Introduction
Renal cell carcinoma (RCC) was the third most common
urological cancer worldwide in 2010 (1). Despite the use of multimodal therapy
(chemotherapy, radiation therapy and surgery), the long-term
disease-free survival rate for patients with RCC remains low
(2). Sunitinib is considered the
standard of care for the first-line therapy of advanced clear cell
RCC (3,4). However, the majority of patients
eventually develop resistance to sunitinib therapy, resulting in
therapy failure. To improve the therapy efficiency of sunitinib,
additional studies are currently being undertaken, including trials
of sunitinib in combination with chemotherapy or molecular targeted
agents (5). Although a number of
combination therapies with sunitinib are being tested in clinical
trials, there are further novel chemotherapy combinations to be
explored (6).
Chloroquine (CQ) has previously been considered as a
potential anti-cancer agent and a chemo-sensitizer in combination
with anti-cancer drugs; it has been demonstrated to inhibit cell
growth and/or induce cell death in various types of cancer
(7,8).
At present, sunitinib-based adjuvant chemotherapy in combination
with CQ for the treatment of RCC is in a phase I clinical trial
(9). However, the understanding of
its antitumor effect and mechanism remain incomplete.
Autophagy is the self-digestive process of the
lysosomal degradation of mature proteins and organelles to maintain
cellular homeostasis (10). A number
of antineoplastic therapies have been observed to induce autophagy
in human cancer cell lines, and autophagy induced by chemotherapy
is considered a mechanism of resistance to therapy-mediated cell
death (11,12). CQ may act as an autophagy inhibitor by
interfering with lysosomal acidification to block the autophagic
process at the final step, which may enhance the antitumor effect
of chemotherapy and induce cell apoptosis (13). Based on these observations, we
hypothesized that sunitinib may induce autophagy in RCC, and CQ may
enhance its antitumor effect by inhibiting autophagy. To the best
of our knowledge, there are no studies concerning the autophagic
potency of sunitinib on RCC and the molecular mechanisms of the
potential synergistic effect of sunitinib and CQ.
In the present study, the combination efficiency of
sunitinib with CQ was investigated in vitro and in
vivo, and the underlying mechanism of their synergistic effect
was examined. These results demonstrated that CQ may enhance the
antitumor effect of sunitinib by inhibiting the autophagy induced
by sunitinib, and enhance the rate of apoptosis, which may be a
promising strategy for adjuvant chemotherapy in RCC.
Materials and methods
Cell culture
The OS-RC-2 human RCC cell line was purchased from
the Cell Bank of Type Culture Collection of Chinese Academy of
Sciences (Shanghai, China). The cells were cultured in RPMI-1640
media containing 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and 1% penicillin and
streptomycin in humidified conditions with 5% CO2 at
37°C.
Materials
Sunitinib, CQ and all fluorescent dyes were
purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The
primary antibodies against Bcl-2 (sc-492), Bax (sc-70407) and
β-actin (sc-47778) were purchased from Santa Cruz Biotechnology,
Inc (Dallas, TX, USA). Primary antibodies against Beclin-1
(ab62557), autophagy related 5/12 (Atg5/12; ab78073) were purchased
from Abcam (Cambridge, MA, USA). Primary antibodies against light
chain 3 (LC3) I/II (no. 4108), p53 (no. 9282), caspase-3 (no.
9662), caspase-9 (no. 9508), phosphorylation of histone H3 (no.
9711), phosphoinositide 3-kinase (PI3K; no. 4255), Akt (no. 4685),
phosphorylated (p)-Akt (no. 4058), tuberous sclerosis complex 2
(TSC2; no. 4308), p-TSC2 (no. 3617), p-mechanistic target of
rapamycin (p-mTOR; no. 5536) and mTOR (no. 4517), p70 ribosomal S6
kinase (p70S6K; no. 2708) and p-p70S6K (no. 9204) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell viability assay
The cell viability was measured using an MTT assay,
as previously described (14).
Briefly, cells were seeded at 3–4×103 cells/well in a
96-well plate, and treated with sunitinib and CQ for 48 h. Then, 20
µl MTT solutions (5 mg/ml) were added to each well for an
additional 2 h at 37°C. Following removal of the culture medium,
dimethyl sulfoxide was added (200 µl/well) and the optical density
was measured at 570 nm with a microplate reader.
Colony formation assay
A total of 1×103 cells/well were seeded
in 6-well plates and treated with sunitinib and 25 µM CQ.
Subsequent to an additional incubation for 14 days, the cells were
stained with 0.2% crystal violet for 10 min at room temperature,
and the number of colonies in each well were counted.
Electron microscopy
Electron microscopy was performed to detect the
induction of autophagic morphology. Following treatment with 25 µM
CQ and 10 µM sunitinib for 48 h, cells were fixed with a solution
containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1 M
cacodylate buffer (pH 7.3) for 60 min. The cells were then
post-fixed in 1% OsO4 buffer for 1 h on ice and
subjected to transmission electron microscopy analysis at room
temperature for 3 h, as previously described (15).
Detection of acidic vesicular
organelles
A total of 1×105 cells/well were plated
in 6-well plates. Following treatment with 25 µM CQ and 10 µM
sunitinib for 48 h, the cells were stained with 1 µg/ml acridine
orange for 15 min, washed with PBS and examined under a
fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Determination of mean red:green
fluorescence ratio with acridine
Cells (1×105 cells/well) were stained
with 0.5 mg/l acridine orange for 10 min, removed from the plate
with trypsin-EDTA, and collected in phenol red-free growth medium.
The number of red-fluorescing (650 nm) cells were measured with a
FACSCalibur flow cytometer (BD Biosciences, San Diego, CA,
USA).
Monodansylcadaverine (MDC)
staining
Following treatment with 25 µM CQ and 10 µM
sunitinib for 48 h, the culture medium was replaced with fresh
medium containing 0.05 mM MDC and the cells were incubated at 37°C
for 30 min in the dark. The cells were then washed three times with
PBS and images were captured with fluorescence microscopy.
Transient transfection
OS-RC-2 cells were plated at a density of
1×105 on a coverslip and cultured until they reached 60%
confluence. Then, the cells were transfected with an enhanced green
fluorescent protein-LC3 (pEGFP-LC3) plasmid using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. When cells reached 90% confluence, drugs
were added into the culture medium. GFP-LC3 fluorescence was then
observed with fluorescence microscopy.
Immunofluorescence analysis
Following treatment with 25 µM CQ and 10 µM
sunitinib for 48 h, cells were fixed with methanol for 5 min on
ice, and then washed with PBS for 10 min. The cells were incubated
with an LC3 antibody for 1 h at 37°C, followed by incubation with a
fluorescein isothiocyanate-conjugated secondary antibody, and
staining with 1 µg/ml Hoechst 33342 for 1 h at 37°C. The change in
LC3 distribution was examined using a laser scanning confocal
microscope (Leica Microsystems GmbH).
Western blot analysis
Following treatment with 25 µM CQ and 10 µM
sunitinib for 48 h, the cells were washed with PBS and lysed in
radioimmunoprecipitation buffer [150 mM NaCl, 1.0% NP-40, 0.5%
sodium deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0), 1%
pentylmethylsulfonyl fluoride and 0.1% cocktail (CT)]. The protein
concentration in the cell lysates was measured using a Bio-Rad
protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Equal amounts of protein (100 µg) were subjected to SDS-PAGE
(10–15% gels) and transferred onto polyvinylidene difluoride
membranes. Subsequent to blocking with 5% non-fat milk at room
temperature for 1 h, incubation with the primary (1:1,000-1:2,000
dilution according to recommendations from antibodies procotol,
overnight at 4°C) and secondary antibodies: Anti-mouse
IgG/horseradish peroxidase (HRP)-conjugated (cat no. ZDR-5307;
ZSGB-BIO, Beijing, China) and anti-rabbit IgG/HRP-conjugated (cat
no. BA1055; Boster Biotechnology Co., Ltd., Wuhan, China), the
reactive band was identified using an enhanced chemiluminescence
substrate with HRP (Amersham; GE Healthcare, Chalfont, UK).
Flow cytometry (FCM) for apoptosis
detection and mitochondrial membrane potential assay
Following treatment with 25 µM CQ and 10 µM
sunitinib for 48 h, the cells were collected, suspended in 50 µg/ml
propidium iodide for 10 min and analyzed using a FACSCalibur flow
cytometer. Additionally, the mitochondrial membrane potential (Δψm)
and the mitochondrial permeability transition were determined by
the retention of Rh123 dye. The cells were incubated with Rh123 (5
g/ml) at 37°C for 30 min, in the dark. Following two washes, the
cells were analyzed with FCM or observed under an inverted
fluorescence microscope (Leica Microsystems GmbH).
In vivo study
Female BALB/c nude mice (6–8 weeks old, 18–22 g;
Beijing Animal Center, Beijing, China) were used in the present
study. The total numbers of mice used in the present study was 160.
The mice were maintained under controlled conditions at 21°C, 55%
humidity, on a 12 h light/dark cycle and had food and water
available ad libitum. The protocol was approved by the Animal
Experimental Ethics Committee of the State Key Laboratory of
Biotherapy, Sichuan University (Sichuan, China). OS-RC-2 cells
(5×106) were subcutaneously injected into the hind flank
of the mice. When the volume of the tumors reached 100
mm3, the mice were randomized into the vehicle control
or treatment groups. Mice were administered with CQ (20 mg/kg/day)
intravenously, sunitinib (20 or 40 mg/kg/day) orally or
co-treatment with CQ and sunitinib for 28 days. Tumor growth and
body weight were measured every 3 days during the treatment. Tumor
volume was calculated using the following formula: Tumor volume
(mm3)=0.52 xaxb2, where
a is the length and b is the width.
Terminal dexynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
A TUNEL assay was performed to measure cellular
apoptosis in vivo, according to the manufacturer's protocol
(Promega Corporation, Madison, WI, USA). The TUNEL-positive cells
were identified using a laser scanning confocal microscope (Leica
Microsystems GmbH).
Immunohistochemistry analysis
The phosphorylation of histone H3 and LC3 were
detected using immunohistochemistry analysis. Paraffin sections
(4-µm thick) of the tumor tissue were incubated overnight at 4°C
with phosphorylation of histone H3 (1:50) and LC3 polyclonal
antibodies (1:100), followed by incubation at 4°C for 2 h with a
biotinylated secondary antibody (HRP-anti rabbit IgG; cat no.
BA1055; 1:300; Boster Biotechnology Co., Ltd., Wuhan, China) for 1
h. Diaminobenzidine was used to visualize the staining.
Statistical analysis
All results were expressed as the means ± standard
deviation. Statistical analysis was performed using SPSS v.13.0
software (SPSS, Inc., Chicago, IL, USA). A post hoc test with
GraphPad Prism 5 (San Diego, California) analysis of variance was
used for the comparison of data. P<0.05 was considered to
indicate a statistically significant difference.
Results
Inhibitory effects of sunitinib on
cell proliferation are enhanced by CQ treatment in RCC cell
lines
In order to investigate the proliferation inhibitory
properties of sunitinib and CQ, the cell viability was determined
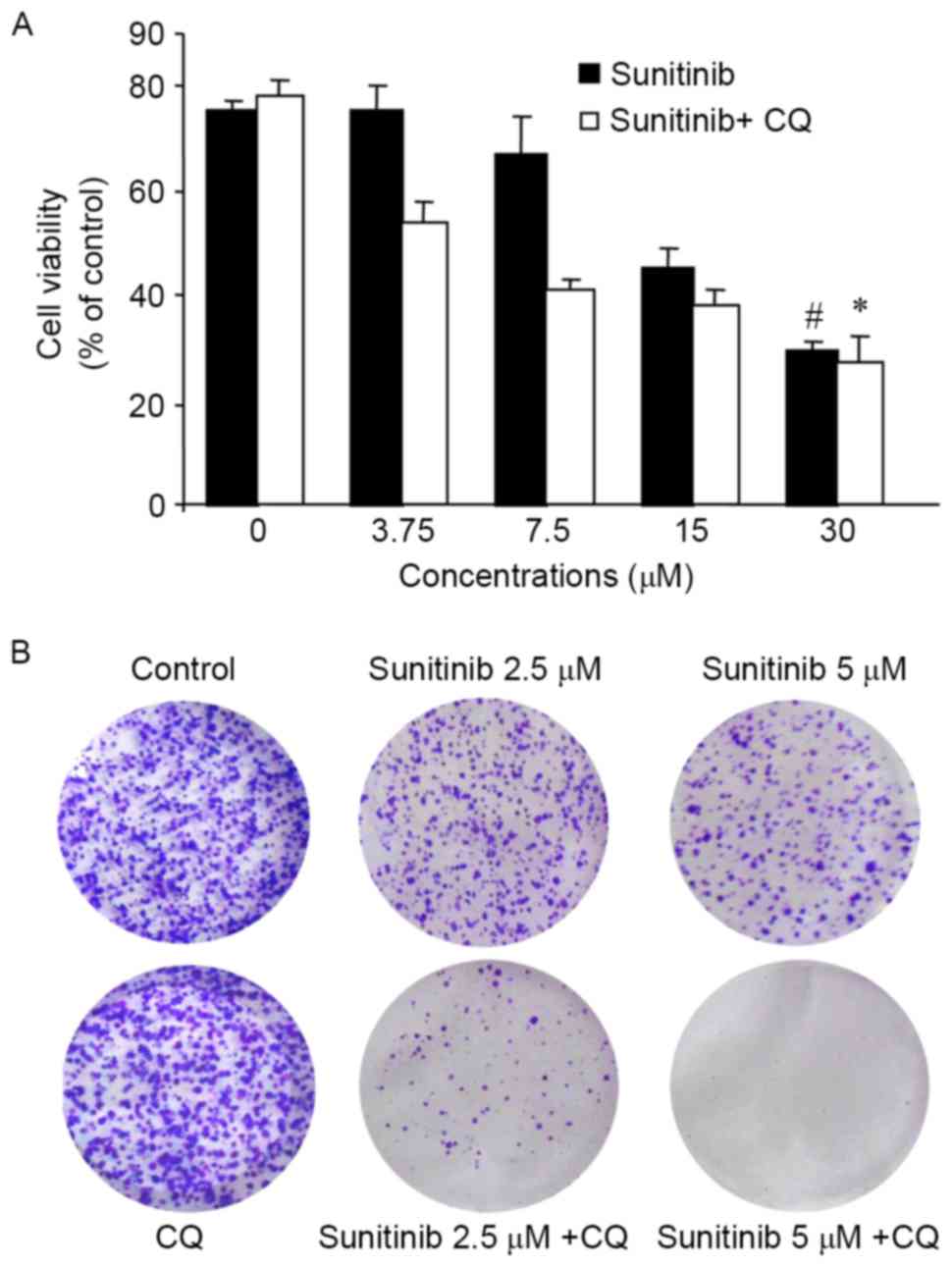
using an MTT assay. As demonstrated in Fig. 1A, OS-RC-2 cell growth was effectively
inhibited by sunitinib in a concentration-dependent manner; CQ at
concentrations of 25 µM exhibited almost no inhibitory effect on
cell proliferation following 24 h treatment (data not shown), so
was selected for the subsequent experiments. The inhibition rates
were 25 and 35% when the cells were treated with 3.75 and 7.5 µM
sunitinib, respectively, whereas the inhibition rate increased to
34 and 58% following co-treatment.
A colony formation assay was used to further
determine whether CQ was able to enhance the proliferation
inhibitory effect. The results clearly demonstrated that CQ
significantly enhanced the inhibitory effect of sunitinib exposure
on clone formation (Fig. 1B). These
results suggested that CQ may enhance sunitinib-inhibited cell
proliferation in OS-RC-2 cells.
CQ inhibits sunitinib-induced
autophagy in OS-RC-2 cells
To examine the mechanism responsible for the
CQ-enhanced inhibitory effect of sunitinib on proliferation,
whether sunitinib may induce autophagy in OS-RC-2 cells and whether
CQ may inhibit sunitinib-induced autophagy were examined. Firstly,
transmission electron microscopy analysis was performed following
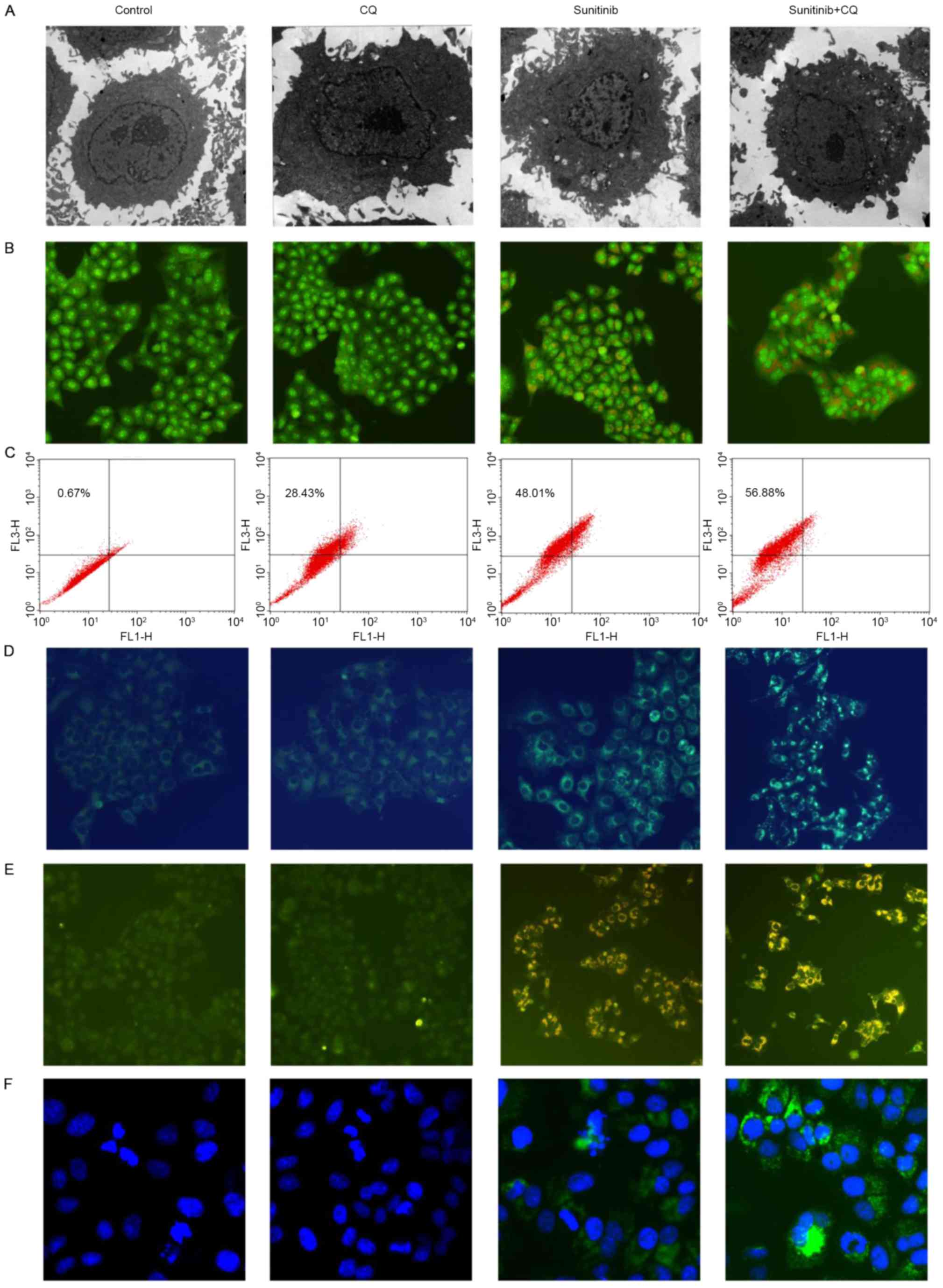
10 µM sunitinib treatment of the OS-RC-2 cells for 48 h. As
demonstrated in Fig. 2A, numerous
autophagosomes were observed in the cytoplasm of sunitinib-treated
cells, whereas the number of membrane-bound vacuoles decreased when
cells were treated with sunitinib and CQ.
To evaluate if the formation of membrane-bound
vacuoles was an autophagic response upon sunitinib treatment, the
formation of the acidic vesicular organelles (AVOs) was analyzed
with acridine orange staining. As indicated by Fig. 2B, sunitinib treatment resulted in the
formation of yellow-orange AVOs in OS-RC-2 cells. Following CQ and
sunitinib treatment, the number of yellow-orange AVOs
increased.
To further quantify the formation of AVOs, FCM
analysis was performed. The results demonstrated that the
percentage of AVOs positive cells increased to 48.0% following
sunitinib treatment, whereas it was 0.76% in control cells.
Subsequent to CQ treatment, the AVOs formation increased to 56.88%;
the results from FCM analysis were consistent with the results from
fluorescence microscopy (Fig.
2C).
MDC staining was used to further detect AVOs;
co-treatment with CQ and sunitinib increased the number of
MDC-labeled green fluorescent particles compared with sunitinib
alone (Fig. 2D). Concurrently, the
typical characteristics of autophagy were also observed; the
formation of autophagic vacuoles was confirmed by GFP-LC3
distribution (Fig. 2E). Co-treatment
increased the extent of GFP-LC3 localization induced by sunitinib.
The formation of autophagosomes was also evaluated using
immunofluorescence, and GFP puncta were detected (Fig. 2F). Taken together, these results
demonstrate that sunitinib may induce autophagy and CQ may inhibit
this effect.
Sunitinib increases the expression of
autophagy-associated proteins
In order to further confirm the sunitinib-induced
autophagy, the expressions of autophagy-associated proteins were
examined using western blot analysis. The increased conversion of
LC3-I to LC3-II is considered to be an autophagosomal marker due to
its localization and aggregation on autophagosomes. It was
demonstrated that native LC3-II accumulation was increased in
sunitinib-treated cells, and the expression of other proteins,
including Atg5-Atg12 conjugations and Beclin-1, also increased
(Fig. 3A). Concurrently, the
sunitinib-induced upregulation of autophagy-associated proteins was
increased by co-treatment.
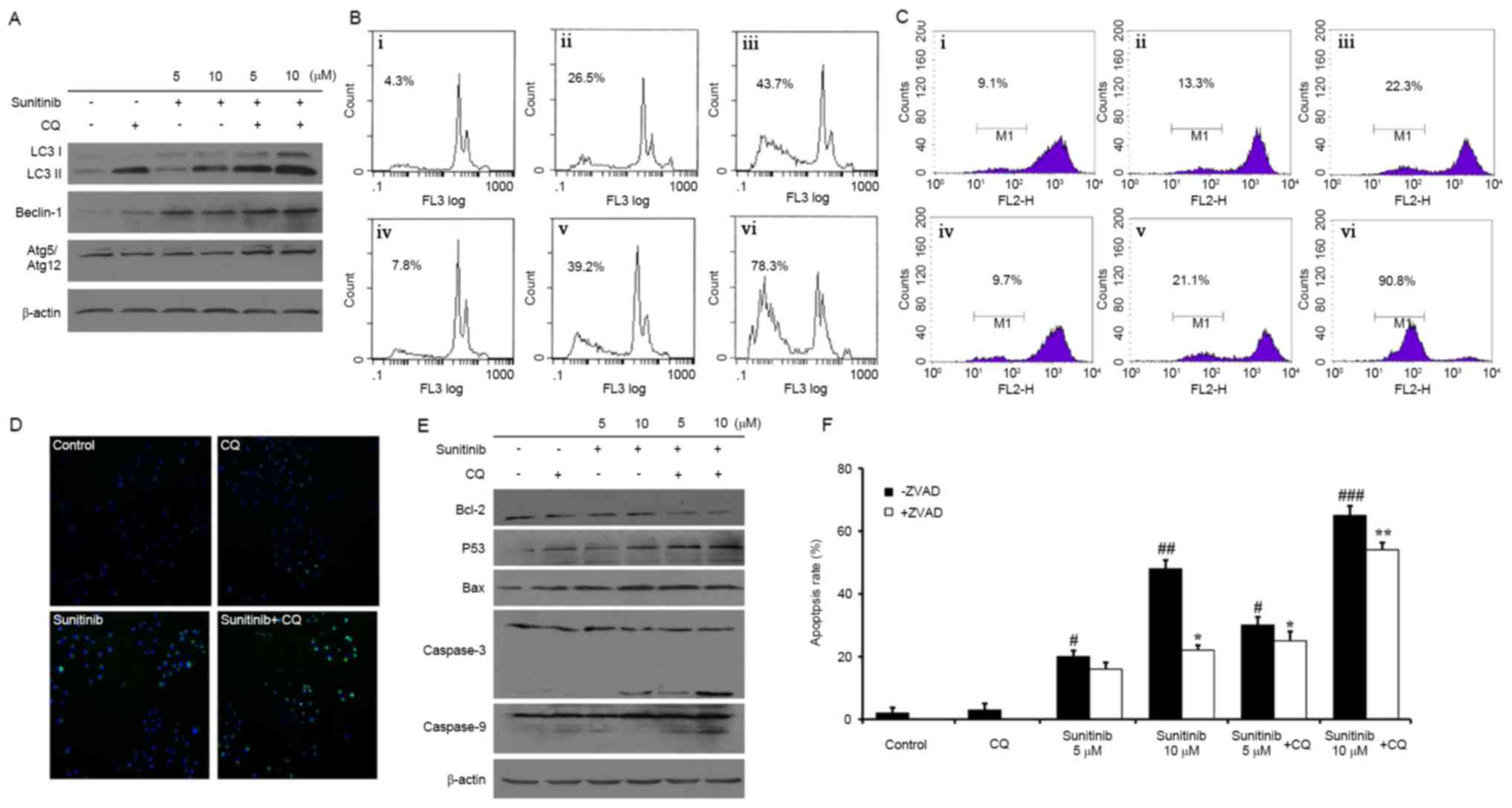 | Figure 3.Expression of autophagy-associated
proteins and sunitinib with CQ induced apoptosis in renal cancer
cells. (A) Protein expression of LC3, Atg5, and Beclin-1 in OS-RC-2
cells treated with various treatment concentrations. (B) Flow
cytometry was used to determine following propidium iodide
staining. i, Control; ii, 5 µM sunitinib; iii, 10 µM sunitinib; iv,
25 µM CQ; v, 5 µM sunitinib + CQ; vi, 10 µM sunitinib + CQ. (C)
Mitochondrial permeability transition in OS-RC-2 cells following
drug treatment. i, control; ii, 5 µM sunitinib; iii, 10 µM
sunitinib; iv, 25 µM CQ; v, 5 µM sunitinib + CQ; vi, 10 µM
sunitinib + CQ. (D) Microscopy images displaying the relative
extent of fluorescence intensity, representing the extent of
mitochondrial permeability following treatment with 10 µM sunitinib
and/or 25 µM CQ. (E) Protein expression of Bcl-2, Bax, caspase-9
and caspase-3 in OS-RC-2 cells. (F) OS-RC-2 apoptosis was
determined following treatment with 2 µM Z-VAD-FMK. All data are
representative of three independent experiments. * and
#, P<0.05; ** and ##, P<0.01;
### P<0.001; #compared with control
(-ZVAD); *compared with control (+ZVAD). CQ, chloroquine; LC3,
light chain 3; Atg5, autophagy protein 5; Bax, Bcl-2-associated
X. |
Combining sunitinib with CQ treatment
induces apoptosis
To determine whether sunitinib- and CQ-treated tumor
cells undergo apoptosis, an FCM apoptosis assay was performed. The
rate of apoptosis was 26.5 and 43.7%, respectively, when cells were
treated with 5 and 10 µM sunitinib. However, the rate of apoptosis
increased to 39.2 and 78.3%, respectively, for co-treated cells
(Fig. 3B). Therefore, it was
concluded that CQ may improve the rate of apoptosis induced by
sunitinib. Additionally, the change in Δψm was detected using an
FCM assay. The loss of Δψm was 13.3 and 22.3%, following treatment
with 5 and 10 µM sunitinib, respectively, whereas the loss of Δψm
increased to 21.1 and 90.8% subsequent to co-treatment (Fig. 3C). Similar results were obtained from
staining the mitochondria membrane (Fig.
3D). A total of >50% of the cells were intensely green
following CQ and sunitinib co-treatment.
In addition, western blot analysis revealed a
potential molecular mechanism of sunitinib-induced apoptosis as the
levels of cleaved caspase-3 and caspase-9 increased in a
concentration-dependent manner. A decrease of Bcl-2 and increases
of Bax and p53 were also observed (Fig.
3E). These phenomena were more apparent following co-treatment.
In addition, to explore whether sunitinib-induced apoptosis was
specifically associated with the caspase family, a caspase
inhibitor, Z-VAD-FMK, was administered to cells undergoing
sunitinib-induced apoptosis. As demonstrated in Fig. 3F, the apoptosis rate was 20 vs. 14%
with 5 µM sunitinib, and 48 vs. 22%, with 10 µM sunitinib, with and
without 2 µM Z-VAD-FMK, respectively. Similar results were obtained
following co-treatment with CQ. Therefore, apoptosis induced by
sunitinib may be partly reversed by a pan-caspase inhibitor,
Z-VAD-FMK.
Sunitinib induced autophagy by
inhibiting the Akt/mTOR/p70S6K signaling pathway in OS-RC-2
cells
As the Akt/mTOR/p70S6K pathway is the main
regulatory pathway that negatively regulates autophagy, whether
sunitinib and CQ treatment induced an alteration in this pathway
was examined using a western blotting assay. The results
demonstrated that sunitinib decreased the phosphorylation of Akt,
TSC2, mTOR and p70S6K, whereas the total levels of PI3K, Akt, TSC2,
mTOR and p70S6K were not altered (Fig.
4A). These results suggest that the upstream pathway of the
Akt/mTOR/p70S6K pathway was inhibited by sunitinib treatment, and
that sunitinib may induce autophagy through this pathway. In
addition, the combination of CQ and sunitinib treatment enhanced
the suppression of this pathway in a concentration-dependent
manner.
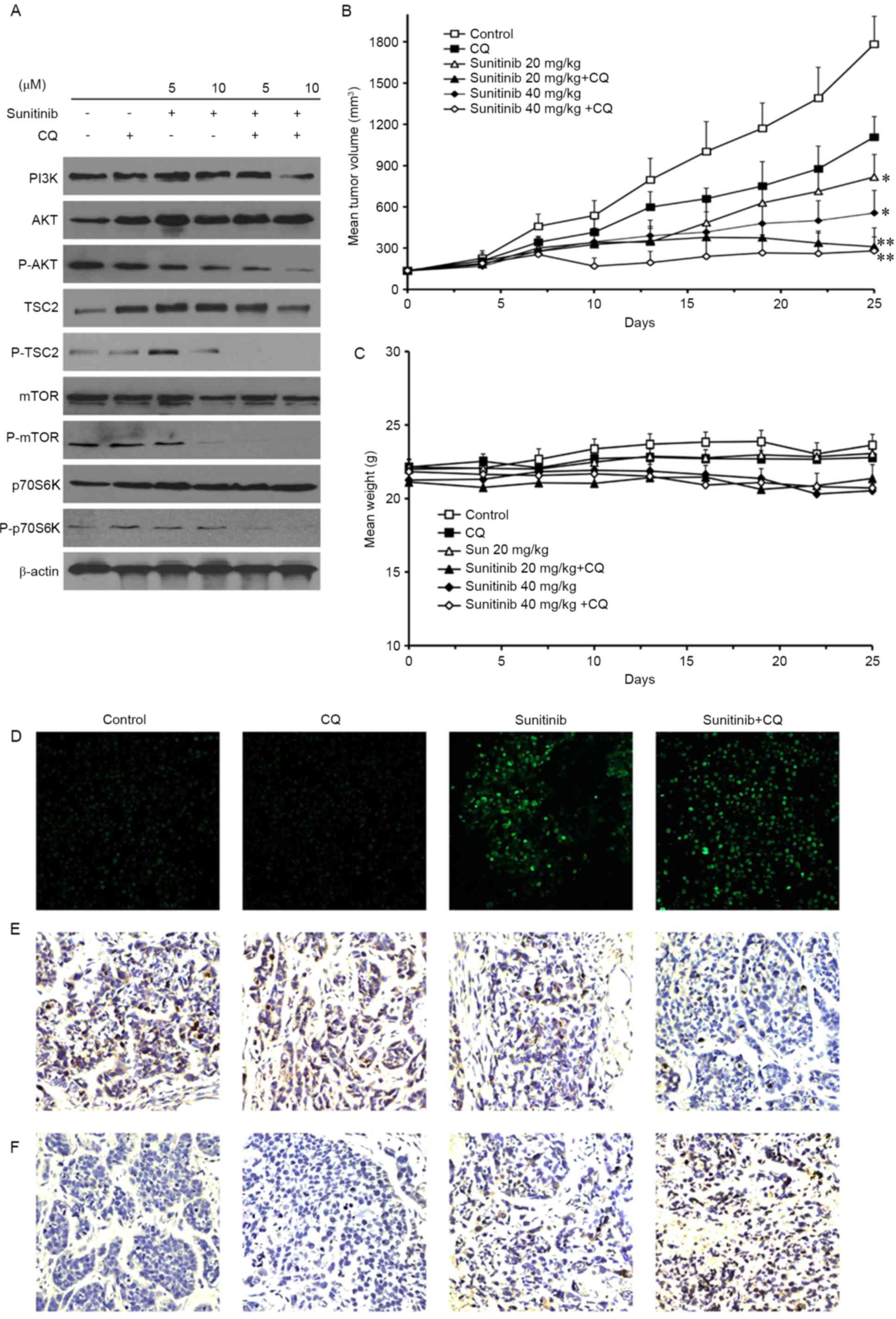 | Figure 4.Sunitinib induces autophagy by
inhibiting the Akt/mTOR/p70S6K signaling pathway, and may inhibit
the growth of OS-RC-2 tumors in vivo. (A) Protein expression
of PI3K, AKT, p-AKT, TSC2, p-TSC2, mTOR, p-mTOR, p70S6K and
p-p70S6K in OS-RC-2 cells. (B) The inhibitory effect on the
proliferation of OS-RC-2 cells established in nude BALB/c mice
following CQ (20 mg/kg/day intravenously), sunitinib (40 mg/kg/day
orally) or both. The data were expressed as the mean ± SD. (C)
Mouse body weight was not significantly altered following sunitinib
and CQ co-treatment. (D) A terminal dexynucleotidyl
transferase-mediated dUTP nick end labeling assay was performed to
measure the induction of apoptosis. (E) Tumor cell proliferation in
different groups was analyzed by the staining of phosphorylated
histone H3. (F) Tumor cell autophagy in different groups was
analyzed by LC3-II staining. Data represent the means ± SD or are
representative of ≥3 independent experiments (magnification, ×200).
*P<0.05 and **P<0.01 compared with control. PI3K,
phosphoinositide 3-kinase; p-, phosphorylated; TSC2, tuberous
sclerosis complex 2; mTOR, mechanistic target of rapamycin; p70S6K,
p70S6 kinase; CQ, chloroquine; SD, standard deviation. |
In vivo antitumor activity of combined
treatment with sunitinib and CQ
In vivo study was performed to validate the
antitumor activity of sunitinib and CQ. The results demonstrated
that the inhibition of tumor volume was 47% (20 mg/kg) and 56% (40
mg/kg) following sunitinib treatment compared with the control
group. However, the inhibition of tumor volume reached 64 and 73%,
respectively, following co-treatment with CQ (Fig. 4B). No loss of body weight was observed
during sunitinib and CQ treatment (Fig.
4C). In addition, the effect on the apoptosis and proliferation
of xenograft tumors in mice was evaluated. As demonstrated in
Fig. 4D, an increase in the levels of
nuclear green fluorescence was observed in the co-treatment group
compared with sunitinib treatment alone in the TUNEL assay. In the
in vivo proliferation assay, the appearance of brown spots,
which indicated the phosphorylation of histone H3, was reduced
following sunitinib treatment compared with the control, and
decreased further following co-treatment with sunitinib and CQ
(Fig. 4E). Therefore, these results
suggested that co-treatment enhanced cell apoptosis and inhibited
cell proliferation. The changes in autophagy in mice tumors
following drug treatment were also investigated.
Immunohistochemistry analysis revealed that the brown punctate
staining, which represented the autophagic marker of LC3-II, was
increased in the co-treatment group compared with the sunitinib
group (Fig. 4F). Taken together,
these results suggested that co-treatment may improve the antitumor
activity of sunitinib though the inhibition of autophagy.
Discussion
Sunitinib is an anticancer drug used for the
treatment of RCC, and sunitinib in combination with CQ for the
treatment of RCC is in clinical trials (16,17).
Understanding the underlying mechanism of sunitinib in combination
with CQ is important for its future use in clinical therapy. To the
best of our knowledge, the present study is the first to
demonstrate that CQ may enhance the anti-RCC effect of sunitinib by
inhibiting the autophagy induced by sunitinib.
Sunitinib is currently considered the standard of
care for the first-line therapy of RCC (18). However, the vast majority of patients
eventually develop resistance to sunitinib therapy (4,19).
Therefore, a complete understanding of the mechanisms of sunitinib
action against RCC is critical in understanding and improving the
treatment of this disease. Previously, autophagy has been
demonstrated as an adaptive mechanism that contributes to tumor
cell survival and resistance to therapy-induced apoptosis. There
are a number of pharmacological mediators that can clinically
induce autophagy. For example, temozolomide, 5-fluorouracil and
tyrosine kinase inhibitors, including imatinib and dasatinib, have
also been demonstrated to induce autophagy, which may decrease the
anticancer efficiency of anticancer drugs (20). Therefore, there is interest in
determining whether sunitinib induces autophagy in human RCC cells.
In the present study, it was demonstrated that sunitinib may induce
the formation of AVOs, GFP-LC3 punctuates and autophagosomes in
OS-RC-2 cells. In addition, pathognomonic autophagy-associated
genes, including LC3, Atg5 and BECN1, were increased following
sunitinib treatment. Based on these results, it may be concluded
that sunitinib induces autophagy in OS-RC-2 cells.
Autophagy is a temporary survival mechanism of
cancer cells to adapt to the stressful conditions caused by
anticancer therapies (12). Previous
studies have revealed that autophagy inhibitors, including
3-methyladenine and CQ, may sensitize cancer cells to chemotherapy
or radiation, and enhance the antitumor effect of anticancer drugs
(21). CQ inhibits lysosome
acidification, therefore blocking the late stages of autophagy, and
is already clinically available as an autophagy inhibitor (7). Combinations of chemotherapeutic agents
with CQ are in clinical trials against breast cancer, multiple
myeloma, prostate cancer and other types of advanced tumor
(22). The combination of sunitinib
and CQ is in phase I clinical trials against RCC (23). In the present study, in vivo
and in vitro antitumor assays demonstrated that CQ may
significantly increase the anti-RCC activity of sunitinib. In
addition, during the 28 days of treatment, CQ combined with
sunitinib was well-tolerated by the mice and no adverse effects
were observed. To gain insight into the molecular mechanism, the
changes in autophagy were investigated. The results of the present
study demonstrated that CQ increased the formation of AVOs, GFP-LC3
puncta and autophagosomes compared with sunitinib treatment alone
in OS-RC-2 cells. The LC3, Atg5 and Beclin-1 protein expression
increased following the co-treatment with sunitinib and CQ. Thus,
it may be concluded that the autophagy induced by sunitinib was
inhibited by CQ.
Autophagy is considered a survival mechanism in
sunitinib-treated cells (24). The
suppression of autophagy leads to apoptosis, thus enhancing the
antitumor effect. The loss of Δψm is one of the hallmarks of
apoptosis. In the mitochondrial-mediated apoptotic pathway, caspase
family proteins caspase-3 and caspase-9 serve a central role.
Co-treatment with sunitinib and CQ increased the levels of
anti-apoptotic Bcl-2 and p53, and decreased the pro-apoptotic Bax
protein expression. This result is in accord with a previous study,
which reported that CQ may improve dasatinib-induced apoptosis
(20). At present, the precise
molecular mechanism that links autophagy and apoptosis is not
clear. Autophagy and apoptosis may be induced in response to
different cellular stresses, and the induction of
autophagy/apoptosis may occur sequentially, simultaneously or in a
mutually exclusive manner (25). The
PI3K-Akt-mTOR signaling pathway is an important negative regulator
of autophagy (26). In the present
study, it was demonstrated that the phosphorylation of Akt, mTOR,
TSC2 and p70S6K was decreased following sunitinib treatment,
suggesting that sunitinib inhibited Akt-mTOR signaling and induced
autophagy. However, according to the results of the present study,
CQ also inhibited this pathway, which suggests that it may be
associated with anti-tumor activity. In addition, apoptosis and
autophagy are not mutually exclusive pathways; they have been
observed to act in synergy or in opposition to each other. They
share a number of the same molecular regulators. Therefore, the
present study indicated that sunitinib may induce autophagy in
vivo and in vitro as a defense mechanism.
To conclude, to the best of our knowledge, the
present study demonstrated for the first time that CQ may enhance
the anti-RCC effect of sunitinib by inhibiting autophagy, as well
as enhancing the rate of apoptosis and inhibiting cell
proliferation. A combination therapy of sunitinib with CQ may be a
promising strategy for adjuvant chemotherapy in RCC.
Acknowledgements
The present study was supported by the National
S&T Major project (grant nos. 2011ZX09102-001-013 and
2012ZX09501001-003), the Project of the National Natural Sciences
Foundation of China (grant nos. 81272459 and 81402947), the Natural
Sciences Foundation of Anhui Province (grant no. 1508085QH162) and
the Grants for Scientific Research of BSKY from Anhui Medical
University (grant nos XJ201315 and XJ201509).
Glossary
Abbreviations
Abbreviations:
|
RCC
|
renal cell carcinoma
|
|
CQ
|
chloroquine
|
|
Δψm
|
mitochondrial membrane potential
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling
|
|
AVOs
|
acidic vesicular organelles
|
|
FCM
|
flow cytometry
|
References
|
1
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu K, Ding Q, Fang Z, Zheng J, Gao P, Lu Y
and Zhang Y: Silencing of HIF-1alpha suppresses tumorigenicity of
renal cell carcinoma through induction of apoptosis. Cancer Gene
Ther. 17:212–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao Y, Xue T, Yang X, Zhu H, Ding X, Lou
L, Lu W, Yang B and He Q: Autophagy plays an important role in
sunitinib-mediated cell death in H9c2 cardiac muscle cells. Toxicol
App Pharmacol. 248:20–27. 2010. View Article : Google Scholar
|
|
4
|
Motzer RJ, Rini BI, Bukowski RM, Curti BD,
George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G,
et al: Sunitinib in patients with metastatic renal cell carcinoma.
JAMA. 295:2516–2524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chairatvit K and Ngamkitidechakul C:
Control of cell proliferation via elevated NEDD8 conjugation in
oral squamous cell carcinoma. Mol Cell Biochem. 306:163–169. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bello CL, Sherman L, Zhou J, Verkh L,
Smeraglia J, Mount J and Klamerus KJ: Effect of food on the
pharmacokinetics of sunitinib malate (SU11248), a multi-targeted
receptor tyrosine kinase inhibitor: Results from a phase I study in
healthy subjects. Anticancer Drugs. 17:353–358. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sasaki K, Tsuno NH, Sunami E, Tsurita G,
Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al:
Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on
colon cancer cells. BMC Cancer. 10:3702010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe cancer
therapies. Euro J Pharmacol. 625:220–233. 2009. View Article : Google Scholar
|
|
9
|
Flanigan RC, Campbell SC, Clark JI and
Picken MM: Metastatic renal cell carcinoma. Curr Trea Option Oncol.
4:385–390. 2003. View Article : Google Scholar
|
|
10
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ng G and Huang J: The significance of
autophagy in cancer. Mol Carcinogen. 43:183–187. 2005. View Article : Google Scholar
|
|
12
|
Notte A, Leclere L and Michiels C:
Autophagy as a mediator of chemotherapy-induced cell death in
cancer. Biochem Pharmacol. 82:427–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carew JS, Nawrocki ST and Cleveland JL:
Modulating autophagy for therapeutic benefit. Autophagy. 3:464–467.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu Y, Lu W, Yang P, Peng W, Wang C, Li M,
Li Y, Li G, Meng N, Lin H, et al: A small molecular agent YL529
inhibits VEGF-D-induced lymphangiogenesis and metastasis in
preclinical tumor models in addition to its known antitumor
activities. BMC Cancer. 15:5252015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Asoro MA, Kovar D and Ferreira PJ: In situ
transmission electron microscopy observations of sublimation in
silver nanoparticles. ACS Nano. 9:7844–7852. 2013. View Article : Google Scholar
|
|
16
|
Nanus DM, Garino A, Milowsky MI, Larkin M
and Dutcher JP: Active chemotherapy for sarcomatoid and rapidly
progressing renal cell carcinoma. Cancer. 101:1545–1551. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sunkara U, Walczak JR, Summerson L, Rogers
T, Eisenberger M, Denmeade S, Pili R, Huff CA, Sinibaldi V and
Carducci MA: A phase II trial of temozolomide and IFN-alpha in
patients with advanced renal cell carcinoma. J Interf Cytok Res.
24:37–41. 2004. View Article : Google Scholar
|
|
18
|
Czarnecka AM, Szczylik C and Rini B: The
use of sunitinib in renal cell carcinoma: Where are we now? Expert
Rev Anticancer Ther. 14:983–999. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili
R, Bjarnason GA, et al: Overall survival and updated results for
sunitinib compared with interferon alfa in patients with metastatic
renal cell carcinoma. J Clin Oncol. 27:3584–3590. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Milano V, Piao Y, LaFortune T and de Groot
J: Dasatinib-induced autophagy is enhanced in combination with
temozolomide in glioma. Mol Cancer ther. 8:394–406. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Hou N, Faried A, Tsutsumi S and
Kuwano H: Inhibition of autophagy augments 5-fluorouracil
chemotherapy in human colon cancer in vitro and in
vivo model. Euro J Cancer. 46:1900–1909. 2010. View Article : Google Scholar
|
|
22
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Levy JM and Thorburn A: Targeting
autophagy during cancer therapy to improve clinical outcomes.
Pharmacol Ther. 131:130–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu J, Fan L, Wang H and Sun G: Autophagy,
a double-edged sword in anti-angiogenesis therapy. Med Oncol.
33:102016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bhutia SK, Dash R, Das SK, Azab B, Su ZZ,
Lee SG, Grant S, Yacoub A, Dent P, Curiel DT, et al: Mechanism of
autophagy to apoptosis switch triggered in prostate cancer cells by
antitumor cytokine melanoma differentiation-associated gene
7/interleukin-24. Cancer Res. 70:3667–3676. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park EJ, Kim SY, Kim SH, Lee CR, Kim IS,
Park JK, Lee SW, Kim BJ, Chun JN and Jeon JH: SK&F 96365
induces apoptosis and autophagy by inhibiting Akt-mTOR signaling in
A7r5 cells. Biochim Biophys Acta. 1813:2157–2164. 2011. View Article : Google Scholar : PubMed/NCBI
|