Introduction
Perivascular epithelioid cell (PEC) neoplasms
(PEComas) are rare mesenchymal tumors composed of PECs. PEC, a cell
type that has no known normal counterpart, was coined by Bonetti
et al (1) in 1992 in order to
describe tumor cells with an epithelioid appearance, perivascular
distribution, and co-expression of myogenic and melanocytic
markers, most classically Human Melanoma Black 45 (HMB45).
Histologically, PEComas typically exhibit both epithelioid and
spindled cell morphology, and can be either benign or malignant
(2).
While the most prevalent tumors in the PEComa family
are angiomyolipoma (AML), lymphangioleiomyomatosis (LAM) and
clear-cell (sugar) tumor (CCST) of the lung, other significantly
less common manifestations of PEComa have been detected at a
variety of visceral and somatic sites throughout the body (3–5). Primary
PEComa of the bone is particularly rare, with only 12 reported
cases in the literature (6–14). The present study investigated a case
of malignant primary bone PEComa presenting in the right distal
femur of a 46-year-old female and described the clinical,
radiologic and histologic malignant features, in addition to a
review of the relevant literature.
Case report
A 46-year-old female with no significant past
medical history presented with a 5-month history of right distal
thigh pain following trauma. The patient presented to urgent care
in 2016 for evaluation following the trauma and obtained
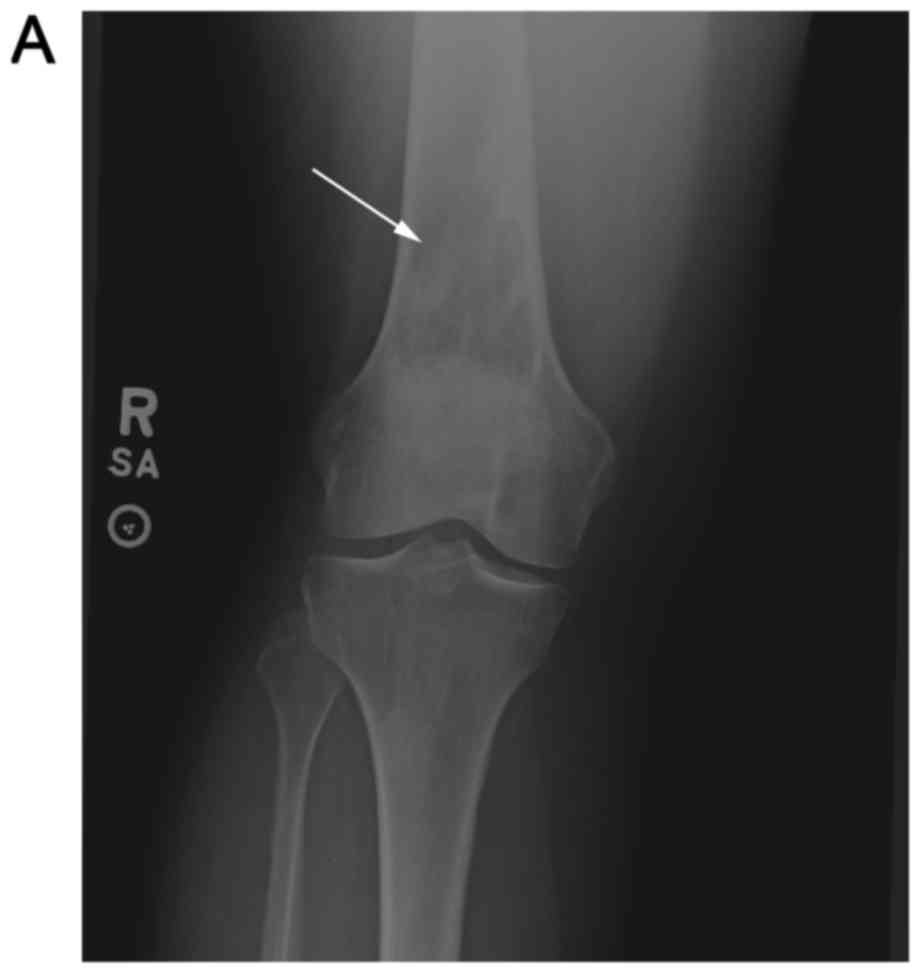
radiographs of the right knee and distal femur (Fig. 1A and B). These radiographs revealed a
mixed, lytic and sclerotic lesion involving the distal femoral
metadiaphysis extending into the epiphysis. The patient's pain
progressed and she returned three months later for a repeat
evaluation. At that time, the patient reported 5/10 distal thigh
pain that was throbbing and aching. The pain was constant. The pain
was worse with activity and relieved with rest and non-steroidal
anti-inflammatory drugs.
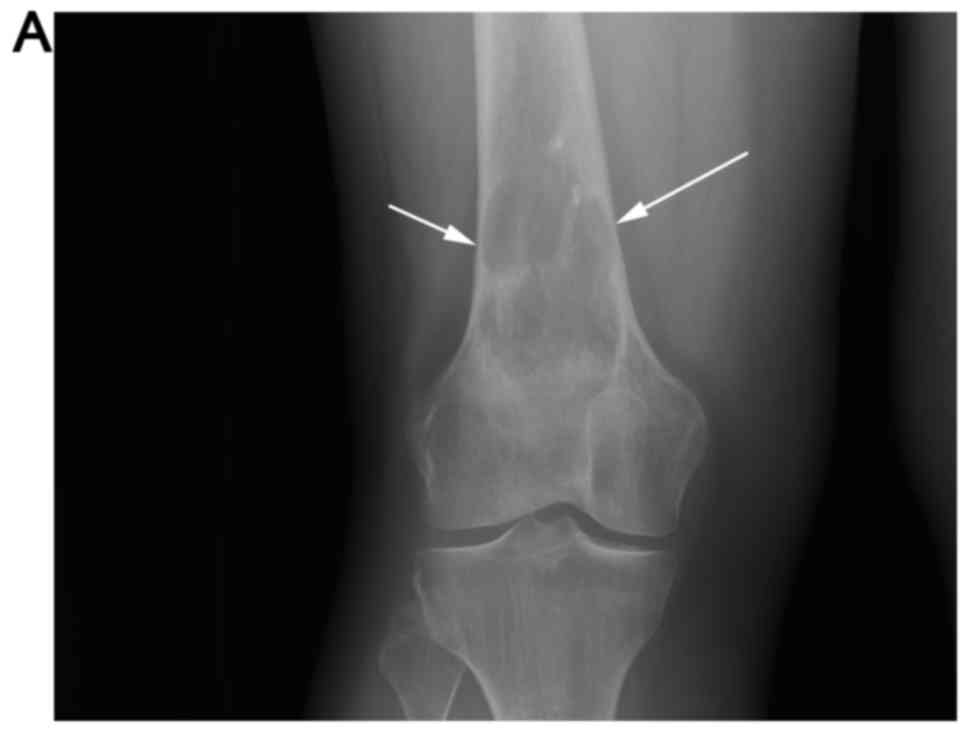
Radiographs of the right distal femur were repeated
(Fig. 2A and B), which revealed the
lesion to be grossly similar in size when compared with prior
radiographs, though with increased cortical scalloping. The patient
was referred for further evaluation with magnetic resonance imaging
(MRI). The MRI was obtained using a 0.3T Hitachi Airis II scanner
(Hitachi Medical Corporation, Tokyo, Japan).
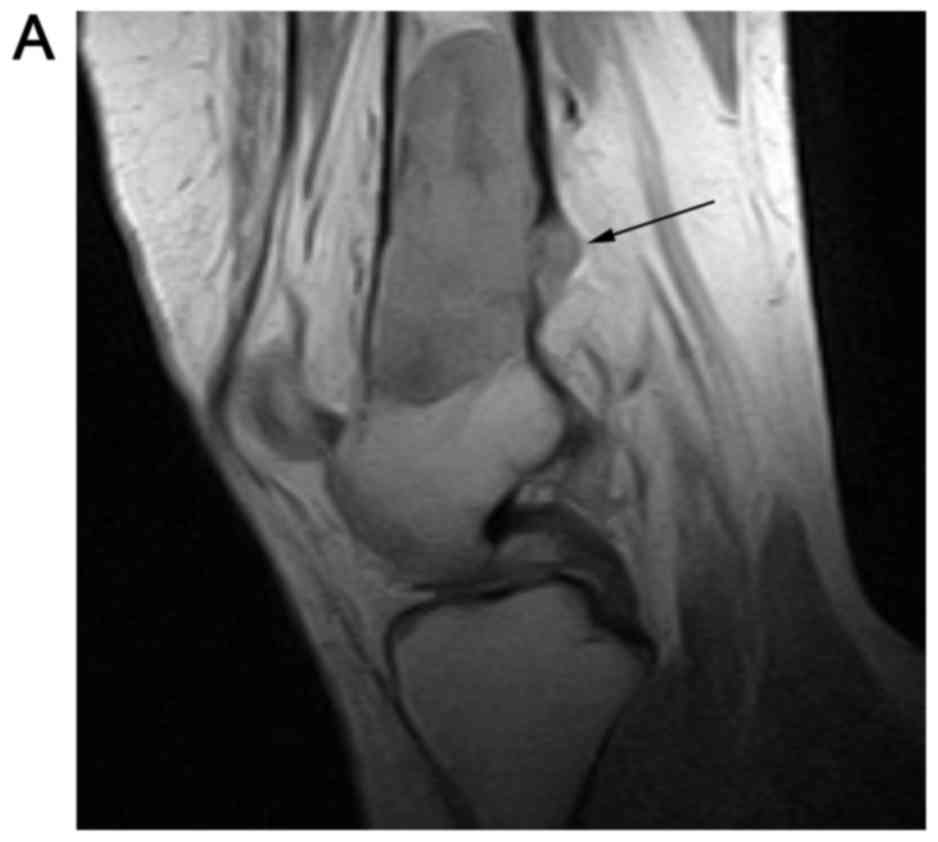
Sagittal T1-weighted sequences were obtained using
repetition time (TR) of 900 ms, echo time (TE) of 12.6 ms, slice
thickness of 4 mm, interslice gap of 1 mm and acquisition matrix of
256×158 (Fig. 3A). T2-weighted
sequences were obtained with TR/TE 6182/125 ms, slice thickness of
4 mm, interslice gap of 1 mm and acquisition matrix of 256×144
(Fig. 3B). The lesion was
heterogeneous in T2 signal with three distinct signal intensities
noted. The MRI demonstrated areas of cortical destruction with soft
tissue extension consistent with an aggressive lesion (Fig. 3B). Coronal T1-weighted sequences
(TR/TE 490/15 ms, slice thickness 4 mm, interslice gap of 0.5 mm
and acquisition matrix 256×160) exhibited a heterogeneously T1
hypointense and isointense lesion (Fig.
3C). The patient was referred for computed tomography
(CT)-guided core-needle biopsy of the lesion at the Hospital of the
University of Pennsylvania (Philadelphia, PA, USA). Histologic
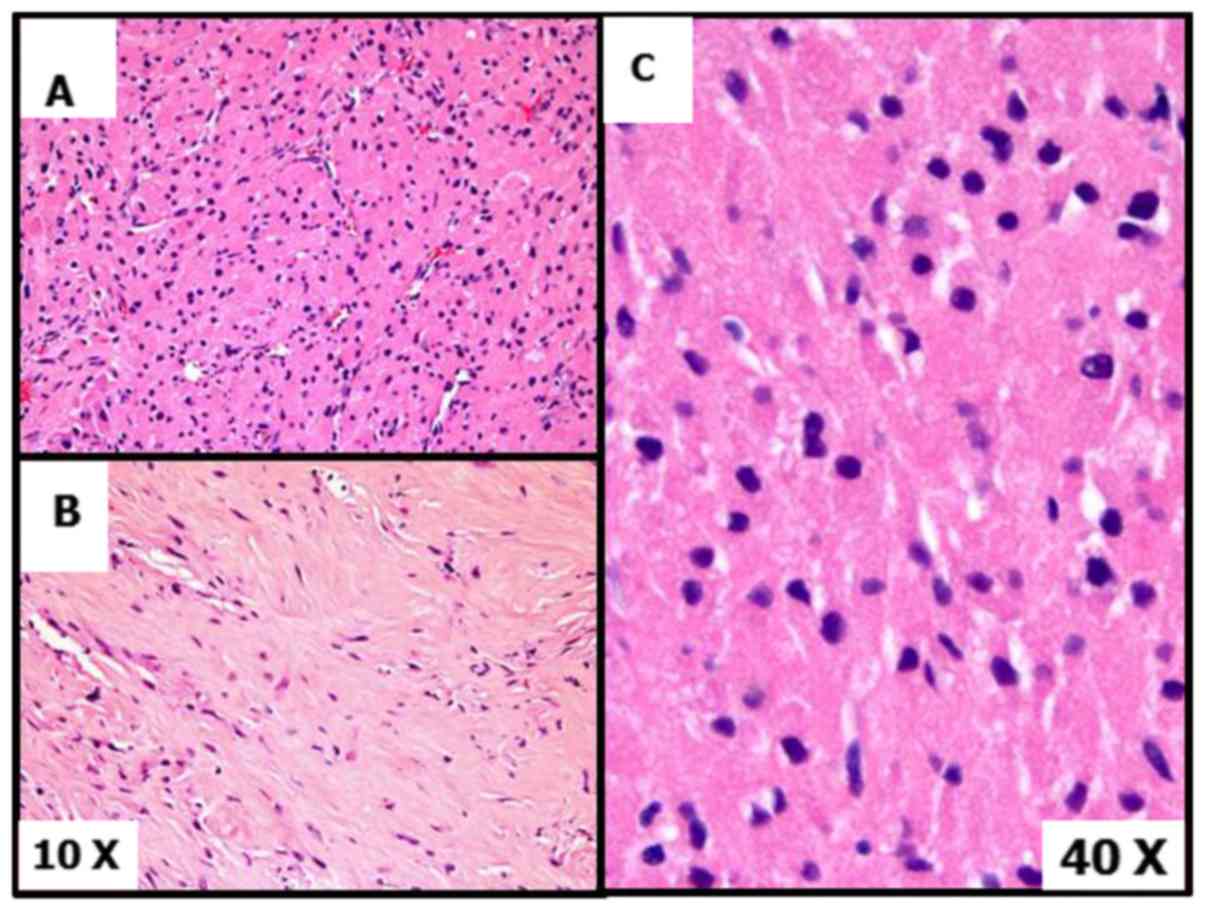
evaluation of the area of the lesion that had been sampled with the
core needle biopsy (Fig. 4A-C)
revealed a cytologically bland proliferation of cells, with ample
bright granular eosinophilic cytoplasm and dark even chromatin with
few scattered enlarged nuclei with inclusions. The cells were in a
nested configuration with a rich vascular stroma, and other areas
had extensive stromal hyalinization. There was no necrosis or
mitotic activity. The morphologic impression was that of a granular
cell tumor, although an S100 immunostain was negative and a
subsequent extensive immunohistochemical panel was unrevealing.
Considering the overall features, the core needle biopsy diagnosis
was favored to be an unusual (S100 negative) granular cell neoplasm
with extensive stromal hyalinization. Since malignant granular cell
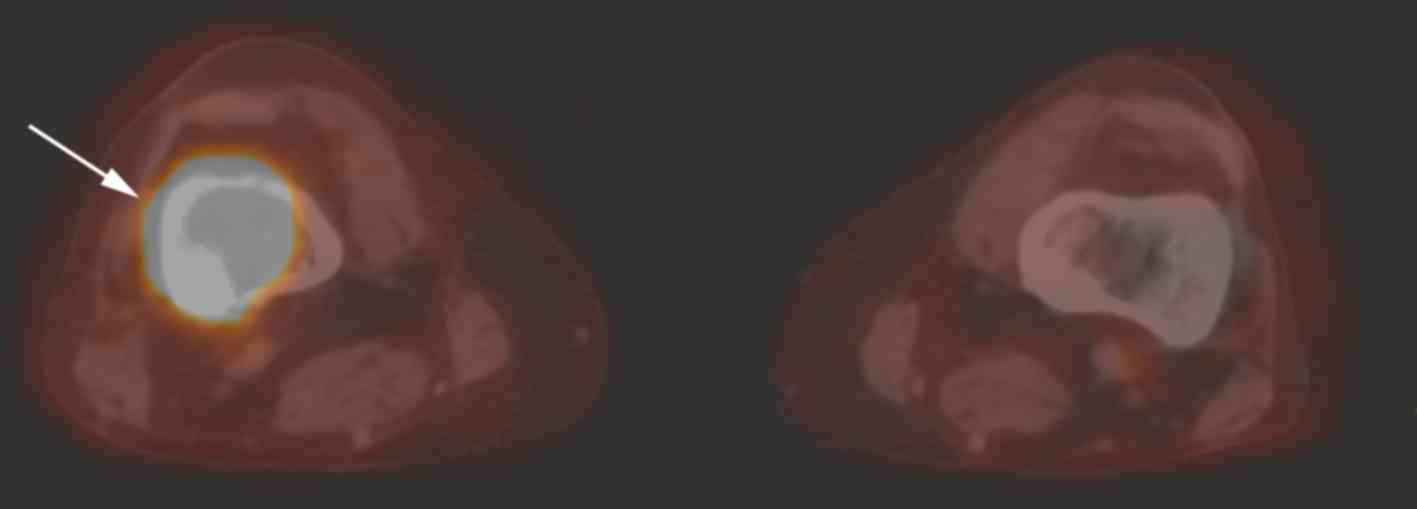
tumors are 18fluorodeoxyglucose (FDG)-avid, a PET/CT was
performed to evaluate FDG-avidity and assess for additional lesions
(Fig. 5). The PET/CT indicated an
FDG-avid lesion with a maximum SUV of 28.6, so the lesion was
treated as a malignant granular cell neoplasm.
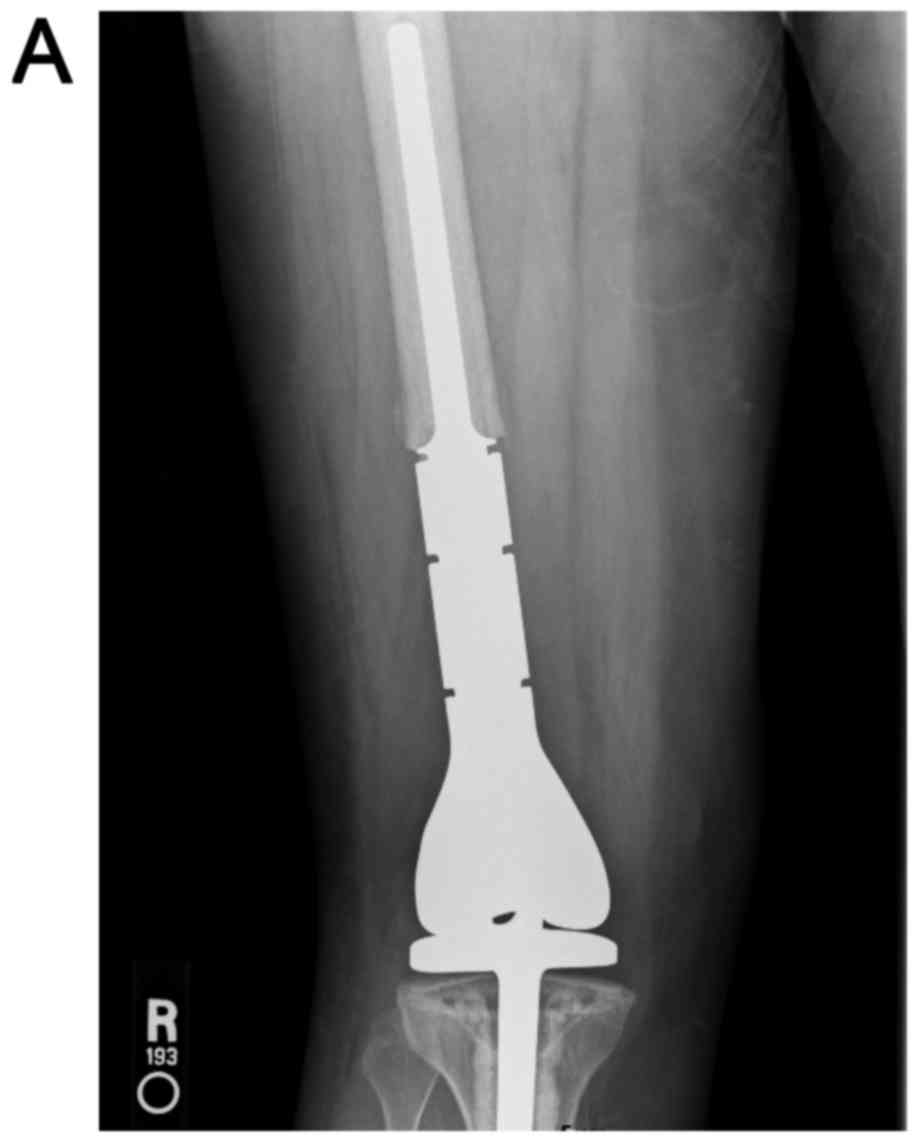
The distal femur lesion was resected with negative
margins and the resection gap reconstructed with a distal femoral
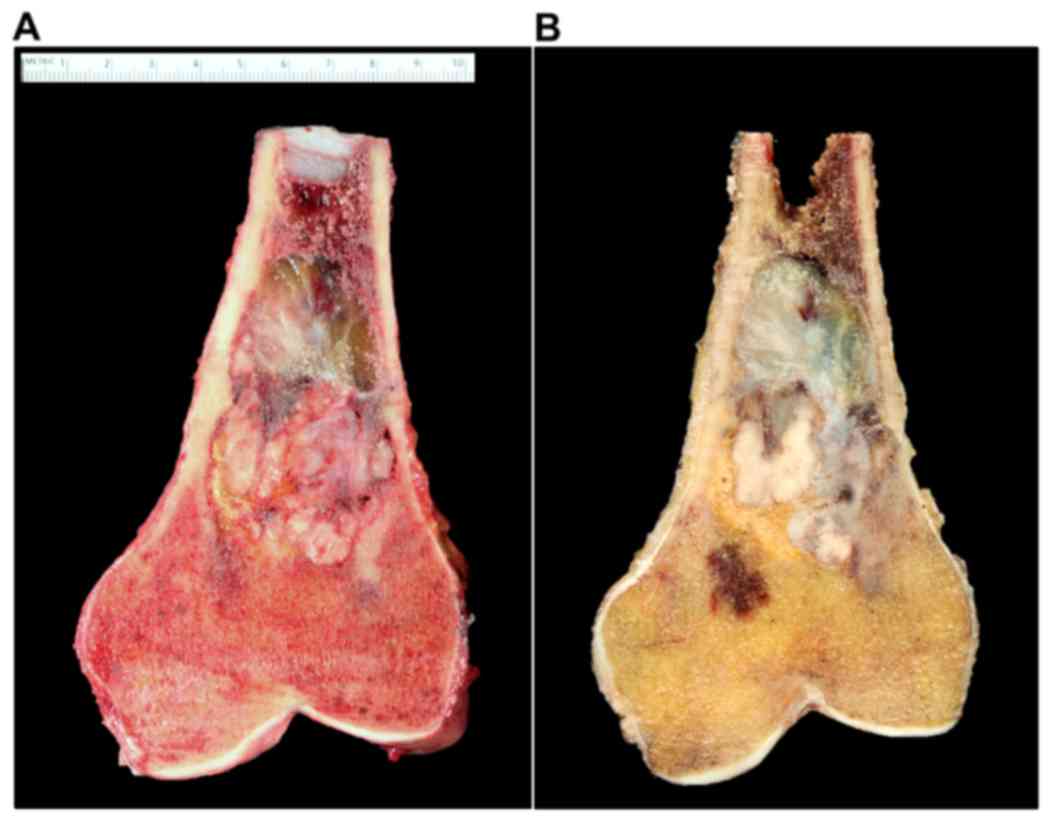
megaprosthesis and hinged knee replacement (Fig. 6A and B). Gross examination of the
tumor revealed a 6.9×3.9×2.9 cm irregular, heterogeneous, firm
lesion with an appearance varying from beige to gray in color
(Fig. 7A and B). The lesion occupied
the majority of the intramedullary cavity with an area of posterior
endosteal scalloping and soft tissue extension through the cortex,
as noted on prior MRI.
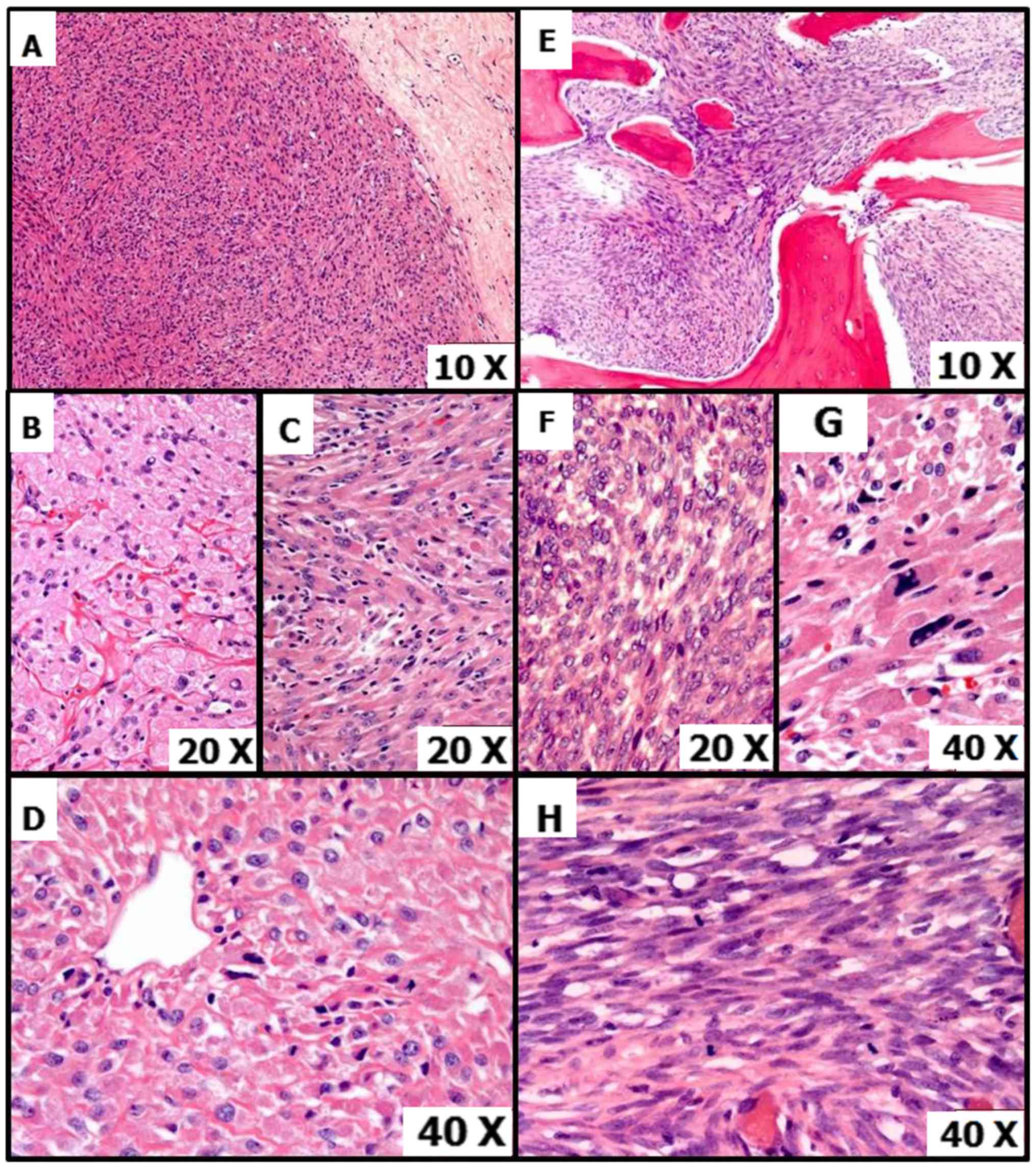
Histology from the resected specimen revealed a
heterogenous morphology with various patterns. A number of areas
had plump, epithelioid cells with granular cytoplasm, round
medium-large nuclei and distinct cell borders. This was concordant
with the previous biopsy (Fig. 8A).
These cells were predominantly arranged in a nested architecture
with individual nests separated from each other by a vascular or
hyalinizing stroma, and in areas with a vaguely storiform pattern
(Fig. 8B and C). At least focally,
characteristic thin vascular spaces were observed, around which
tumor cells were arranged tightly or in a radiating manner
(Fig. 8D). Other regions had an
infiltrative pattern with a spindled morphology, with medium sized
spindled cells arranged in interweaving fascicles and containing
fibrillar or granular eosinophilic cytoplasm with elongated, ovoid
nuclei with prominent nucleoli (Fig.
8E). There were cells present that exhibited marked nuclear
pleomorphism (Fig. 8F). Mitotic
activity was highest in very cellular epithelioid and spindled
areas with >5 mitoses per high power field (Fig. 8G and H). Focal coagulative tumor
necrosis was also present.
Tumor cells revealed positivity for smooth muscle
actin, Caldesmin, melanoma antigen, epithelial membrane antigen and
thyroid transcription factor-1 (TTF-1; nuclear), while being
negative for pan-cytokeratin, cytokeratin AE1/AE3, cytokeratin
Cam5.2, S100, CKIT, discovered on gastrointenstinal stromal tumors
protein 1, DOG-1, Desmin, Paired box 8, Signal transducer and
activator of transcription 6, Inhibin, Thyroglobulin, Cyclin D1,
microphthalmia associated transcription factor 2 and Human Melanoma
Black (HMB45). There was abnormal transcription factor E3 (TFE3;
nuclear) expression, although fluorescent in situ
hybridization (FISH) did not demonstrate a TFE3 translocation. Due
to the negative cytokeratin expression, the TTF-1 reactivity was
considered as aberrant reactivity rather than evidence of an
epithelial tumor. There was no abnormal loss of SDHB. CD34 and CD31
indicated an extensive vascular network. Based on the morphologic
and immunohistochemical findings (co-expression of smooth muscle
markers and melanocytic markers), the final pathologic diagnosis on
the resected specimen was of a malignant PEComa.
All procedures performed involving human
participants were in accordance with the ethical standards of the
University of Pennsylvania Health System (Philadelphia, PA, USA)
and with the Declaration of Helsinki declaration (1964) and its
later amendments or comparable ethical standards. The requirement
for informed consent was waived by the local institutional review
board.
Discussion
We present a novel case of primary bone PEComa,
localized to the right distal femur. The concept of PEComa is
relatively recent, arising within the last 25 years: Following
Bonetti's description of PECs in 1992 (1), Zamboni et al (15) coined the term ‘PEComa’ in 1996 and the
World Health Organization first recognized PEComa in 2002 (16). As a result, numerous rare neoplastic
disorders, including renal AML (prevalence ~1–2/1000 individuals)
(17), pulmonary LAM (prevalence
~1–2/1,000,000 individuals) (2,18) and CCST
of the lung (unknown prevalence, ~50 reported cases) (19–21), now
fall within the umbrella term of PEComa. Other forms of PEComa,
known as PEC tumor not otherwise specified (PEComa-NOS), are even
less common with <300 reported cases (22).
Notably, only 12 cases of primary PEComa of the bone
have been reported to date, not including the current case
(6–14), with 7 of these classified as
malignant. They include a wide age range of patients (mean age 49,
range 26–93), with tumors most commonly localized to lower
extremities (tibia, fibula, femur and acetabulum) but also
occurring in the vertebrae and ribs. Table I summarizes the salient details of
each case, including presentation, diagnostic features, treatment
and outcomes. Table I also includes
details of the present case for comparison.
 | Table I.Reported cases of primary PEComa of
the bone, to date. |
Table I.
Reported cases of primary PEComa of
the bone, to date.
| Year | Age/sex | Location | Presenting
symptom | Size (cm) | Radiologic
features | Histology-cell
types | IHC markers | Treatment | Follow-up | Folpe's
classification | (Refs.) |
|---|
| 2002 | 30/M | R proximal tibia | Pain | 2 | Osteolytic with
cortical destruction | Epithelioid | HMB45 | Local resection | 12 mo NED | Benign | (6) |
| 2008 | 52/F | R midshaft
fibula | Swelling | 6.3 | Extension through
cortex forming a soft-tissue mass | Epithelioid | HMB45, CyclinD1 | Wide excision | 3 mo NED | Malignant | (7) |
| 2008 | 28/M | R 6th rib | Pain | 2 | Osteolytic | Epithelioid, clear,
spindle | HMB45, SMA | Local resection | NR | Benign | (8) |
| 2008 | 92/F | R fibula | NR | NR | NR | Epithelioid | HMB45, CD10,
TFE3 | Local resection | NR | Benign | (9) |
| 2010 | 35/M | 7th thoracic
vertebra | Bilateral leg
weakness, back pain | 1.8 | Osteolytic,
destructive enhancing lesion | Epithelioid,
spindle | HMB45, Melan-A,
SMA | CRT | 12 mo pelvic bone
met | Malignant | (10) |
| 2010 | 39/F | R proximal
tibia | Pain | 6.5 | Enhancing mass
w/extension through cortex forming a soft-tissue mass | Epithelioid,
spindle | HMB45, Melan-A,
SMA | Excision + RT | 34 mo NED | Malignant | (10) |
| 2010 | 48/F | R distal tibia | Pain | Very small | Permeable
destructive lesion w/soft tissue extension | Epithelioid,
spindle | HMB45, Melan-A,
SMA | Excisional biopsy,
amputation | 36 mo NED (with
recurrence ×3) | Unknown malignant
potential | (10) |
| 2012 | 29/M | L acetabulum | Pain | 5 | Extensive lytic
w/soft tissue expansion | Epithelioid,
spindle | Melan-A, desmin,
vimentin | Resection +
Adjuvant C | 8 mo death (lung
met) | Malignant | (11) |
| 2012 | 93/F | R distal
fibula | Pain/swelling | NR | Expansile lytic
lesion | Epithelioid | HMB45 | Local
resection | 24 mo NED | Benign | (11) |
| 2012 | 26/M | 5th lumbar
vertebra | Lower back pain,
left leg weakness | Large | Destructive lesion
w/extra-osseous mass | Epithelioid | HMB45, S-100 | Conservative | NR | Unknown malignant
potential | (12) |
| 2015 | 47/M | L distal femur | Pain/swelling | 5.2 | Osteolytic mass,
destruction of cortex forming a soft-tissue mass | Epithelioid,
clear | HMB45 PNL2, TFE3,
vimentin, SMA | Curretage, CRT | 42 mo lung met | Malignant | (13) |
| 2016 | 65/M | R distal thigh | Pain/swelling | 11.5 | Expansile lytic
lesion w/soft tissue expansion | Epithelioid,
spindle | HMB45, Melan-A,
SMA | Wide excision | 28 mo NED | Malignant | (14) |
| 2017 | 46/F | R distal femur | Pain/swelling | 7.3 | Mixed lytic and
sclerotic lesion w/soft tissue expansion | Epithelioid,
spindle | Melan-A, SMA
TFE3 | Wide excision | 6 mo NED | Malignant | (Present case) |
The etiology of PEComa is unclear. There is no
normal cell type analogous to the PEC and no identified precursor
lesion. Genetically, AML, LAM and CCST are strongly associated with
tuberous sclerosis complex (TSC), an autosomal dominant
neurocutaneous disorder caused by TSC1/TSC2 gene mutations and
characterized by seizures, intellectual disability and cellular
proliferations (23–25). However, no similar genetic association
has been established in primary bone PEComa. AML, LAM, CCST and
numerous forms of PEComa-NOS have a well-documented marked female
predominance (15,17,24,26,27),
which suggests a potential role of hormones in pathogenesis.
However, this trend is not evident in primary bone PEComa (6/13
existing cases, including the present case, are female), though the
total number of cases is too few to be conclusive.
In general, PEComas may be asymptomatic or present
with nonspecific pain. All existing cases of primary bone PEComa
presented with pain and/or swelling, with PEComas of the vertebral
column also causing leg weakness due to cord compression (10,12).
Diagnostic workup of bone PEComa involves a combination of imaging,
histology, and immunohistochemistry. Based on the existing case
studies, there appears to be no consistent protocol in terms of
imaging workup, though the majority of studies include at least a
radiograph, often followed by CT and/or MRI (11,12). The
most common radiographic finding is a mixed lytic and sclerotic
lesion, with more aggressive tumors also presenting with soft
tissue expansion and cortical destruction on CT or MRI. MRI is
likely to exhibit heterogeneous T1 signal hypointensity on
T1-weighted imaging and heterogeneous T2 signal hyperintensity on
T2-weighted images (10,11,13).
Following analysis of local radiologic findings, patients often
undergo systemic clinical and radiologic testing for staging and
exclusion of metastatic disease.
In addition to imaging, histological and
immunohistochemical studies are crucial for the diagnosis of
PEComa. These can be carried out following biopsy, intralesional
curettage or wide resection of the tumor. Cytologic findings are
significant for epithelioid cells with clear and eosinophilic
cytoplasm, intermixed with spindle cells. The cells lie adjacent to
thin-walled blood vessels. Other findings common in more aggressive
cases are high mitotic activity, nuclear pleomorphism,
multinucleated giant cells, stromal hyalinization and focal
necrosis (7,8,10,11,13,14).
PEComas typically exhibit reactivity to both melanocytic (e.g.,
HMB45, melan-A) and myogenic (e.g., smooth muscle actin, desmin)
markers, but not to epithelial markers. The present case is one of
only two reported primary bone PEComas with negative staining for
HMB45 but positive staining for melan-A, another melanocytic
marker. Also, the current case also positively expressed at least
one smooth muscle marker, similar to 7/12 of the prior cases.
Notably, this case also stained positively for TFE3, a member of
the MiT family of transcription factors, albeit FISH did not
demonstrate a translocation. TFE3 positivity has been observed in
two prior primary bone PEComa cases and other forms of PEComa. In
fact, in a study of TFE3 positivity in PEComa cases, Righi et
al (9) proposed that PEComa be
added to the MiT family of human tumors, which also includes
melanoma, alveolar soft part sarcoma, translocation-associated
renal cell carcinomas and clear cell sarcoma of soft tissue
(9).
The histologic differential diagnosis for bone
PEComa includes metastatic melanoma, metastatic carcinoma (e.g., of
renal origin), and leiomyosarcoma. These lesions can have similar
cytologic presentations, such as mixed epithelioid and spindle cell
morphology, though they lack a perivascular distribution. Certain
forms of renal cell carcinoma also exhibit TFE3 nuclear
immunoreactivity (9). Alveolar soft
part sarcoma (ASPS), which is also commonly reactive to TFE3 and
presents with eosinophilic cytoplasm and hypervascularity (28), should also be considered in the
differential. Immunohistochemistry is crucial for distinguishing
primary bone PEComa from other metastatic disease. In contrast to
PEComa, leiomyosarcoma and ASPS lack expression of melanocytic
markers, metastatic melanoma lacks expression of smooth muscle
markers and expresses S-100, and carcinoma expresses epithelial
markers (2,10,16,26,28).
Therefore, the trifecta of imaging, histology and
immunohistochemistry is essential in PEComa evaluation: While
radiologic studies are often the first step of the workup and
microscopic examination can provide further insights,
immunohistochemistry is required for a definitive diagnosis.
In 2005, Folpe and Kwiatkowski (2) established histologic criteria to
classify PEComas as ‘benign’, ‘uncertain malignant potential’ or
‘malignant’. Specifically, they suggest that a PEComa be classified
as malignant if it exhibits at least two of the following features:
i) Tumor size >5 cm; ii) infiltrative growth pattern; iii) high
nuclear grade; iv) high cellularity; v) necrosis; vi) mitotic
activity >1/50 HPF with subsequent aggressive clinical behavior.
Neoplasms fulfilling only one of the six criteria may be
characterized as having uncertain malignant potential, while those
with none of the features may be considered benign. Based on this
system, the current patient's PEComa would be classified as
malignant, given its large size, high mitotic rate, infiltrative
growth pattern, high nuclear grade and cellularity, and the
presence of necrosis. Table I
includes malignancy status based on Folpe's criteria for all prior
primary bone PEComa cases. It is important to note that malignancy
status based on Folpe's histological criteria may not reflect
malignancy based on clinical aggressiveness, though the prior cases
that involved metastatic disease were all also histologically
classified as malignant.
In terms of treatment, resection of bone PEComa is
most common and often curative, though chemo- and/or radiotherapy
may be necessary in the case of metastatic disease. Prognosis is
variable and benign lesions are cured with local resection and
malignant lesions develop metastasis in 42.9% (3/7) of cases from
Table I. As evident from Table I, long-term follow-up information is
not consistently reported, which can make conclusions regarding
treatment outcomes challenging. Additional and longer-term studies
of patients with primary PEComa of the bone can further
understanding of this exceptionally rare but fascinating
disorder.
References
|
1
|
Bonetti F, Pea M, Martignoni G and Zamboni
G: PEC and sugar. Am J Surg Pathol. 16:307–308. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Folpe AL and Kwiatkowski DJ: Perivascular
epithelioid cell neoplasms: Pathology and pathogenesis. Hum Pathol.
41:1–15. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martignoni G, Pea M, Reghellin D, Zamboni
G and Bonetti F: PEComas: The past, the present and the future.
Virchows Arch. 452:119–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pan CC, Yang AH and Chiang H: Malignant
perivascular epithelioid cell tumor involving the prostate. Arch
Pathol Lab Med. 127:E96–E98. 2003.PubMed/NCBI
|
|
5
|
Vang R and Kempson RL: Perivascular
epithelioid cell tumor (‘PEComa’) of the uterus: A subset of
HMB-45-positive epithelioid mesenchymal neoplasms with an uncertain
relationship to pure smooth muscle tumors. Am J Surg Pathol.
26:1–13. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Insabato L, De Rosa G, Terracciano LM,
Fazioli F, Di Santo F and Rosai J: Primary monotypic epithelioid
angiomyolipoma of bone. Histopathol. 40:286–290. 2002. View Article : Google Scholar
|
|
7
|
Lian DW, Chuah KL, Cheng MH and Yap WM:
Malignant perivascular epithelioid cell tumour of the fibula: A
report and a short review of bone perivascular epithelioid cell
tumour. J Clin Pathol. 61:1127–1129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Torii I, Kondo N, Takuwa T, Matsumoto S,
Okumura Y, Sato A, Tanaka F, Nishigami T, Hasegawa S and Tsujimura
T: Perivascular epithelioid cell tumor of the rib. Virchows Arch.
452:697–702. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Righi A, Dimosthenous K and Rosai J:
PEComa: Another member of the MiT tumor family? Int J Surg Pathol.
16:16–20. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamashita K and Fletcher CD: PEComa
presenting in bone: Clinicopathologic analysis of 6 cases and
literature review. Am J Surg Pathol. 34:1622–1629. 2010.PubMed/NCBI
|
|
11
|
Desy NM, Bernstein M, Nahal A, Aziz M,
Kenan S, Turcotte RE and Kahn LB: Primary perivascular epithelioid
cell neoplasm (PEComa) of bone: Report of two cases and review of
the literature. Skeletal Radiol. 41:1469–1474. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kazzaz D, Khalifa M, Alorjan M, Shaw M,
Rezajooi K and Saifuddin A: Malignant PEComa of the lumbar
vertebra: A rare bone tumour. Skeletal Radiol. 41:1465–1468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lao IW, Yu L and Wang J: Malignant
perivascular epithelioid cell tumor (PEComa) of the femur: A case
report and literature review. Diagn Pathol. 10:542015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu H, Zhu X, Sheng H, Gao H, Xiao W and
Wang C: Case report primary perivascular epithelioid cell neoplasm
of thigh bone: A case report and literature review. Int J Clin Exp
Pathol. 9:2487–2491. 2016.
|
|
15
|
Zamboni G, Pea M, Martignoni G, Zancanaro
C, Faccioli G, Gilioli E, Pederzoli P and Bonetti F: Clear cell
‘sugar’ tumor of the pancreas. A novel member of the family of
lesions characterized by the presence of perivascular epithelioid
cells. Am J Surg Pathol. 20:722–730. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fletcher CDM, Unni KK and Mertens F: World
Health Organization classification of tumours: Pathology and
genetics of tumours of soft tissue and bone. Cancer. 177:1365–1376.
2002.
|
|
17
|
Eble JN: Angiomyolipoma of kidney. Semin
Diagn Pathol. 15:21–40. 1998.PubMed/NCBI
|
|
18
|
Urban T, Lazor R, Lacronique J, Murris M,
Labrune S, Valeyre D and Cordier JF: Pulmonary
lymphangioleiomyomatosis. A study of 69 patients. Groupe d'Etudes
et de Recherche sur les Maladies ‘Orphelines’ Pulmonaires. Medicine
(Baltimore). 78:321–337. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gaffey MJ, Mills SE, Askin FB, Ross GW,
Sale GE, Kulander BG, Visscher DW, Yousem SA and Colby TV: Clear
cell tumor of the lung. A clinicopathologic, immunohistochemical,
and ultrastructural study of eight cases. Am J Surg Pathol.
14:248–259. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gaffey MJ, Mills SE, Zarbo RJ, Weiss LM
and Gown AM: Clear cell tumor of the lung. Immunohistochemical and
ultrastructural evidence of melanogenesis. Am J Surg Pathol.
15:644–653. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bonetti F, Pea M, Martignoni G, Doglioni
C, Zamboni G, Capelli P, Rimondi P and Andrion A: Clear cell
(‘sugar’) tumor of the lung is a lesion strictly related to
angiomyolipoma-the concept of a family of lesions characterized by
the presence of the perivascular epithelioid cells (PEC).
Pathology. 26:230–236. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bleeker JS, Quevedo JF and Folpe AL:
‘Malignant’ perivascular epithelioid cell neoplasm: Risk
stratification and treatment strategies. Sarcoma. 2012:5416262012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rakowski SK, Winterkorn EB, Paul E, Steele
DJ, Halpern EF and Thiele EA: Renal manifestations of tuberous
sclerosis complex: Incidence, prognosis, and predictive factors.
Kidney Int. 70:1777–1782. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Costello LC, Hartman TE and Ryu JH: High
frequency of pulmonary lymphangioleiomyomatosis in women with
tuberous sclerosis complex. Mayo Clin Proc. 75:pp. 591–594. 2000;
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Flieder DB and Travis WD: Clear cell
‘sugar’ tumor of the lung: Association with
lymphangioleiomyomatosis and multifocal micronodular pneumocyte
hyperplasia in a patient with tuberous sclerosis. Am J Surg Pathol.
21:1242–1247. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Folpe AL, Mentzel T, Lehr HA, Fisher C,
Balzer BL and Weiss SW: Perivascular epithelioid cell neoplasms of
soft tissue and gynecologic origin: A clinicopathologic study of 26
cases and review of the literature. Am J Surg Pathol. 29:1558–1575.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Armah HB and Parwani AV: Perivascular
epithelioid cell tumor. Arch Pathol Lab Med. 133:648–654.
2009.PubMed/NCBI
|
|
28
|
Folpe AL and Deyrup AT: Alveolar soft-part
sarcoma: A review and update. J Clin Pathol. 59:1127–1132. 2006.
View Article : Google Scholar : PubMed/NCBI
|