Introduction
Bladder cancer is the fifth most common type of
cancer in men and ninth in women (1).
Various factors have been investigated for their involvement in the
pathogenesis of bladder cancer. Such factors include the epidermal
growth factor (EGF) family, which consists of four transmembrane
tyrosine kinase receptors, human epidermal growth factor receptor
(HER)1, HER2, HER3 and HER4, and a dozen ligands (2). While HER1, HER2 and HER4 activate
intracellular signals following ligand binding, HER3 (also named
ErbB3 and encoded by the ErbB3 gene) lacks a kinase domain,
and activates intracellular signals by forming heterodimers with
other EGF receptors (3). HER3 also
exists as soluble HER3 (sHER3), a truncated secreted form of either
45 kDa (p45-sHER3) or 85 kDa (p85-sHER3) (4). p85-sHER3 is a potent negative regulator
of HER2, HER3 and HER4 activation (5).
We previously demonstrated that increased tumour
expression of HER3 mRNA was associated with a more improved
survival rate in bladder cancer (6,7). The aim
of the present study was to explore the role of sHER3 as a
potential non-invasive predictor of prognosis in patients with
bladder cancer and to identify the possible mechanisms
involved.
Materials and methods
Patients
Eighty-two patients with primary bladder cancer were
included with a median age of 70 years (range, 53–88). A total of
66 patients were male and 16 were female. Biopsies were
consecutively collected between November 1995 and June 2002 as part
of the Molecular Oncology of the Bladder Bio-bank (Aarhus,
Denmark). No patients were excluded from the present study. The
tumours were staged according to the Union for International Cancer
Control Tumour Node Metastasis staging system (7th edition)
(8) and graded according to the World
Health Organisation/International Society of Urological Pathology
2004 classification system (9).
Patients were allocated to one of three groups according to tumour
stage: Ta, superficial tumours; T1, superficial invasive tumours;
and T2-T4, muscle-invasive tumours. At the time of inclusion, 18
patients had previously received treatment in the form of radical
radiotherapy, chemotherapy or intravesical bacillus Calmette-Guérin
therapy. The follow-up period was between the date of biopsy and
the day of mortality or the end of the study. Patients were
censored if they were alive at the time of analysis. The median
follow-up was 54 months (range, 1–141 months). The Regional
Committee of Scientific Ethics in Aarhus (Aarhus, Denmark) approved
the study (approval no. 1994/2920) and written informed consent was
provided by all participants in the study. All the procedures were
performed in accordance with the Declaration of Helsinki. The same
cohort was previously used in multiple studies (6,7,10).
ELISA
Whole-blood samples were collected into EDTA tubes
and centrifuged at 2,000 × g for 15 min at 4°C to collect the
plasma. The samples were then aliquoted and stored at −80°C for
ELISA analysis as described below.
A sandwich ELISA was developed employing monoclonal
anti-human HER3 antibody (dilution, 1:1,000; catalogue no. MAB3481;
R&D Systems Europe, Ltd., Abingdon, UK) as the capture antibody
and the polyclonal goat anti-human HER3 antibody (dilution, 1:200;
catalogue no. AF234; R&D Systems Europe, Ltd.) as the detection
antibody. A total of 0.1 µg capture antibody was diluted in 100 µl
of 15 mmol/l sodium carbonate and 35 mmol/l sodium bicarbonate (pH
9.6), and added to each well of Nunc MaxiSorp F96 immunoplates
(catalogue no. 442404; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Following incubation for 20 h at 4°C, the supernatant was
removed from the wells and the samples were blocked with 200 µl of
1 mol/l ethanolamine for 20 h at 4°C. The plates were stored at
−20°C until use in subsequent experiments.
The detection antibody was biotinylated by
dissolving 80 µg polyclonal goat anti-human HER3 antibody in 800 µl
of dilution buffer (10 mM phosphate, 145 mM sodium chloride, pH
7.4) and dialysing it against 1 litre of sodium hydrogen carbonate
(0.1 M, pH 8.3) for 24 h at 4°C with one change of dialysis buffer
after 3 h. The dialysed sample was incubated with 8 µl of 4.4 mM
biotinaminocaproate N-hydroxysuccinimidyl ester in dimethyl
sulfoxide both (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in
the dark with gentle agitation for 4 h at room temperature.
Following the addition of 8 µl of 100 mM lysine-HCl (Fluka Chemie
GmbH, Sigma-Aldrich; Merck KGaA), the samples were incubated for 15
min at room temperature. Finally, 8 µl of λ globulin [5% rabbit λ
globulin (Calbiochem; Merck KGaA) and 10% bovine IgG
(Sigma-Aldrich; Merck KGaA)] dissolved in dilution buffer was
added, and the sample was dialysed for 3 days against 1 litre of
dilution buffer with three changes of buffer, and with 0.1% sodium
azide in the last volume of buffer. The biotinylated antibody was
stored at −20°C until further use.
WM266 melanoma cells were obtained from American
Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured
in Dulbecco's Modified Eagle's Medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2. Conditioned media
from these cells was used as a calibrator, and assigned a value of
100 arbitrary units to the highest calibrator. When comparing the
signal to that of recombinant HER3 (catalogue no. 348-RB; R&D
Systems Europe, Ltd.) 100 arbitrary units corresponded to ~300
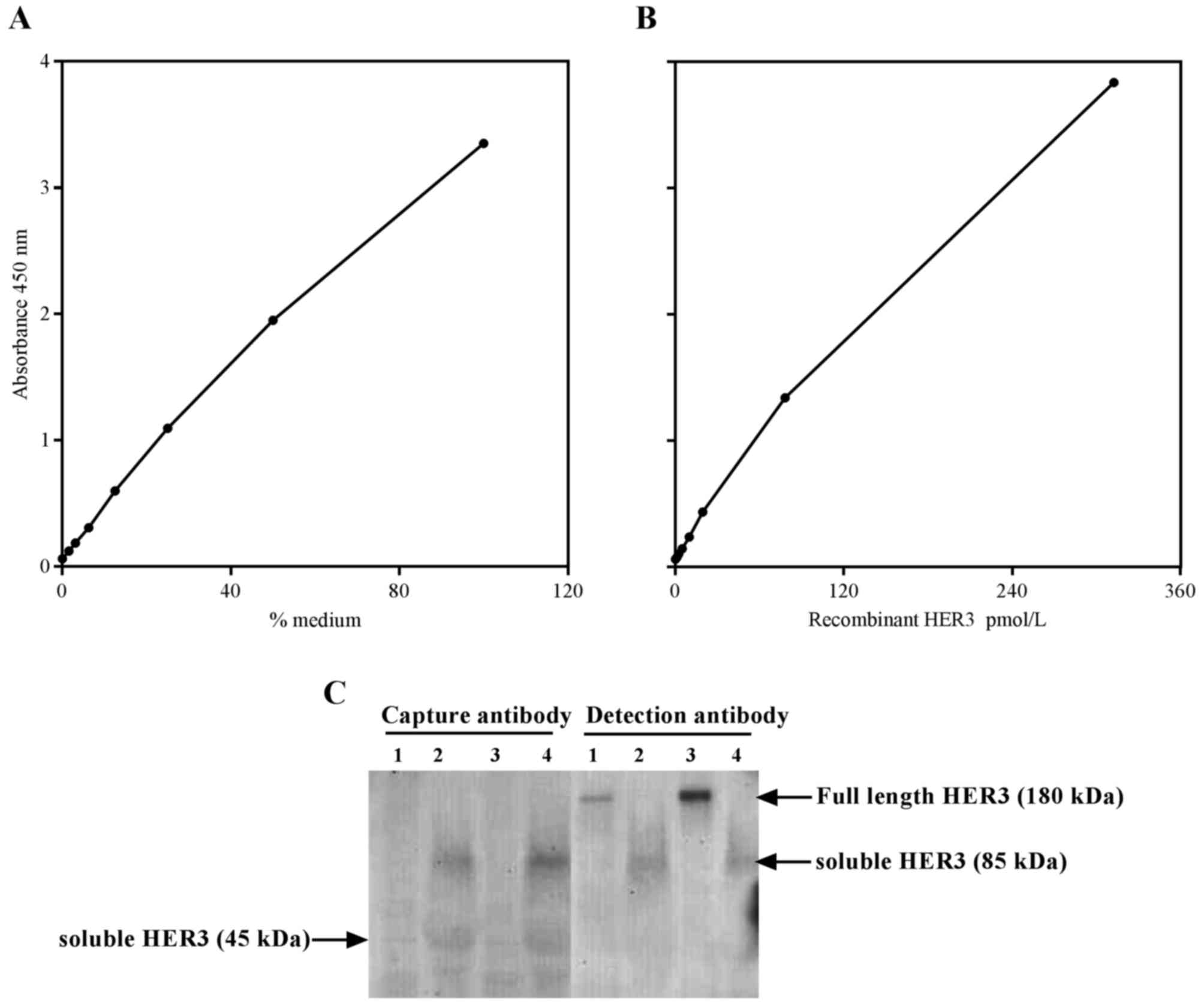
pmol/l (Fig. 1A). The calibration
curve covered 0.1–100 arbitrary units (Fig. 1B).
Each assay was performed as follows. First, the
plates were washed three times with washing buffer [10 mmol/l
sodium phosphate buffer containing 145 mmol/l NaCl and 1 g/l Tween
20 (pH 7.4); VWR International, Radnor, PA, USA; catalogue no.
AMPQ15265)]. Then, 100 µl of calibrator or sample diluted (1:7) in
assay buffer [0.1 M phosphate, 0.15 bovine serum albumin (catalogue
no. A7030; Sigma-Aldrich; Merck KGaA), pH 8.0] was added to each
well. After incubation for 2 h at room temperature, the plates were
washed three times with washing buffer. The detection antibody was
diluted to a concentration of 485 µg/l in assay buffer and 100 µl
of diluted antibody was added to each well. The plates were then
incubated for 2 h at room temperature. The plates were washed three
times with washing buffer, and 100 µl of horseradish
peroxidase-avidin (Dako; Agilent Technologies, Inc., Santa Clara,
CA, USA; catalogue no. P0364) diluted 1:2,000 in 10 mmol/l sodium
phosphate (pH 7.4), 400 mmol/l NaCl and 0.2 g/l lysozyme (catalogue
no. L6876; Sigma-Aldrich; Merck KGaA) was added to each well. The
plates were incubated for 30 min at room temperature and were
washed three times with washing buffer. Next, 100 µl of TMB ONE
ready-to-use substrate (catalogue no. 4380A; Kem-En-Tec
Diagnostics, Taastrup, Denmark) was added. The colour reaction was
stopped after 18 min by adding 100 µl of 1 mol/l phosphoric acid at
room temperature. The colour developed was measured photometrically
at a wavelength of 450 nm and corrected for absorbance at 620 nm.
The calibration curve was computed by plotting the absorbance of
the calibrators and constructing a cubic spline curve using
GraphPrism for Windows version 7 (GraphPad Software Inc., La Jolla,
CA, USA). The imprecision of the assay was 14% (n=82; mean, 4.5
arbitrary units) as determined from running the patient samples
twice ~1 month apart. The antibodies were tested using western
blotting with cell lysate and conditioned media from DU145 (ATCC)
prostate cancer cells which were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C with 5% CO2, and WM266
melanoma cells.
Cell culture and reagents
HCV-29 bladder cancer cells were obtained from the
American Type Culture Collection (Manassas, VA, MD). Cells were
seeded in T25 culture flasks (Nalge Nunc International, Penfield,
NY, USA) and maintained at 37°C in a humidified atmosphere of 5%
CO2 in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% foetal calf serum. Cells were checked
routinely for Mycoplasma infection. At 80–90% confluence, cells
were treated with 10–40 nM sHER3 (R&D Systems Europe, Ltd.).
Cells untreated with sHER3 were used a control. Control and treated
cells were washed with cold PBS [137 mM NaCl, 2.7 mM KCl, 10 mM
Na2HPO4, 2 mM KH2PO4
(pH 7.4)] and harvested after 24 h with scraping buffer (PBS
containing 4 mM iodoacetic acid, 1 mM orthovanadate, 1 µg/ml
aprotinin, 1 µg/ml chymostatin, 1 µg/ml leupeptin and 1 µg/ml
pepstatin). Cell pellets were stored at −80°C for western blotting
analysis.
Western blotting
Cell pellets were incubated on ice for 30 min in
RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% sodium
dodecylsulfate, 50 mM Tris-HCl, pH 7.2, supplemented with 4 mM
iodoacetate, 1 mM orthovanadate, 1 mg/ml eachpepstatin,
chymostatin, leupeptin, and aprotinin), homogenised by gentle
vortexing and cleared by centrifugation at 18,500 × g at 4°C for 10
min (11). Protein concentration was
determined using BCA reagent (Pierce; Thermo Fisher Scientific,
Inc.). Equal amounts of protein (25 µg) were resolved by 8–12%
SDS-PAGE. Resolved proteins were transferred onto polyvinylidene
difluoride membranes, which were blocked overnight at 4°C with 5%
(w/v) non-fat dry milk in TBS-T solution [25 mM Tris (pH 7.5), 150
mM NaCl, 0.05 % (w/v) Tween 20]. After washing, the membranes were
incubated with specific primary and secondary antibodies according
to the manufacturer's protocol. Primary antibodies against the
following proteins were used: Phosphorylated-HER3; total-HER3; and
β-actin (Sigma-Aldrich; Merck KGaA). Immunoreactive bands were
detected using ECL reagent (GE Healthcare, Chicago, IL, USA).
Secondary horseradish peroxidase-conjugated goat anti-rabbit
antibody (dilution, 1:2,000; catalogue no. P0448; Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) for 40 min at room
temperature followed by washing and enhanced chemiluminescence
(ECL) for 2 min. Immunoreactive bands were detected by a
Biospectrum® AC Imaging System (UVP, Inc., Upland, CA,
USA). This experiment was repeated at least twice.
Proliferation and migration
assays
The proliferation of HCV-29 cells was assessed using
xCELLigence technology as described previously (9). Briefly, a total of 5×103
HCV-29 cells were seeded in quadruplets overnight in DMEM in a
96-well microelectronic cell sensor system plate (Roche
Diagnostics, Basel, Switzerland). They were then treated with the
recombinant sHER3 peptide (0 or 20 nM). After treatment, the plate
was inserted into an xCELLigence RTCA proliferation instrument
(Roche Diagnostics) connected to a computer. This system measures
cell growth by analysing changes in electrical impedance in
microelectrodes under the 96-well plate when cells attach to the
bottom of the plate (12). Cell
growth was analysed in real time for 96 h. Values are expressed as
a cell index, normalised for differences in cell density
immediately prior to treatment. The experiment was performed in
triplets (n=3) and repeated at least three times.
HCV-29 cell migration was also assessed using
xCELLigence technology. Cells were treated with recombinant sHER3
peptide (0, 20 or 40 nM) and their migration relative to untreated
control cells was measured at 24 h. The experiment was run in
quadruplet (n=4) and repeated at least twice.
Statistical analysis
Kruskal-Wallis test following by Bonferonni post hoc
test was performed to compare overall differences in sHER3 levels
in T-stages and migration assay results. The multiple comparisons'
criterion was employed to identify differences between groups.
P<0.05 were considered to indicate a significant difference
(two-tailed). Kaplan-Meier survival curves were used to estimate
the survival rate of the patients. The log-rank test was used to
compare the survivals. The significance of various variables for
survival was analysed by the Cox proportional hazards model in the
multivariate analysis. Statistical analyses were performed using
SPSS software version 23.0 (IBM Corp., Armonk, NY, USA). Graphs and
figures were made using GraphPad Prism software version 7.0
(GraphPad Software, Inc., La Jolla, CA, USA).
Results
ELISA assay
An ELISA was developed for HER3 employing commercial
antibodies (Fig. 1). To determine
which isoform was detected by this novel ELISA, western blotting
was performed using capture and detection antibodies in cell
lysate, and conditioned media samples obtained from WM266 and DU145
cell lines. Results from the conditioned medium experiments
(Fig. 1C; lane 3 and 4) demonstrated
that both cell lines expressed the 85 kDa protein. However, a faint
band at 45 kDa was also detected in conditioned media, but not by
the capture antibody. Full length HER3 was detected by the
detection antibody only in cell lysate (Fig. 1C; Lane 1 and 3). Therefore, based on
these results, it was concluded that this ELISA detects the 85 kDa
isoform of the HER3 receptor. The WM266 cell line has been used
previously to investigate full length HER3 via western blotting
with a different antibody compared with that used in this study and
in agreement with the results of the present study they also
demonstrated that HER3 is expressed in these cells (13).
sHER3 levels in bladder cancer
patients
ELISA was used to measure sHER3 in 82 patients with
bladder cancer. Patient characteristics and sHER3 levels according
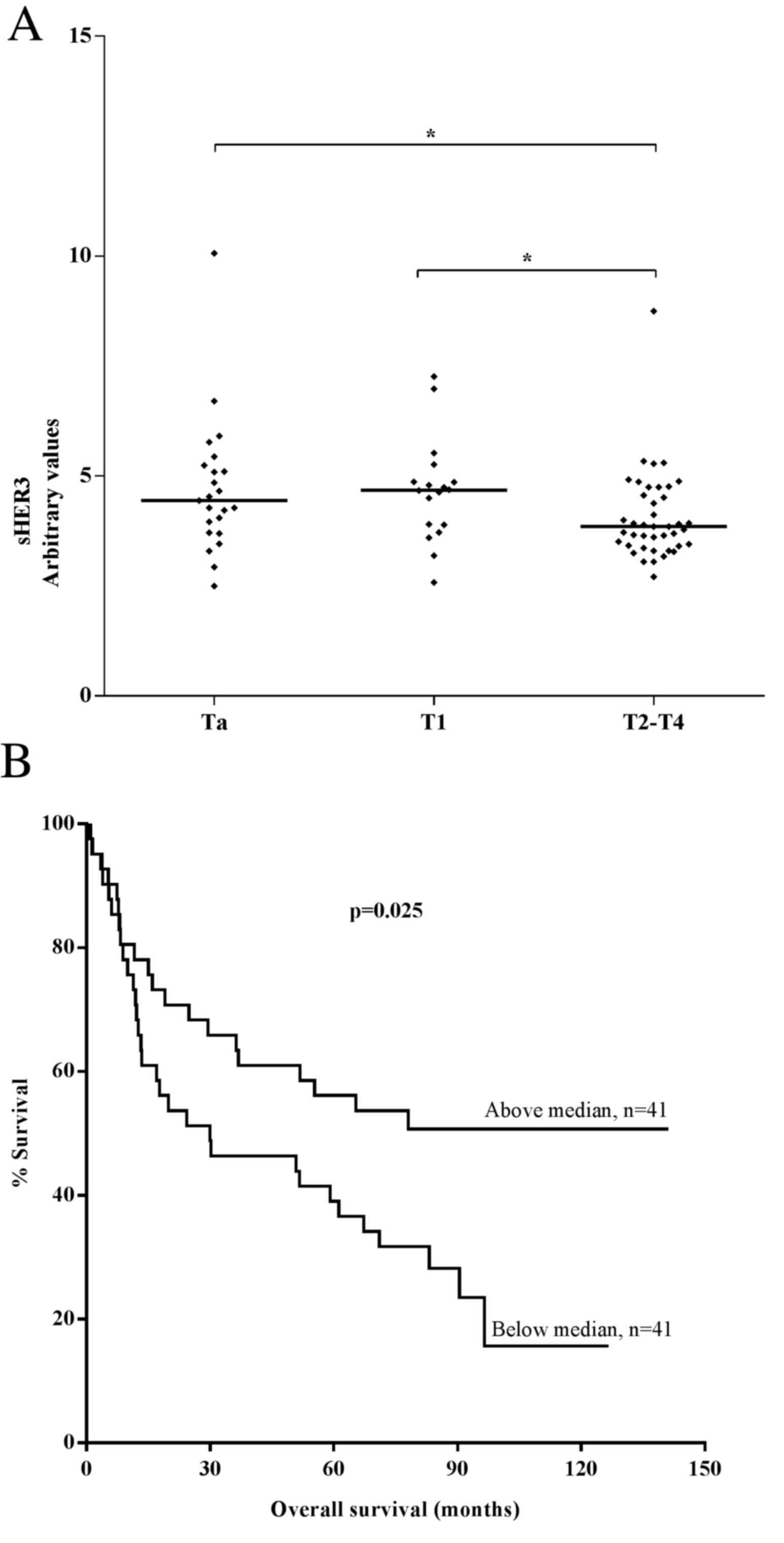
to tumour stages of bladder cancer are presented in Table I. Plasma sHER3 levels were
significantly higher in patients with superficial tumours compared
with in those with superficial invasive and muscle-invasive tumours
(P<0.05 for Ta vs. T2-T4; Fig.
2A). No significant differences in sHER3 levels were identified
between patients with Ta and T1 cancer (P=0.80).
 | Table I.Baseline data and distribution of
sHER3 according to T stage of bladder cancer. |
Table I.
Baseline data and distribution of
sHER3 according to T stage of bladder cancer.
| T stage | Ta | T1 | T2-4 |
|---|
| Patients, n (%) | 23 (28) | 18 (22) | 41 (50) |
| Sex, n (%) |
|
|
|
| Male | 15 (65) | 16 (89) | 35 (85) |
|
Female | 8
(35) | 2
(11) | 6
(15) |
| Age (years) |
|
|
|
|
Median | 69 | 74 | 68 |
|
Range | 53–88 | 60–88 | 54–83 |
| sHER3, n (%) |
|
|
|
|
≤Median | 8
(35) | 6
(33) | 27 (66) |
|
>Median | 15 (65) | 12 (67) | 14 (34) |
Plasma sHER3 levels were dichotomised as ‘high’
(above median, >4.0 arbitrary units) and ‘low’ (median or below,
≤4.0 arbitrary units) for all subjects. Patients with high sHER3
levels demonstrated significantly longer overall survival times
compared with those with low sHER3 levels in the Kaplan-Meier
analysis (P=0.025; log-rank test; Fig.
2B). However, multivariate logistic regression analysis of the
data revealed that sHER3 levels were not an independent factor for
survival (data not shown). The clinical stage and the size of the
tumour were identified as independent predictors of survival (data
not shown).
Role of sHER3 in bladder cancer
cells
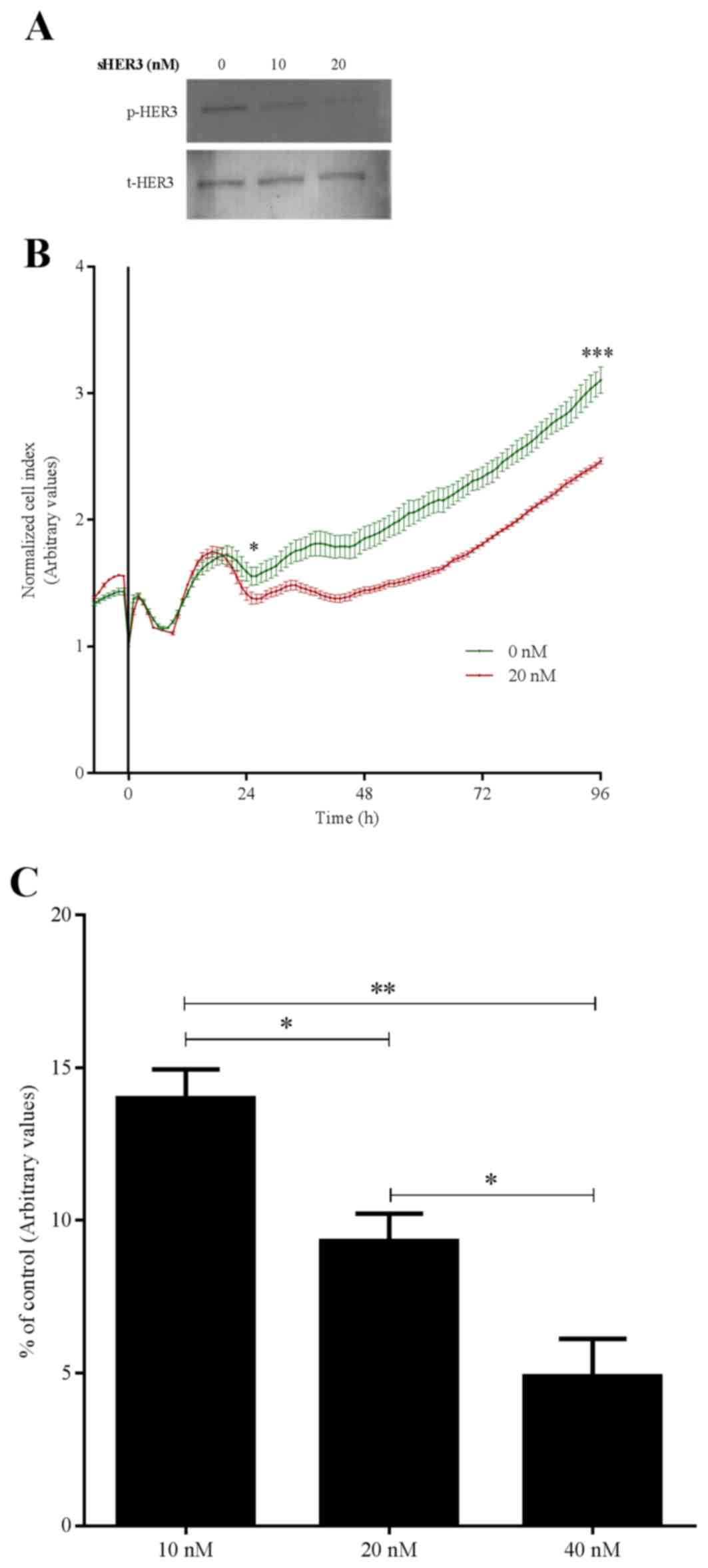
To investigate possible mechanisms of increased
survival in patients with higher sHER3 levels, HCV-29 cells were
treated with a recombinant sHER3 peptide. Peptide treatment
resulted in a dose-dependent reduction in the phosphorylation of
full-length HER3 (Fig. 3A). The time
point 24 h was selected based on our previous experiments on EGFRs,
where it was evident that compared with chemical inhibitors,
including drugs targeting the EGF receptors, other types of
inhibitors, including calcium (14)
and as in this study (HER3 peptide) may take longer to affect the
inhibition of HER3 phosphorylation. Furthermore, western blotting
clearly demonstrated that HER3 phosphorylation was partially
inhibited at lower concentrations (10 nM) and it was only with
higher concentrations (20 nM) that a significant inhibition was
observed even after 24 h. Therefore, it was suggested that at an
earlier point, no inhibition with 10 nM HER3 peptide may have been
observed. Furthermore, the real-time proliferation assay revealed
differences in cell growth from after 24 h treatment of sHER3
peptide. This inhibition of HER3 was accompanied by decreased
growth (Fig. 3B) and migration
(Fig. 3C) of HCV-29 cells.
Discussion
We previously revealed a dual role of HER in bladder
cancer (6), but the role of sHER3 in
patients with bladder cancer remains unclear. In the present study,
an ELISA assay was developed to determine the levels of circulating
sHER3 and investigate its role in bladder cancer. The results
demonstrated that sHER3 is associated with a longer survival time
of patients with bladder cancer, and that it inhibits the growth
and migration of bladder cancer cells in vitro.
Transcripts of the extracellular domain of sHER3 of
various lengths have been previously reported (4). Although it is not clear whether all of
these transcripts are translated into proteins, p45-HER3 and
p85-HER3 are well-characterised naturally occurring secreted
products of the ErbB3 gene (5). A cell culture study demonstrated that
p45-HER3 and p85-HER3 inhibits ligand-dependent stimulation of HER3
tyrosine phosphorylation, and that p85-HER3 is a more potent
inhibitor of HER3 phosphorylation compared with p45-HER3 (5). The ELISA protocol used in the present
study was hypothesised to detect the various forms of sHER3 likely
to be present in the samples; however, western blotting analysis
using conditioned media, and capture and detection antibodies only
detected the 85 kDa form of HER3. However, additional controls,
including the use of recombinant protein for competition
experiments and verification of the protein identity by mass
spectrometry, are required to validate these results.
It was revealed that significantly higher levels of
sHER3 were present in early-stage (Ta-T1) bladder cancers compared
with in late-stage (T2-T4) bladder cancer. Furthermore, higher
levels of sHER3 were associated with a longer overall survival
time, suggesting a protective role for sHER3 in bladder cancer. To
the best of our knowledge, no previous studies have analysed the
role of sHER3 in patients with bladder cancer. In agreement with
the findings in the current study, a study analysing the role of 45
kDa sHER3 in prostate cancer reported that higher levels of sHER3
were associated with longer progression-free survival times
(15). However, another study
suggests that sHER3 increased the invasiveness of PC-3 prostate
cancer cells (16). Previously, using
same cohort, we demonstrated that a higher expression of the full
length HER3 was significantly associated with a more improved
survival rate of patients with bladder cancer (6,7). Taken
together, these results indicate that the full length and soluble
forms of the HER3 receptor are associated with a more improved
survival rate of patients with bladder cancer. However, further
studies on a larger cohort and on other isoforms of the soluble
HER3 are required to confirm these results.
To understand the role of sHER3 in bladder cancer,
the effects of recombinant sHER3 were also investigated on bladder
cancer cell growth and migration. Incubation of HCV-29 bladder
cancer cells with recombinant sHER3 inhibited tyrosine
phosphorylation of HER3. Cell growth and migration analyses
revealed a significant decrease in the growth, and migration of
HCV-29 cells treated with recombinant sHER3 compared with untreated
cells. A previous study on sHER3 reported that it binds to
heregulin (HRG), a ligand for HER3 and HER4, with an affinity
equivalent to the affinity of HRG for full-length HER3 (5). This binding can effectively limit
binding of HRG to its receptors on the cell surface (5). Furthermore, exposure of cells to high
levels of sHER3 inhibited HRG-mediated activation of other HERs
(HER2 and HER4) and thereby inhibited HRG-stimulated cell growth
(5). Together these results suggest
that sHER3 may be a negative regulator of HER3-mediated
signalling.
In conclusion, the results of the current study
suggest that higher levels of sHER3 are associated with a longer
survival time of patients with bladder cancer, and that sHER3
decreases the growth and migration abilities of bladder cancer
cells. These results identify sHER3 as a candidate biomarker in
patients with bladder cancer and provide a foundation for further
studies to explore the possible use of sHER3 in bladder cancer
treatment.
Acknowledgements
The present study was supported by the Danish
Research Medical Council, Danish Cancer Society (grant no.
DP08162), Danish Cancer Foundation and Novo Nordisk Foundation
awarded to Dr Ashfaque Memon. ALF funding from Region Skåne and
Swedish Research Medical Council awarded to Jan Sundquist (grant
no. 2012-2378) and Kristina Sundquist (grant no. 2014-2517) and the
Swedish Freemasons Foundation awarded to Jan Sundquist.
References
|
1
|
Droller MJ: Bladder cancer:
State-of-the-art care. CA Cancer J Clin. 48:269–284. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Riese DJ II, van Raaij TM, Plowman GD,
Andrews GC and Stern DF: The cellular response to neuregulins is
governed by complex interactions of the erbB receptor family. Mol
Cell Biol. 15:5770–5776. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee H and Maihle NJ: Isolation and
characterization of four alternate c-erbB3 transcripts expressed in
ovarian carcinoma-derived cell lines and normal human tissues.
Oncogene. 16:3243–3252. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee H, Akita RW, Sliwkowski MX and Maihle
NJ: A naturally occurring secreted human ErbB3 receptor isoform
inhibits heregulin-stimulated activation of ErbB2, ErbB3 and ErbB4.
Cancer Res. 61:4467–4473. 2001.PubMed/NCBI
|
|
6
|
Memon AA, Sorensen BS, Meldgaard P, Fokdal
L, Thykjaer T and Nexo E: The relation between survival and
expression of HER1 and HER2 depends on the expression of HER3 and
HER4: A study in bladder cancer patients. Br J Cancer.
94:1703–1709. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Memon AA, Sorensen BS, Melgard P, Fokdal
L, Thykjaer T and Nexo E: Expression of HER3, HER4 and their ligand
heregulin-4 is associated with better survival in bladder cancer
patients. Br J Cancer. 91:2034–2041. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nielsen TO, Borre M, Nexo E and Sorensen
BS: Co-expression of HER3 and MUC1 is associated with a favourable
prognosis in patients with bladder cancer. BJU Int. 115:163–165.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fisher C: TNM classification of malignant
tumours. J Clin Pathol. 51:84–85. 1998.
|
|
10
|
Yin H and Leong AS: Histologic grading of
noninvasive papillary urothelial tumors: Validation of the 1998
WHO/ISUP system by immunophenotyping and follow-up. Am J Clin
Pathol. 121:679–687. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Munk M, Memon A, Poulsen SS, Borre M, Nexo
E and Sorensen BS: The HER4 isoform JM-a/CYT2 relates to improved
survival in bladder cancer patients but only if the estrogen
receptor α is not expressed. Scand J Clin Lab Invest. 73:503–513.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schooler K and Wiley HS: Ratiometric assay
of epidermal growth factor receptor tyrosine kinase activation.
Anal Biochem. 277:135–142. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Atienza JM, Zhu J, Wang X, Xu X and Abassi
Y: Dynamic monitoring of cell adhesion and spreading on
microelectronic sensor arrays. J Biomol Screen. 10:795–805. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fattore L, Marra E, Pisanu ME, Noto A, de
Vitis C, Belleudi F, Aurisicchio L, Mancini R, Torrisi MR, Ascierto
PA and Ciliberto G: Activation of an early feedback survival loop
involving phospho-ErbB3 is a general response of melanoma cells to
RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J
Transl Med. 11:1802013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Memon AA, Munk M, Nexo E and Sorensen BS:
Calcium-induced apoptosis is delayed by HER1 receptor signalling
through the Akt and PLCγ pathways in bladder cancer cells. Scand J
Clin Lab Invest. 71:45–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin SH, Lee YC, Choueiri MB, Wen S, Mathew
P, Ye X, Do KA, Navone NM, Kim J, Tu SM, et al: Soluble ErbB3
levels in bone marrow and plasma of men with prostate cancer. Clin
Cancer Res. 14:3729–3736. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen N, Ye XC, Chu K, Navone NM, Sage EH,
Yu-Lee LY, Logothetis CJ and Lin SH: A secreted isoform of ErbB3
promotes osteonectin expression in bone and enhances the
invasiveness of prostate cancer cells. Cancer Res. 67:6544–6548.
2007. View Article : Google Scholar : PubMed/NCBI
|