Introduction
Breast cancer is the most common type of invasive
cancer in females worldwide (1).
Despite diverse screening programs and novel therapeutic strategies
implemented to markedly decrease mortality rates, the physiological
mechanism underlying the pathogenesis of breast cancer remains
unknown (2). Therefore, the
identification of biomarkers is required to improve the early
diagnosis of breast cancer and decrease cancer mortality rates.
Phosphatase of regenerating liver-3 (PRL-3), also
known as PTP4A3, is a member of the protein tyrosine phosphatase
superfamily (3). Protein tyrosine
phosphatases exert crucial functions in modulating cellular
processes, including cell growth, cell cycle progression and
apoptosis, by regulating a number of proteins (4,5). PRL-3 is
associated with tumor proliferation and metastasis, and is
overexpressed in various types of cancer, including colorectal
(6) and gastric cancer (7,8), ovarian
carcinoma (9,10), multiple myeloma (11) and squamous cell carcinoma of the
uterine cervix (12). Wang et
al (13) demonstrated that PRL-3
was overexpressed in breast cancer and predicts a poor clinical
outcome for patients. In the present study, PRL-3 was identified to
function in tumor proliferation by acting on the
p14ARF-p53 axis, suggesting that PRL-3 may be involved
in the tumorigenesis of breast cancer.
Materials and methods
Tissues specimens and cell lines
A total of 24 breast cancer and adjacent normal
tissues were obtained from the Sichuan Cancer Hospital (Chengdu,
China). The tissues from each individual were frozen in liquid
nitrogen and stored at −80°C until use. The tissues were fixed with
10% formalin overnight at room temperature and the fixed-tissues
were embedded in paraffin. The 4-µm-thick sections of the tissues
were validated using H&E staining and immunohistochemical
detection. Briefly, sections were stained for nuclei with
hematoxylin for 5 min at room temperature, and then sections were
washed with tap water and counterstained with eosin for 1 min and
washed. The following primary antibodies were used to perform
immunohistochemistry: anti-estrogen receptor (cat. no. ab180900;
dilution, 1:200; Abcam, Cambridge, UK), anti-progesterone receptor
(cat. no. ab32085; dilution, 1:100; Abcam) and anti-HER2 (cat. no.
ab194979; dilution, 1:200; Abcam). Briefly, sections were blocked
using 2% PBS + bovine serum albumin for 15 min at room temperature.
Subsequently, primary antibody incubation was performed overnight
at 4°C. Next, a biotinylated anti-rabbit immunoglobulin (Ig)G (cat.
no. ab150088; dilution, 1:2,000; Abcam) was used as a secondary
antibody. The antibody staining in the tissue sections was observed
using a light microscope (magnification, ×40). Written informed
consent was obtained from all patients and all protocols were
approved by the Sichuan Cancer Hospital Ethics Committee (Chengdu,
China).
Two breast cancer cell lines (MDA-MB-231 and MCF-7)
were purchased from the Chinese Academy of Sciences (Shanghai,
China) and cultured in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) at 37°C in a humidified chamber containing 5%
CO2.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
TRIzol® reagent (Invitrogen, Thermo
Fisher Scientific, Inc.) was used to extract total RNA from the
breast cancer tissues or two breast cancer cell lines (MDA-MB-231
and MCF-7) and subsequently reverse-transcribed into cDNA using a
First Strand cDNA Synthesis kit (Roche Diagnostics GmbH, Mannheim,
Germany) according to the manufacturer's protocol with the
following temperature protocol: −25°C for 10 min and then at 42°C
for 60 min, followed by 99°C for 5 min and then cooling to 4°C for
5 min. Subsequently, RT-qPCR was performed on the ABI 7500 Fast
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with FastStart™ Universal SYBR® Green Master (ROX)
reagents (Roche Diagnostics GmbH). The temperature protocols for
the qPCRs were as follows: 95°C for 10 min, 40 cycles of 95°C for
30 sec (inactivation) and 56°C for 1 min (annealing). A melt curve
was constructed following each reaction and data were analyzed
using a 7500 Fast System SDS (version 1.4.0.25; Applied Biosystems;
Thermo Fisher Scientific, Inc.). The primer sequences for PRL-3
were as follows: Forward, 5′-CTTCCTCATCACCCACAACC-3′; reverse,
5′-GTCTTGTGCGTGTGTGTGGGTC-3′. The primer sequences for
p14ARF were as follows: Forward,
5′-GCCACATTCGCTAAGTGCTC-3′ and reverse, 5′-GCCACATTCGCTAAGTGCTC-3′.
The primer sequences for GAPDH were as follows: Forward,
5′-AACGACCCCTTCATTGAC-3′ and reverse, 5′-TCCACGACATACTCAGCAC-3′.
All reactions were conducted in triplicate. GAPDH was used as the
normalization control and the relative levels were quantified using
the 2−ΔΔCq method (14).
Plasmid construction and
transfection
Total RNA was extracted from the MDA-MB-231 cells
using TRIzol buffer (Invitrogen; Thermo Fisher Scientific, Inc.).
Then the reverse transcription was performed using a Transcript
First Strand cDNA Synthesis kit (Roche Diagnostics GmbH). The cDNA
of PRL-3 was amplified by PCR using the following primers: Forward,
5′-TACCGGACTCAGATCTCGAGCGCCACCATGGCTCGGATGAACCGC-3′ and reverse,
5′-GATCCCGGGCCCGCGGTACCGTCATAACGCAGCACCGGGTCT-3′. The reverse
transcription temperature protocol used was as follows: 42°C for 60
min and 70°C for 5 min. The PCR program for the amplification of
cDNA was started at 94°C for 5 min, followed by 40 cycles at 94°C
for 30 sec, 58°C for 30 sec, 72°C for 5 min and completed with a
final extension at 72°C for 5 min. The amplified product and pcDNA3
vector (Invitrogen, Thermo Fisher Scientific, Inc.) were digested
with EcoRI and XhoI enzymes to construct
pcDNA3/PRL-3. The short hairpin (sh)RNAs against PRL-3 (target
sequence, 5′-AAATCTCGTTTCTCTTGGACA-3′) and p14ARF
(target sequence, 5′-GAACAUGGUGCGCAGGUUCTT-3′) were annealed and
cloned into the BamHI and HindIII restriction sites
of the pSilencer vector (Ambion; Thermo Fisher Scientific, Inc.) to
construct pSilencer/ShR-PRL-3, and pSilencer/ShR-p14ARF.
Empty vectors with a scramble shRNA sequence were used as a
negative controls (pSilencer/NC). The shRNA sequences were as
follows: PRL-3shRNA sense, 5′-ACAAACACATGCGCTTCCTCA-3′ and
antisense, 5′-TGAGGAAGCGCATGTGTTTGT-3′; p14ARFshRNA sense,
5′-CCGATTGAAAGAACCAGAGAG-3′ and antisense,
5′-CTCTCTGGTTCTTTCAATCGG-3′; and scrambled shRNA (pSilencer/NC)
sense, 5′-UAAUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. The transfection was performed with
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, MDA-MB-231 or MCF-7 cells were seeded into 12-well plates
(3×105 or 4×105 cells/well) the day prior to
transfection and then transfected with 100 nM constructed
pcDNA3/PRL-3, pcDNA3, pSilencer/ShR-PRL-3, pSilencer negative
control (pSilencer/NC) and pSilencer/ShR-p14ARF. At 48 h
after transfection, the mRNA/protein were collected and subject to
RT-qPCR (as aforementioned) and western blot analysis.
Western blot analysis
Proteins were extracted from the cell lines using
Radio-immunoprecipitation Assay lysis buffer (Thermo Fisher
Scientific, Inc.) for 20 min at 4°C with occasional agitation.
Equal amounts of protein extracts (determined using the BCA method)
were subjected to 10% SDS-PAGE and subsequently transferred to a
polyvinylidene fluoride membrane (0.45 µm pore size; EMD Millipore,
Billerica, MA, USA). Followed by blocking with 5% skimmed milk for
1.5 h at room temperature, the membrane was incubated with primary
antibodiesanti-PRL-3 (dilution, 1:500; cat. no. ab50276; Abcam),
anti-p53 (dilution, 1:500; cat. no. ab31333; Abcam),
anti-p14ARF (dilution, 1:500; cat. no. ab3642; Abcam)
and anti-GAPDH antibody (dilution, 1:500; cat. no.
SAB4300645-100UG; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at
4°C overnight and rinsed twice with PBS, followed by incubation
with peroxidase-conjugated secondary anti-rabbit (dilution,
1:2,000; cat. no. ab6721; Abcam) or anti-mouse IgG antibodies
(dilution, 1:2,000; cat. no. ab6709; Abcam) for 1 h at room
temperature. The membrane was subsequently incubated with an
enhanced chemiluminescent substrate kit (Thermo Fisher Scientific,
Inc.) and exposed to X-ray film. ImageJ (version 1.41; National
Institutes of Health, Bethesda, MD, USA) was used for the
quantitative analysis of band densities. GAPDH was used as a
control (primers as aforementioned).
Colony formation assay
MDA-MB-231 and MCF-7 cells were seeded into 12-well
plates (3×105 or 4×105 cells/well) the day
prior to transfection and were then transfected with 100 nM
constructed pcDNA3/PRL-3, pSilencer/ShR-PRL-3,
pSilencer/ShR-p14ARF and corresponding controls
(Psilencer/NC). After 2 weeks of culture, 0.2% crystal violet was
used to stain the cell colonies at room temperature for 30 min.
Subsequently, the colonies were counted using an inverted
fluorescence microscope (magnification, ×40; IX71; Olympus
Corporation, Tokyo, Japan). Each experiment was performed in
triplicate (1 colony, >50 cells) and images of MCF-7 or
MDA-MB-231 cell colonies were captured.
MTS assay to determine cell
proliferation
Transfected MDA-MB-231 and MCF-7 cell lines
[(3–5)
×103 cells/well] were plated in 96-well plates for 48
and 72 h and subsequently, the proliferation was determined using
the CellTiter 96® Aqueous cell proliferation assay kit
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. The absorbance was determined at a
wavelength of 490 nm using a Tecan microplate reader (Tecan Group,
Ltd., Mannedorf, Switzerland).
Statistical analysis
Experiments were performed in triplicate. The
results are presented as the mean ± standard deviation. Differences
between two groups were determined with unpaired Student's t-test
using SPSS (version 12.0; SPSS, Inc., Chicago, IL, USA). One-way
analysis of variance was used for multiple comparisons followed by
a Bonferroni Comparison post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PRL-3 is overexpressed in breast
cancer tissue samples
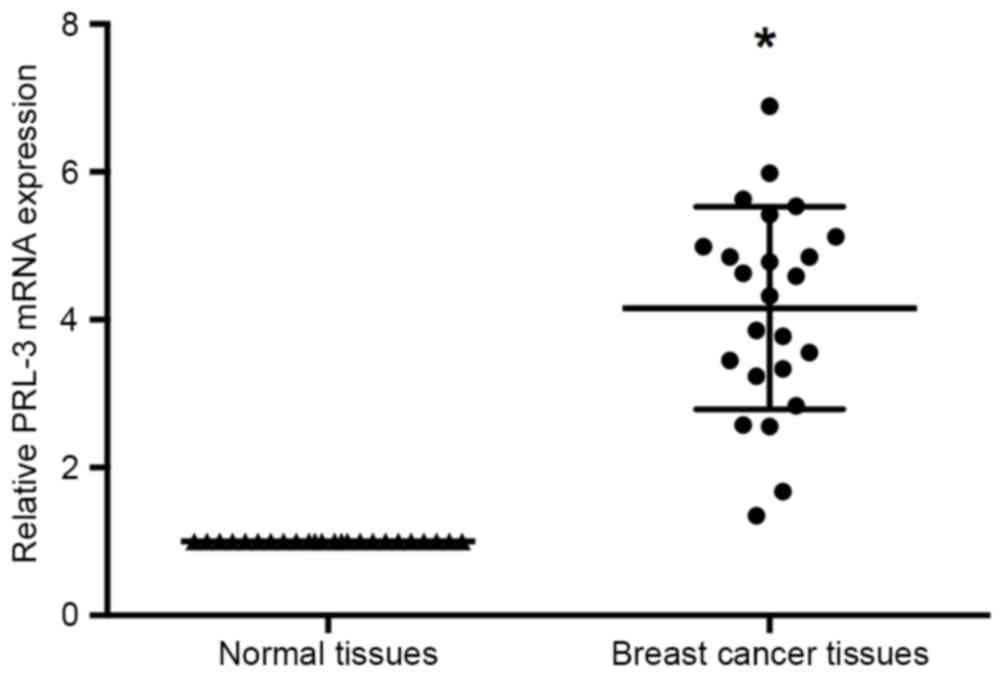
RT-qPCR was performed to investigate the expression
manner in the 24 pairs of breast cancer and adjacent non-cancerous
tissues. The results of RT-qPCR identified that PRL-3 expression
levels were significantly increased in the tumor tissues compared
with adjacent normal tissues (P<0.05; Fig. 1).
PRL-3 promotes the proliferation of
breast cancer cells
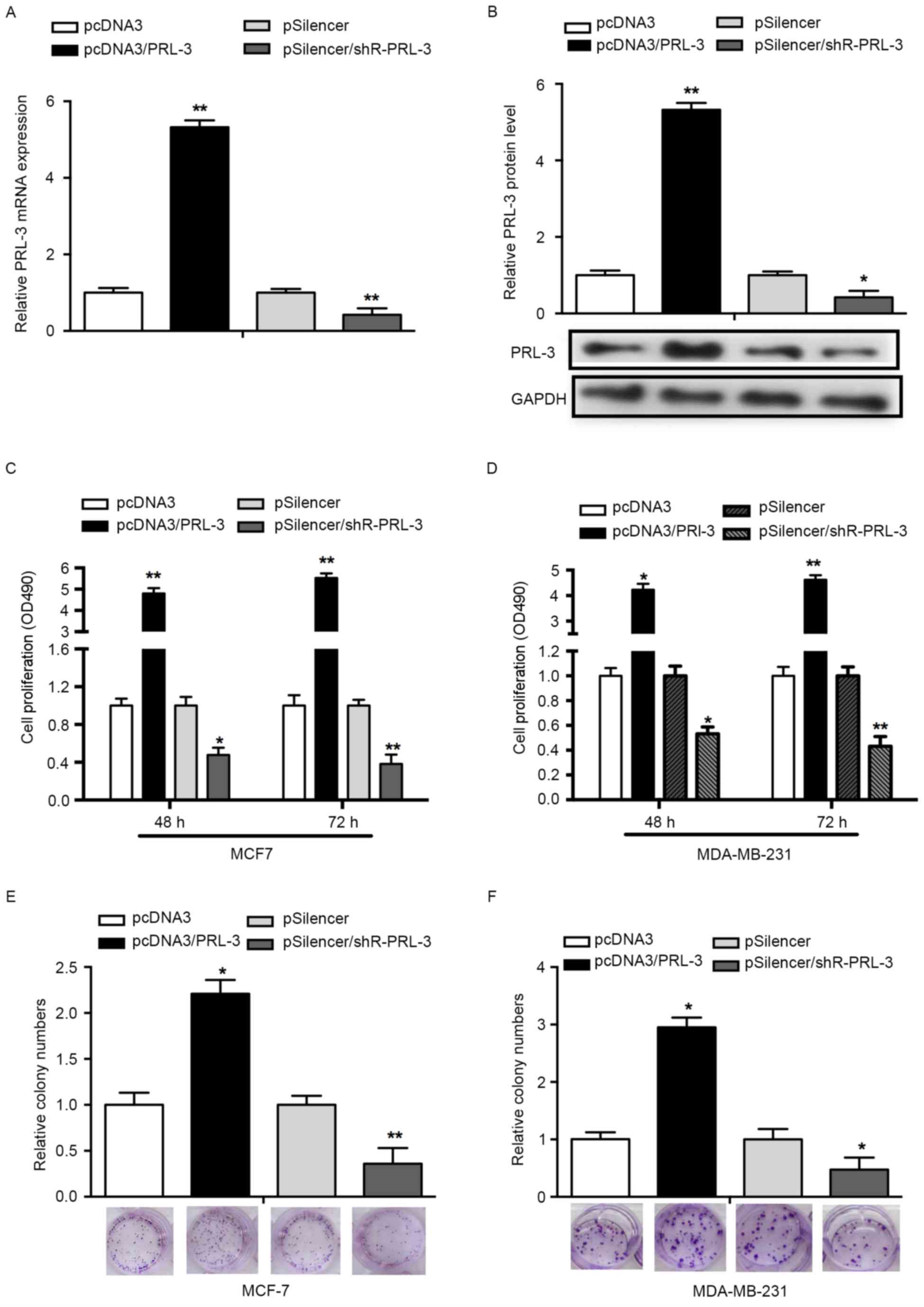
To determine whether PRL-3 affects cell
proliferation, an MTS assay was performed using MCF-7 and
MDA-MB-231 cells. First, overexpression of PRL-3 was achieved via
transfection with pcDNA3/PRL-3, whereas the suppression of PRL-3
was achieved by transfection with pSilencer/PRL-3. RT-qPCR and
western blot analysis was used to validate the efficiency of the
vectors. The expression of PRL-3 mRNA was significantly increased
in the pcDNA3/PRL-3group while decreased in the pSilencer/PRL-3
group, compared with the control groups (P<0.05; Fig. 2A). The expression of PRL-3 protein was
consistent with the expression of PRL-3 mRNA (P<0.01; Fig. 2B). An MTS assay results revealed that
PRL-3 overexpression promoted cell proliferation 3.8- and 4.5-fold
at 48 and 72 h post-transfection, respectively, compared with NCs.
Downregulation of PRL-3 with pSilencer/PRL-3 in MCF-7 cells
decreased the rate of cell proliferation by 52 and 68% at 48 and 72
h post-transfection, respectively (P<0.05; Fig. 2C). Concordant with the results
observed in the MCF-7 cell line, PRL-3 served a similar role in the
MDA-MB-231 cell line (P<0.05; Fig.
2D).
To assess the effects of PRL-3 on cell growth, a
colony formation assay was performed. Overexpression of PRL-3 in
MCF-7 cells revealed a significant increase in the number of
colonies, compared with the control group (P<0.05; Fig. 2E). Knockdown of PRL-3
(pSilencer/shR-PRL-3) in MCF-7 cells resulted in a significant
decrease in the number of colonies, compared with the control group
(Fig. 2E). In addition, a similar
effect was observed in MDA-MB-231 cells (P<0.05; Fig. 2F). The results of the present study
indicated that PRL-3 promotes the proliferation of breast cancer
cell lines.
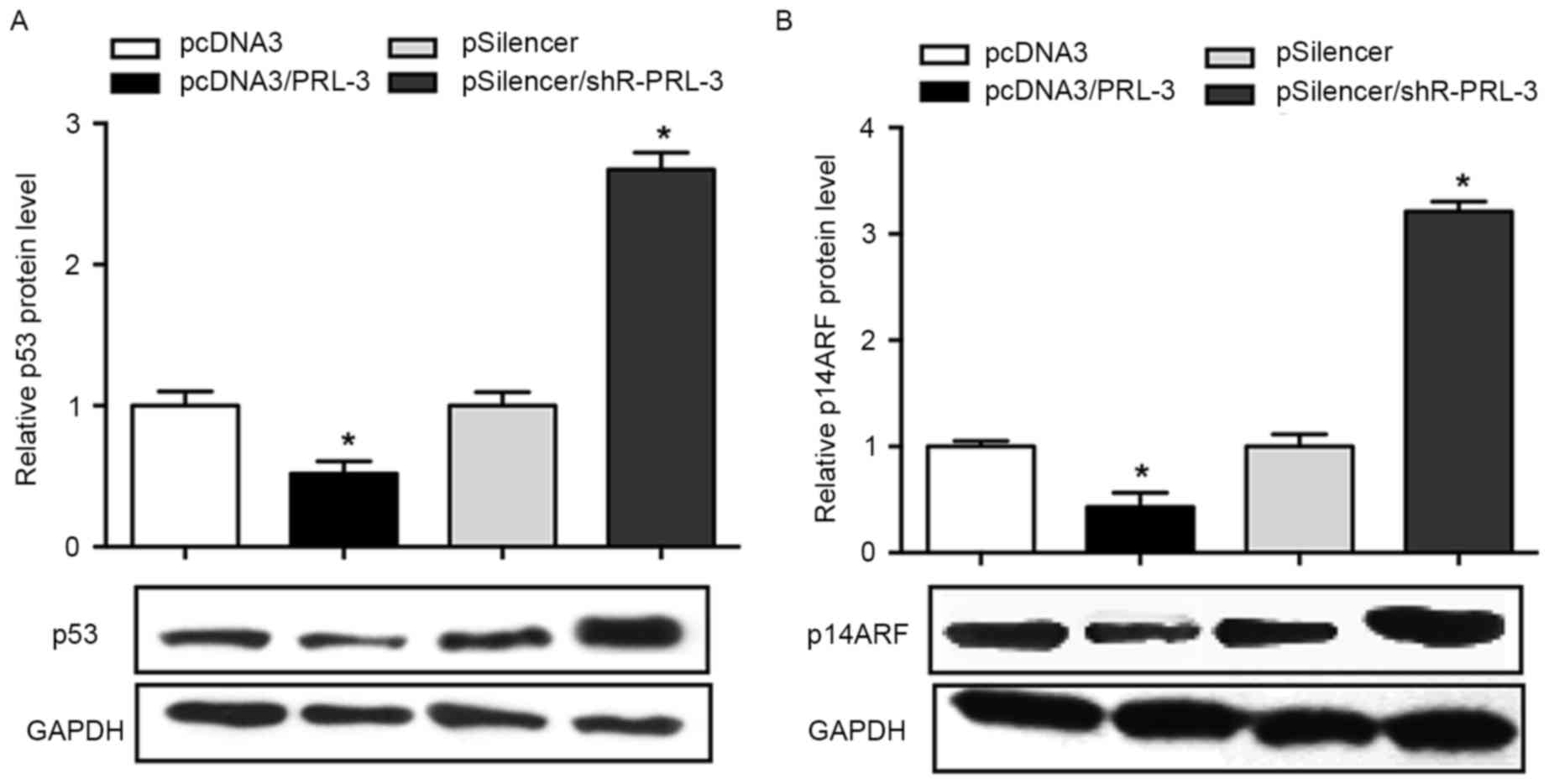
PRL-3 negatively regulatesp53 and
p14ARFprotein expression inMCF-7 cells
As previous studies have demonstrated that PRL-3 was
involved in the regulation of p53 and p14ARF (14–16), the
present study aimed to determine the association between PRL-3, p53
and p14ARF in breast cancer cells. The results of the
present study revealed that the overexpression of PRL-3 in MCF-7
cells results in an inhibited p53 expression (48% decrease)
compared with the negative control, whereas the inhibition of PRL-3
promoted the level of p53 1.8-fold (P<0.05; Fig. 3A). The effect of PRL-3 on the
expression level of p14ARF was investigated. The results
demonstrated that p14ARF was downregulated when PRL-3
was overexpressed, whereas inhibition of PRL-3 promoted the level
of p14ARF in MCF-7 cell lines (P<0.05; Fig. 3B).
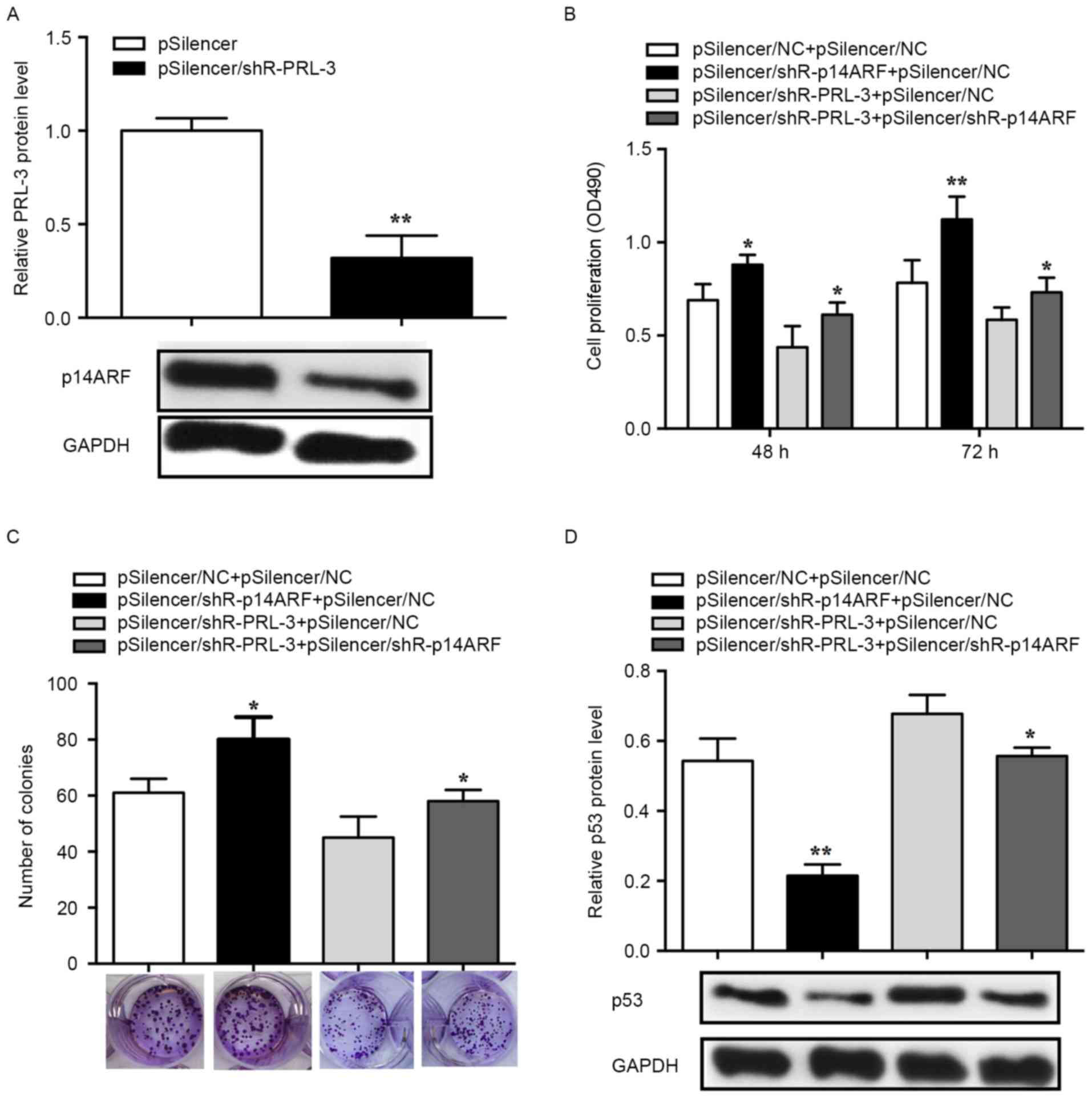
PRL-3 promotes cell proliferation via
the p14ARF-p53 axis
An MTS assay was performed to determine whether
PRL-3 affected cell proliferation via p14ARF-p53 and the
knockdown efficiency of pSilencer/shR-p14ARF was
validated in MCF-7 cells. The RT-qPCR results demonstrated that the
p14ARF expression levels in the pSilencer/shR-PRL-3
group decreased by ~70% (P<0.01; Fig.
4A). Suppression of p14ARF with
pSilencer/shR-p14ARF
(pSilencer/shR-p14ARF+pSilencer NC) promoted cell
proliferation and inhibition of PRL-3 (pSilencer/shR-PRL-3+
pSilencer NC) lead to suppressed cell proliferation in comparison
with 3×102 or 4×102 cells/well controls
(pSilencer NC+ pSilencer NC). The promoted cell proliferation
induced by suppression of p14ARF can be rescued by
co-transfection with pSilencer/shR-PRL-3
(pSilencer/shR-p14ARF+ pSilencer/shR-PRL-3) (72 h,
P<0.05; Fig. 4B). Furthermore, the
colony formation assay was used to evaluate the effects of
p14ARF and PRL-3 on cell proliferation. MCF-7 cells
transfected with pSilencer/shR-p14ARF revealed a
significant increase in the number of the colonies, whereas cells
transfected with pSilencer/shR-PRL-3 revealed a significant
decrease in the number of colonies compared with the control
(pSilencer NC+ pSilencer NC). Additionally, promoted cell
proliferation induced by the knockdown of p14ARFcan be
rescued by co-transfection with pSilencer/shR-PRL-3
(pSilencer/shR-p14ARF+ pSilencer/shR-PRL-3) (P<0.05;
Fig. 4C).
To determine the function of p53 in PRL-3-induced
cell proliferation, p53 protein expression was evaluated in human
breast cancer cells. The results revealed that the p53 level was
significantly decreased in cells transfected with
pSilencer/shR-p14ARF while significantly increased in
cells transfected with pSilencer/shR-PRL-3. The decreased
expression of p53 induced by pSilencer/shR-p14ARF can be
rescued by the knockdown of PRL-3 (P<0.05; Fig. 4D). Therefore, the results of the
present study demonstrated that PRL-3 downregulated p53 by
affecting the expression of p14ARF in the progression of
breast cancer cell proliferation.
Discussion
PRL-3 has previously been identified as an oncogene
(8). The present study hypothesized
that PRL-3 participates in p53-dependentcell proliferation. The
results of the present study suggested that PRL-3 promotes breast
cancer progression via the p14ARF-p53 axis that is
concordant with the hypothesis. In addition, the results of the
present study demonstrated that PRL-3 inhibits p53 and
p14ARF protein expression, indicating that PRL-3 may
serve a key function in breast cancer progression.
Previous studies have identified that PRL-3 is
associated with breast tumor progression (15), which prompted the present study to
investigate the function of PRL-3 in breast cancer through
examining the mRNA levels of PRL-3 in breast cancer and adjacent
normal tissues. The results of the present study revealed that the
expression of PRL-3 was increased in breast tumors compared with
adjacent normal tissues. p14ARF and p53 protein have
been previously identified to function as tumor suppressors
(16–18); in the present study, overexpression of
PRL-3 resulted in decreased levels of p14ARF and p53
protein in breast cancer cell lines. Studies have demonstrated that
p14ARF induces apoptosis to inhibit cancer progression
and that it is downregulated during breast cancer progression
(19–21). The results of the present study
revealed that suppression of p14ARF promotes cell
proliferation, which may be rescued by co-transfection with
pSilencer/shR-PRL-3. Additionally, p14ARF has been
identified to regulate the levels of p53 in breast cancer cells
(22,23). In the present study, it was revealed
that the suppression of p14ARF inhibits the expression
of p53 in breast cancer cell lines. Furthermore, p53 is regulated
by the expression of p14ARF (24–26) and
PRL-3 (27), via mouse double minute
2 homolog. The results of the present study revealed that PRL-3
decreases the p14ARF-mediated expression of p53 in
breast cancer cells. In summary, PRL-3 regulates the level of
p14ARF in order to inhibit the expression of p53. These
results may indicate an underlying molecular mechanism involved in
breast cancer development, and enable the identification of novel
therapeutic targets for breast cancer.
References
|
1
|
McGuire A, Brown JA, Malone C, McLaughlin
R and Kerin MJ: Effects of age on the detection and management of
breast cancer. Cancers (Basel). 7:908–929. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Youlden DR, Cramb SM, Dunn NA, Muller JM,
Pyke CM and Baade PD: The descriptive epidemiology of female breast
cancer: An international comparison of screening, incidence,
survival and mortality. Cancer Epidemiol. 36:237–248. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou J, Cheong LL, Liu SC, Chong PS,
Mahara S, Bi C, Ong KO, Zeng Q and Chng WJ: The pro-metastasis
tyrosine phosphatase, PRL-3 (PTP4A3), is a novel mediator of
oncogenic function of BCR-ABL in human chronic myeloid leukemia.
Mol Cancer. 11:722012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stephens BJ, Han H, Gokhale V and Von Hoff
DD: PRL phosphatases as potential molecular targets in cancer. Mol
Cancer Ther. 4:1653–1661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kato H, Semba S, Miskad UA, Seo Y, Kasuga
M and Yokozaki H: High expression of PRL-3 promotes cancer cell
motility and liver metastasis in human colorectal cancer: A
predictive molecular marker of metachronous liver and lung
metastases. Clin Cancer Res. 10:7318–7328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mollevi DG, Aytes A, Padullés L,
Martínez-Iniesta M, Baixeras N, Salazar R, Ramos E, Figueras J,
Capella G and Villanueva A: PRL-3 is essentially overexpressed in
primary colorectal tumours and associates with tumour
aggressiveness. Br J Cancer. 99:1718–1725. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miskad UA, Semba S, Kato H and Yokozaki H:
Expression of PRL-3 phosphatase in human gastric carcinomas: Close
correlation with invasion and metastasis. Pathobiology. 71:176–184.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Z, Cai SR, He YL, Zhan WH, Zhang CH,
Wu H, Peng JJ, Xu JB, Zhang XH, Wang L and Song W: Elevated PRL-3
expression was more frequently detected in the large primary
gastric cancer and exhibits a poor prognostic impact on the
patients. J Cancer Res Clin Oncol. 135:1041–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polato F, Codegoni A, Fruscio R, Perego P,
Mangioni C, Saha S, Bardelli A and Broggini M: PRL-3 phosphatase is
implicated in ovarian cancer growth. Clin Cancer Res. 11:6835–6839.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu H, Al-aidaroos AQ, Wang H, Guo K, Li
J, Zhang HF and Zeng Q: PRL-3 suppresses c-Fos and integrin α2
expression in ovarian cancer cells. BMC Cancer. 13:802013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fagerli UM, Holt RU, Holien T, Vaatsveen
TK, Zhan F, Egeberg KW, Barlogie B, Waage A, Aarset H, Dai HY, et
al: Overexpression and involvement in migration by the
metastasis-associated phosphatase PRL-3 in human myeloma cells.
Blood. 111:806–815. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Y and Li B: Expression of phosphatase
of regenerating liver-3 in squamous cell carcinoma of the cervix.
Med Oncol. 28:775–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang L, Peng L, Dong B, Kong L, Meng L,
Yan L, Xie Y and Shou C: Overexpression of phosphatase of
regenerating liver-3 in breast cancer: association with a poor
clinical outcome. Ann Oncol. 17:1517–1522. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ustaalioglu BB, Bilici A, Barisik NO,
Aliustaoglu M, Vardar FA, Yilmaz BE, Seker M and Gumus M: Clinical
importance of phosphatase of regenerating liver-3 expression in
breast cancer. Clin Transl Oncol. 14:911–922. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Min L, Ma RL, Yuan H, Liu CY, Dong B,
Zhang C, Zeng Y, Wang L, Guo JP, Qu LK and Shou CC: Combined
expression of metastasis related markers Naa10p, SNCG and PRL-3 and
its prognostic value in breast cancer patients. Asian Pac J Cancer
Prev. 16:2819–2826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sherr CJ: Principles of tumor suppression.
Cell. 116:235–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Procopio MG, Laszlo C, Al Labban D, Kim
DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et
al: Corrigendum: Combined CSL and p53 downregulation promotes
cancer-associated fibroblast activation. Nat Cell Biol.
17:13702015. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma H, Lu Y, Malone KE, Marchbanks PA,
Deapen DM, Spirtas R, Burkman RT, Strom BL, McDonald JA, Folger SG,
et al: Mortality risk of black women and white women with invasive
breast cancer by hormone receptors, HER2 and p53 status. BMC
Cancer. 13:2252013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Milojkovic A, Hemmati PG, Müer A, Overkamp
T, Chumduri C, Jänicke RU, Gillissen B and Daniel PT: p14ARF
induces apoptosis via an entirely caspase-3-dependent mitochondrial
amplification loop. Int J Cancer. 133:2551–2562. 2013.PubMed/NCBI
|
|
21
|
Silva J, Dominguez G, Silva JM, García JM,
Gallego I, Corbacho C, Provencio M, España P and Bonilla F:
Analysis of genetic and epigenetic processes that influence p14ARF
expression in breast cancer. Oncogene. 20:4586–4590. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maglic D, Zhu S, Fry EA, Taneja P, Kai F,
Kendig RD, Sugiyama T, Miller LD, Willingham MC and Inoue K:
Prognostic value of the hDMP1-ARF-Hdm2-p53 pathway in breast
cancer. Oncogene. 32:4120–4129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wazir U, Jiang WG, Yasaei H, Linne H,
Newbold RF and Mokbel K: P14ARF is down-regulated during tumour
progression and predicts the clinical outcome in human breast
cancer. Anticancer Res. 33:2185–2189. 2013.PubMed/NCBI
|
|
24
|
Xia L, Paik A and Li JJ: p53 activation in
chronic radiation-treated breast cancer cells: Regulation of
MDM2/p14ARF. Cancer Res. 64:221–228. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei J, Noto JM, Zaika E, Romero-Gallo J,
Piazuelo MB, Schneider B, El-Rifai W, Correa P, Peek RM and Zaika
AI: Bacterial CagA protein induces degradation of p53 protein in a
p14ARF-dependent manner. Gut. 64:1040–1048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Ding S, Duan Z, Xie Q, Zhang T,
Zhang X, Wang Y, Chen X, Zhuang H and Lu F: Role of p14ARF-HDM2-p53
axis in SOX6-mediated tumor suppression. Oncogene. 35:1692–1702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lv H, Liu R, Fu J, Yang Q, Shi J, Chen P,
Ji M, Shi B and Hou P: Epithelial cell-derived periostin functions
as a tumor suppressor in gastric cancer through stabilizing p53 and
E-cadherin proteins via the Rb/E2F1/p14ARF/Mdm2 signaling pathway.
Cell Cycle. 13:2962–2974. 2014. View Article : Google Scholar : PubMed/NCBI
|