-induced angiogenesis. Cir Res.
106:1731–1742. 2010. View Article : Google Scholar
|
|
31
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prasad G, Sottero T, Yang X, Mueller S,
James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT, et al:
Inhibition of PI3K/mTOR pathways in glioblastoma and implications
for combination therapy with temozolomide. Neuro Oncol. 13:384–392.
2011. View Article : Google Scholar : PubMed/NCBI
|