Introduction
Radiotherapy has been identified as the most common
cancer treatment over the past decades; it has been estimated that
over half of all cancer patients will receive radiotherapy during
the course of their treatment (1).
Unfortunately, the efficacy of this treatment modality is low in
several malignancies due to the resistance of cancer to radiation
(2,3).
Ionizing radiation primarily eradicates cancer cells by damaging
the DNA of irradiated cells (2).
Nevertheless, the radio-responsiveness of cancer cells is modulated
by multiple mechanisms, including cell cycle checkpoint function,
DNA repair and cell death pathways (2,4,5).
Cell cycle checkpoints function as the ‘guardians’
of the cell in response to DNA damage and serve a critical role for
cell survival following exposure to radiation (6,7).
Radiation-induced DNA damage triggers cell cycle checkpoints to
halt cell cycle progression. This allows for the repair of damage
or promotes death of cells with unrepaired DNA damage (7). The majority of cancer types exhibit
defects in cell cycle checkpoints, leading to resistance to
radiotherapy (8). The proficiency of
the cell cycle checkpoints determines the sensitivity of cancer
cells to anticancer treatment. Thus, it is highly likely that the
proficiency of cell cycle checkpoints is a potential biomarker for
predicting radiation and drug responses of tumors. Targeting cell
cycle checkpoint defects is being discussed as the next generation
of anticancer therapy (9). This
therapeutic approach relies on defective checkpoints in cancer
cells and sensitizes them to radiation therapy (10–12).
Investigating checkpoint defects and the synthetic lethal targeting
of defective checkpoints in cancer cells may represent a promising
approach for increasing the efficacy of radiotherapy for individual
cancer patients.
Recently, it was reported by the present authors
that three human cholangiocarcinoma (CCA) cell lines, with
variances in cell cycle defects, differed markedly in their
radiation sensitivities. The different radiation sensitivities were
associated with existing G1 or G2 checkpoint
defects in the analyzed cells (10).
CCA cells with an intact G1 checkpoint were the most
sensitive cells to radiation. CCA cells with a defective
G1 checkpoint but intact G2 checkpoint were
the most radio-resistant cells. Furthermore, inhibition of
checkpoint kinase 1/2 (Chk1/2) selectively enhanced the radiation
sensitivity of CCA cells with a defect in the G1
checkpoint. This indicated that defective cell cycle checkpoints
might be used as biomarkers for predicting the responsiveness to
radiation in individual CCA patients.
Frequently, cancer cells encompass defects in
G1 checkpoints due to a loss of p53 function, resulting
in resistance to radiation treatment (13). The targeting of G2
checkpoint functions effectively enhanced the radio-sensitivity of
cancer cells with defective G1 checkpoints but intact
G2 checkpoints (11,12). Our
hypothesis is that cancer cells defective in the G1 and
G2 checkpoints would not be radiosensitized by targeting
of the G2 checkpoint. Thus, it is crucial to identify
the cell cycle checkpoint defects for the application of synthetic
lethal targeting of cell cycle checkpoints. However, the
identification of checkpoint defects using individual patient
tissue is cumbersome and not clinically applicable. Thus, the
present study sought a substance capable of sensitizing
G1 checkpoint-defective cell lines independent of the
status of their G2 checkpoints.
Etoposide has been widely used as an anticancer
chemotherapeutic drug (14).
Etoposide induces DNA double-strand breaks during the S and
G2 phases of the cell cycle via inhibition of DNA
topoisomerase II activity (15). It
is highly likely that etoposide-induced DNA damage prior to
irradiation renders cancer cells more vulnerable to the effects of
radiation. Therefore, the present study investigated whether
etoposide enhances radiation sensitivity of cells from two
p53-defective CCA cell lines with G1 checkpoint defects,
which differ in their G2 checkpoint status.
Materials and methods
Cell culture
The two CCA cell lines KKU-M055 (JCRB1551) and
KKU-M214 (JCRB1556), and MMNK1 (JCRB1554) were obtained from
Japanese Collection of Research Bioresources Cell Bank (Tokyo,
Japan). KKU-M055 was established from a poorly differentiated
adenocarcinoma tumor of a CCA patient. KKU-M214 was established
from the moderately differentiated adenocarcinoma of a CCA patient.
MMNK1 was established from immortalized normal human cholangiocytes
(16). The human cervical carcinoma
cell line (SiHa; ATCCHTB-35) was a gift from Dr D. Nantajit
(Chulabhorn Hospital, Bangkok, Thailand). MMNK1 and SiHa were used
as reference cell lines, due to their expression of full-length p53
(16,17).
The cells were cultured in Dulbecco's modified Eagle
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
containing 2.5 mM L-glutamine, 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), 0.25% sodium bicarbonate
supplemented with 100 units/ml penicillin, 100 µg/ml streptomycin,
and 0.25 µg/ml Amphotericin B (Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The cells were incubated in a humidified
incubator at 37°C and 5% CO2.
Cell irradiation and treatments
Exponentially growing KKU-M055 and KKU-M214 cells
were seeded into 6-well plates, ~5×104 cells/well. The
cells were irradiated with a single dose of 0, 2, 4 or 6 Gy of
X-rays generated by a 6 MV linear accelerator (Varian 2100CD,
Varian Medical Systems, Palo Alto, CA, USA). The source-to-sample
distance was 100 cm. Following irradiation, cells were incubated at
37°C, 5% CO2 in a humidified atmosphere. For etoposide
treatment, the cells were treated with 0.025 or 0.05 µg/ml
etoposide (Selleck Chemicals, Houston, TX, USA) for 24 h. Next, the
cells were irradiated as aforementioned and subsequently collected
at different time points as indicated in the applicable figures for
further analysis.
Clonogenic cell survival assays
KKU-M055 and KKU-M214 cells were seeded in
triplicate into wells of 6-well plates. The number of cells seeded
per well varied with the dose of radiation administered. The number
of cells for radiation doses of 0, 2, 4, or 6 Gy were 100, 200,
400, or 600, respectively. The cells were irradiated with a single
dose of 0, 2, 4 or 6 Gy. The cells were then allowed to grow for
10–14 days until the surviving cells produced macroscopically
visible colonies. The cells were then fixed with 95% ethanol for 10
min at room temperature and then stained with 0.4% Giemsa solution
for 10 min at room temperature. Colonies were analyzed using light
microscopy (magnification, ×10). Colonies containing >50 cells
were counted, and survival fractions were calculated as ratios of
the number of colonies formed from treated and untreated cells,
corrected for the plating efficiency of unirradiated cells.
Cell cycle analysis
A total of ~8×104 cells/well were
seeded into 6-well plates and incubated in a humidified incubator
at 37°C and 5% CO2 for 12 h. At 24 h prior to
irradiation, the cells were pretreated with 0.05 µg/ml etoposide.
Next, the pretreated cells were irradiated with a single dose of 4
Gy of X-rays as aforementioned and collected at 24 or 48 h
following irradiation. Propidium iodide (PI) staining of isolated
nuclei for cell cycle analysis was performed as described
previously (18). The suspension of
PI-stained, isolated nuclei was analyzed with a flow cytometer
(Cytomics FC500-MCL with CXP 2.2 software; Beckman Coulter, Inc.,
Indianapolis, USA).
Western blot analysis
Total protein was extracted from cells at indicated
time points following each treatment as described previously
(18). A total of 30 µg protein per
lane from each sample were separated by electrophoresis using 8, 10
or 12% SDS-PAGE and electroblotted onto polyvinylidene difluoride
membranes. The membranes were blocked in TBST containing 5% skimmed
milk for 1 h at room temperature. Next, the membranes were probed
with a primary antibody diluted in 3% bovine serum albumin (Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) in TBST overnight at
4°C. Subsequent to washing three times with TBST, the membranes
were incubated with a horseradish peroxidase-labeled secondary
antibody diluted in TBST containing 3% skimmed milk at room
temperature for 1 h. The membranes were washed three times with
TBST, and the immunoreactivity was detected using chemiluminescence
(Luminata Crescendo Western HRP substrate; Merck KGaA, Darmstadt,
Germany) with a digital phosphorimager (Chemi Doc™
XRS+Image Lab™ 5.1 software, Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The following antibodies were used: Anti-p53
(A01767; 1:1,000; Genscript, Piscataway, NJ, USA); anti-actin
(sc1616; 1:1,000) and anti-goat Immunoglobulin (Ig)G horseradish
peroxidase (HRP)-conjugated (sc2020; 1:3,000) were both purchased
from Santa Cruz Biotechnology, Inc; anti-phospho-Chk2 Thr68 (2197;
1:1,000), anti-phospho-Wee1 Ser642 (4910; 1:1,000),
anti-phospho-p53 Ser15 (9286; 1:1,000), anti-phospo-Cdc2 Tyr15
(4539; 1:1,000), anti-p21 (2947; 1:1,000), anti-PARP (9542;
1:1,000), anti-cleaved PARP (5625; 1:1,000), anti-Caspase-3 (9665;
1:1,000), Cleaved Caspase-3 (9664; 1:1,000), anti-mouse IgG
HRP-linked (7076; 1:3,000), anti-rabbit IgG HRP-linked (7074;
1:3,000) were purchased from Cell Signaling Technology, Inc.,
Danvers, MA, USA.
Nuclear staining and fluorescence
microscopy
Approximately 1×104 KKU-M055 and KKU-M214
cells were seeded on glass cover slips and cultured in 6-well
plates overnight at 37°C with 5% CO2. The cells were
pretreated with 0.05 µg/ml etoposide for 24 h. Next, the cells were
irradiated with 4 Gy of X-rays or not irradiated. At each indicated
time point, the cells seeded on cover slips were washed briefly
with PBS and stained with 8 µM Hoechst 33342 in darkness at room
temperature for 5 min. The stained cells were washed with PBS and
then mounted with anti-fade solution for fluorescence microscopy
(Zeiss HBO100 microscope Illuminating System Axiovision Rel 4.8;
Carl Zeiss AG, Oberkochen, Germany). The mode of cell death was
evaluated according to the characteristic nuclear morphologies.
Apoptotic cells were scored from cells containing bright nuclear
staining with apoptotic bodies, nuclear condensation and
fragmentation, as previously described (19). Mitotic catastrophic cells were scored
from cells containing multiple micronuclei, multinucleated and
multilobulated nuclei, as previously described (19,20).
Senescent cells were scored from cells containing nuclei with
senescence-associated heterochromatic foci, as previously described
(18). The number of apoptotic,
mitotic catastrophic or senescent cells was quantified by counting
≥800 cells for each experiment. The relative number of cells for
each mode of cell death was expressed as percentage of the total
number of counted nuclei.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent experiments. Mean and standard
deviation were calculated using the integrated functions in
Microsoft Excel 2016 (Microsoft Corporation, Redmond, WA, USA).
Trend lines were also generated using integrated functions in
Microsoft Excel.
Comparisons of D37 values (radiation dose at which
37% of cells survive compared to untreated cells) for each
etoposide pre-treatment group and etoposide untreated control group
were performed using SPSS (version 17.0; SPSS, Inc., Chicago, IL,
USA). Means of D37 values were calculated from three independent
experiments for each treatment group and each cell line. One way
analysis of variance (ANOVA) with Bonferroni post hoc testing was
used for P-value calculations. P<0.05 was considered to indicate
a statistically significant difference.
Results
Sensitivity of KKU-M055 and KKU-M214
cell lines to radiation
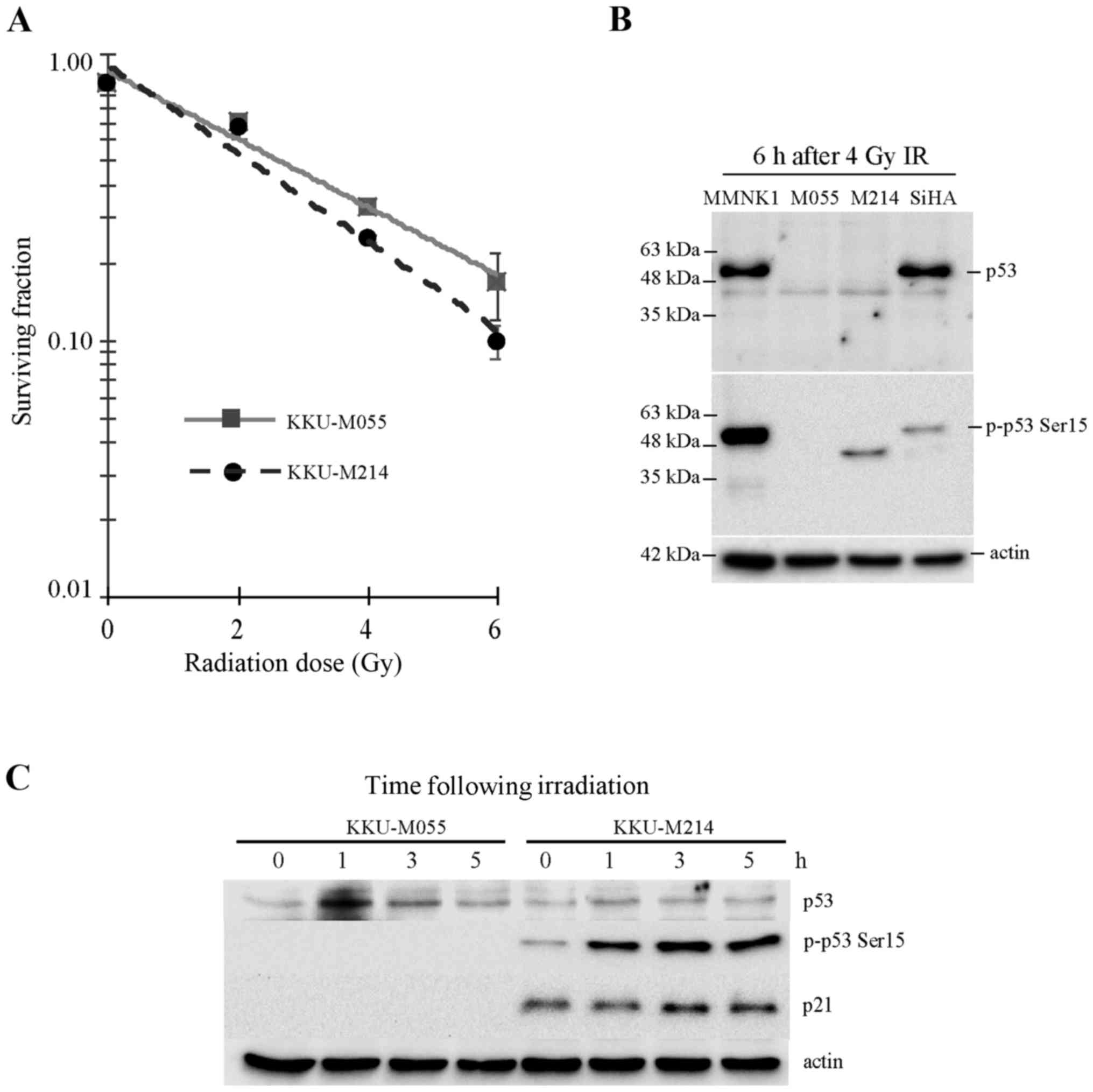
The present study assessed the radiosensitivity of
the KKU-M055 and KKU-M214 CCA cell lines. Clonogenic survival
assays were performed following X-ray irradiation. Cell survival
curves were plotted and the D37 values were calculated. As shown in
Fig. 1A, the D37 values of KKU-M055
and KKU-M214 cells were 3.61 and 2.92 Gy, respectively. This result
indicates that the poorly differentiated adenocarcinoma cell line
KKU-M055 is more resistant to radiation compared with the
moderately differentiated adenocarcinoma cell line KKU-M214.
p53 protein status in KKU-M055 and
KKU-M214 cell lines in response to radiation
The levels of p53, a crucial protein involved in DNA
repair, cell cycle arrest and cell death pathways, were evaluated
(Fig. 1B and C). It was previously
reported by the present authors that KKU-M055 and KKU-M214 CCA cell
lines express truncated, not full-length p53 (10). The result of the present study is
consistent with this previous report (Fig. 1B). Accumulation and activation of p53
are crucial for the function of p53. In response to DNA damage, p53
is phosphorylated at Ser15 by serine-protein kinase ATM. Ser15
phosphorylation contributes to the stabilization of p53 and
initiates additional phosphorylation of p53 that contribute further
to p53 induction and activation (21). An accumulation of the truncated p53
protein in KKU-M055 cells following irradiation was hardly observed
(Fig. 1C). The phosphorylation of p53
at serine 15, which is crucial for p53 stabilization and
activation, was undetectable in KKU-M055 cells. Similarly, the p53
target gene product, p21, which is crucial for the induction of
cell cycle arrest, was also undetectable in KKU-M055 cells
(Fig. 1C). Collectively, the absence
of full-length p53 expression, p53 accumulation, induction of Ser15
phosphorylation of p53 and expression of p21 in response to
radiation indicates that p53 is non-functional in response to
radiation damage in KKU-M055 cells.
The level of truncated p53 protein in KKU-M214 cells
was very low, and the accumulation of p53 following radiation
damage was not observed (Fig. 1B and
C). By contrast, the phosphorylation of p53 at Ser15 was
markedly increased in KKU-M214 cells following radiation damage.
Nevertheless, induction of p21 was hardly noticed. This observation
indicates the partial activation of the p53-p21 axis in response to
radiation.
Proficiencies of G2
checkpoints in KKU-M055 and KKU-M214 cells in response to
radiation
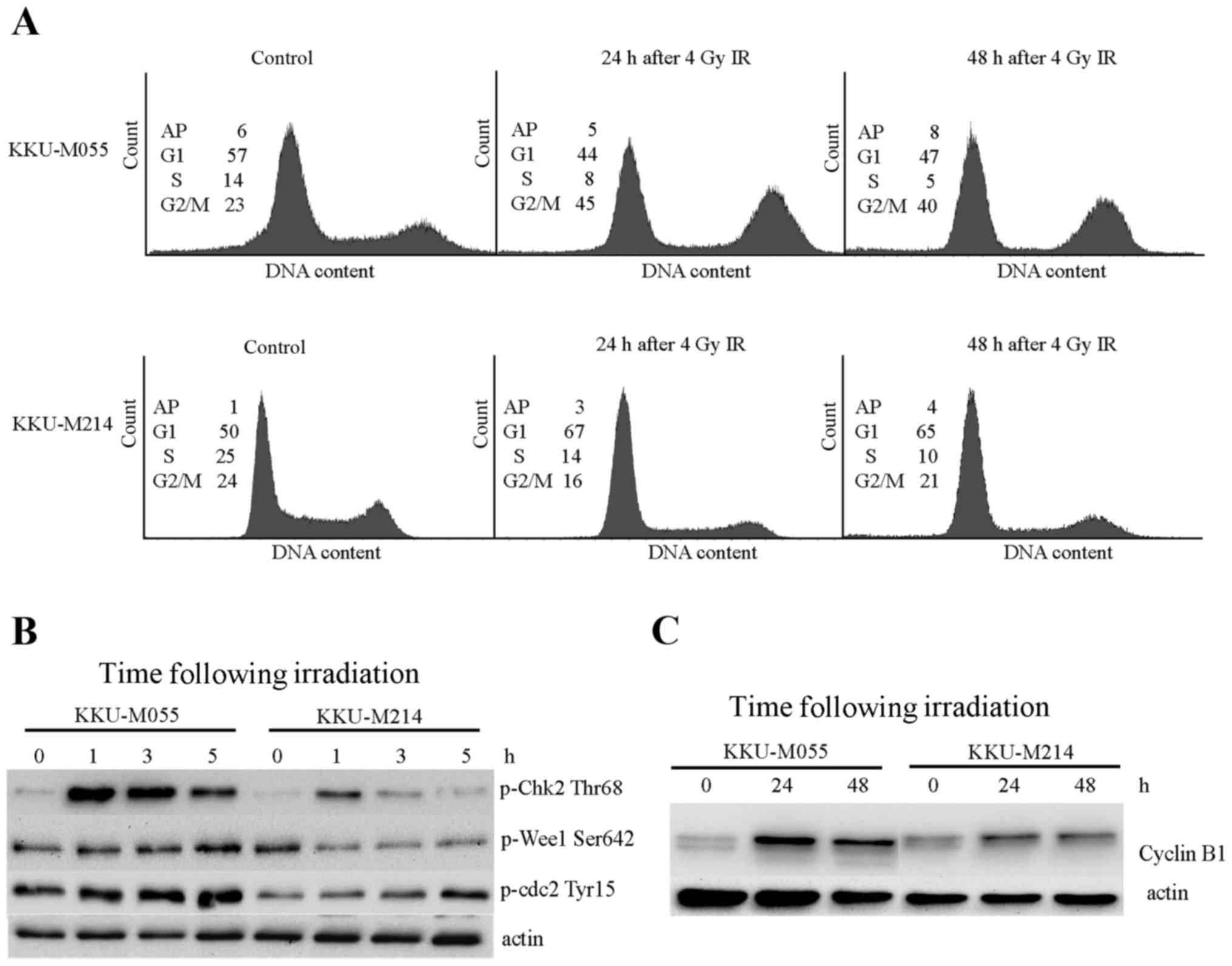
Proficiencies of G2 checkpoints in
KKU-M055 and KKU-M214 cells were evaluated by cell cycle analysis
and determination of the levels of proteins involved in
G2 checkpoint signaling. The doubling times of KKU-M055
and KKU-M214 cells were ~23–25 h. Obchoei et al (22) and Wattanawongdon et al
(23) had reported similar doubling
times of KKU-M055 and KKU-M214 cells, respectively (22,23).
Therefore, the cell cycle distribution profiles of the two cell
lines were analyzed at 24 and 48 h following irradiation (Fig. 2A). A radiation-induced G2/M
block was clearly demonstrated in KKU-M055 cells by an increase of
the G2/M population from 23 to 45% at 24 h following
irradiation. The G2/M population of KKU-M055 cells
slightly decreased from 45 to 40% at 48 h following irradiation,
which remained markedly higher compared with the control cells.
Phosphorylation of Chk2 at Thr68, Wee1-like protein kinase (Wee1)
at Ser642 and Cdc2 at Tyr15 were clearly observed in KKU-M055 cells
(Fig. 2B). Notably, the level of
cyclin B1, which is expressed predominantly during G2/M
phase, markedly increased at 24 h following irradiation in KKU-M055
cells. After 24 h (48 h after irradiation), protein levels slightly
decreased (Fig. 2C). These findings
support the results of the cell cycle analyses. Together with the
p53 and p21 expression data, this indicates the presence of an
intact radiation-induced G2 checkpoint independent of
the p53-p21 axis in KKU-M055 cells.
By contrast, the proportion of KKU-M214 cells in the
G2/M phase was not increased, as determined at 24 and 48
h following irradiation (Fig. 2A).
This result indicates a defective G2 checkpoint in
KKU-M214 cells in response to radiation damage. Slight inductions
of phospho-Chk2 Thr68, phospho-Cdc2 Tyr15 and cyclin B1 were
observed in KKU-M214 cells (Fig. 2B and
C). The induction of phosphorylation of Wee1 at Ser642 was not
observed. These findings indicated a defect in the G2
checkpoint in KKU-M214 cells. It is unlikely that the partial
activation of the p53-p21 axis in response to radiation is
associated with the G2 checkpoint functions of KKU-M214
cells.
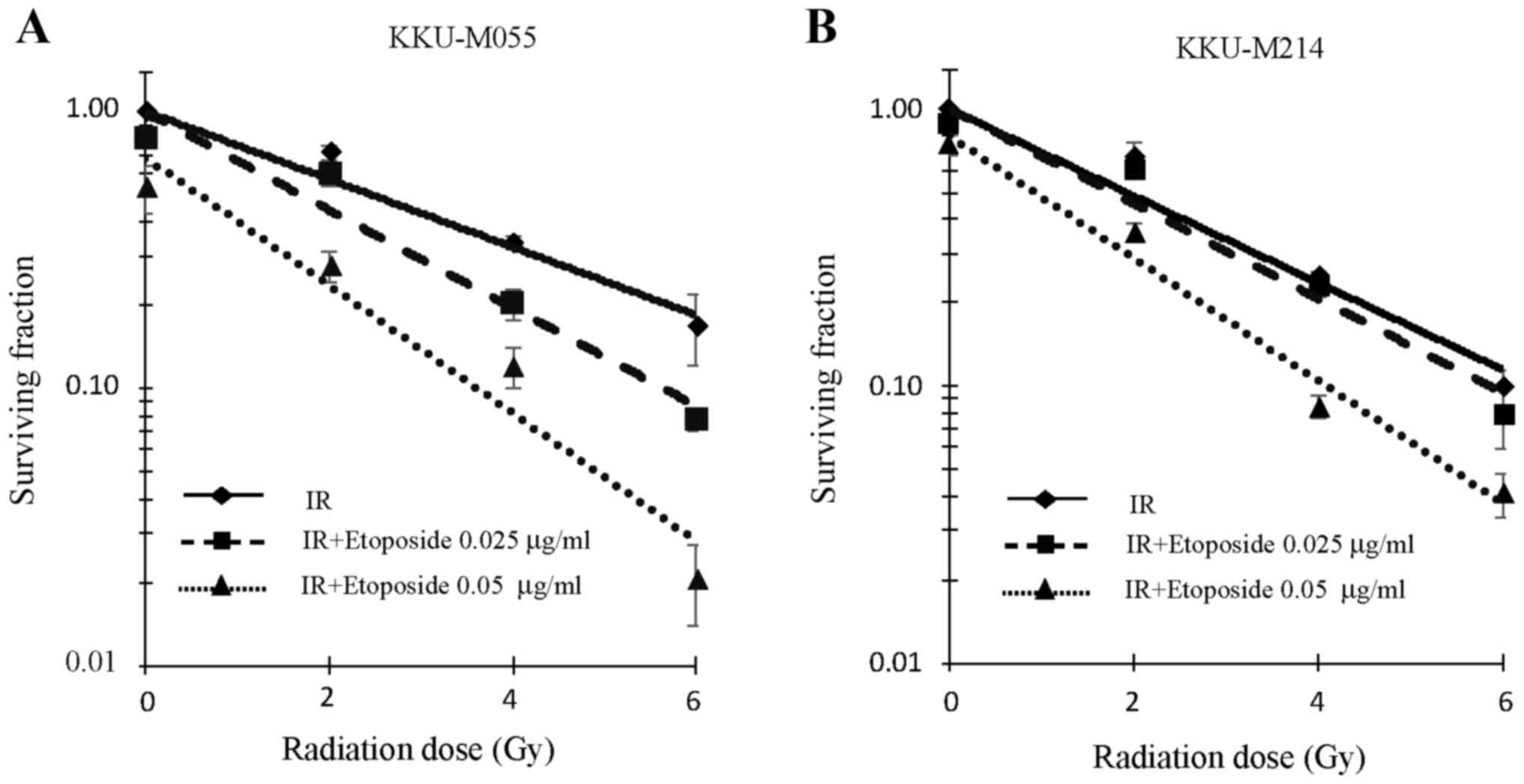
Effect of etoposide on the radiation
sensitivity of KKU-M055 and KKU-M214 cells
The aforementioned results indicate the presence of
an effective G2 checkpoint in KKU-M055 cells, but a
defective G2 checkpoint in KKU-M214 cells. The effect of
etoposide on the radiation sensitivity of KKU-M055 and KKU-M214
cells was therefore investigated. The y-intercepts of the survival
curves (fitted trend lines) of KKU-M055 cells for irradiation
alone, irradiation with 0.025 µg/ml etoposide, and irradiation with
0.05 µg/ml etoposide were 1.00, 0.99 and 0.68, respectively
(Fig. 3A). The y-intercepts of the
survival curves (fitted trend lines) of KKU-M214 cells for
irradiation alone, irradiation with 0.025 µg/ml etoposide, and
irradiation with 0.05 µg/ml etoposide were 1.00, 1.00 and 0.80,
respectively (Fig. 3B).
The clonogenic survival of KKU-M055 cells following
irradiation was decreased by pre-treatment with etoposide at
concentrations of 0.025 and 0.05 µg/ml (Fig. 3A). A D37 value of 3.62 Gy was observed
in KKU-M055 cells that were not pre-treated with etoposide.
Pre-treatment of KKU-M055 cells with 0.025 or 0.05 µg/ml of
etoposide reduced the D37 value to 2.42 or 1.05 Gy, respectively.
Thus, etoposide pre-treatment significantly enhanced
radiosensitivity of KKU-M055 cells (ANOVA test for the presence of
a difference in D37 mean values, P=0.00002; post hoc values for
0.025 µg/ml etoposide or 0.05 µg/ml etoposide pre-treatment groups
vs. irradiation alone treatment group, P=0.00122 or P=0.00002,
respectively). A D37 value of 2.92 Gy was observed in KKU-M214
cells that were not pre-treated with etoposide. Pre-treatment of
KKU-M214 cells with 0.025 or 0.05 µg/ml of etoposide reduced the
D37 value to 2.61 or 1.51 Gy, respectively (Fig. 3B). These cells were also significantly
radiosensitized by etoposide pre-treatment (ANOVA test for the
presence of a difference in D37 mean values, P=0.00001; post hoc
values of 0.025 or 0.05 µg/ml etoposide pre-treatment groups vs.
irradiation alone, P=0.04458 or P=0.00002, respectively).
Data from the cell survival assay indicated that the
radiosensitization activity of etoposide was most potent and
significant at a concentration of 0.05 µg/ml. Therefore, a
concentration of 0.05 µg/ml etoposide was used in cell cycle
analysis, western blot analysis and nuclear staining
experiments.
Effect of etoposide on the DNA damage
response pathway of KKU-M055 and KKU-M214 cells
The aforementioned results demonstrate the
radiosensitizing property of etoposide on KKU-M055 and KKU-M214
cells. Thus, the effect of etoposide on the DNA damage response
pathway of KKU-M055 and KKU-M214 cells was investigated (Fig. 4A and B). Treatment of KKU-M055 cells
with 0.05 µg/ml etoposide did not induce expression of p53 or p21,
or the phosphorylation of p53 at Ser15. This result indicates a
non-functional p53-p21 axis in response to etoposide in KKU-M055
cells. Treatment of KKU-M214 cells with etoposide did not induce
p53 expression. However, the induction of phosphorylation of p53 at
Ser15, phosphorylation of Chk2 at Thr68 and expression of p21 were
evident. These findings indicated a partial activation of the
p53-p21 axis in KKU-M214 cells in response to etoposide treatment.
The induction of phosphorylation of Chk2 at Thr68 was clearly
observed in the two cell lines. However, the levels of phospho-Wee1
Ser642, and phospho-Cdc2 did not markedly increase following
etoposide treatment in the two cell lines. Therefore, it is
unlikely that etoposide contributed to G2 checkpoint
activation in these cells.
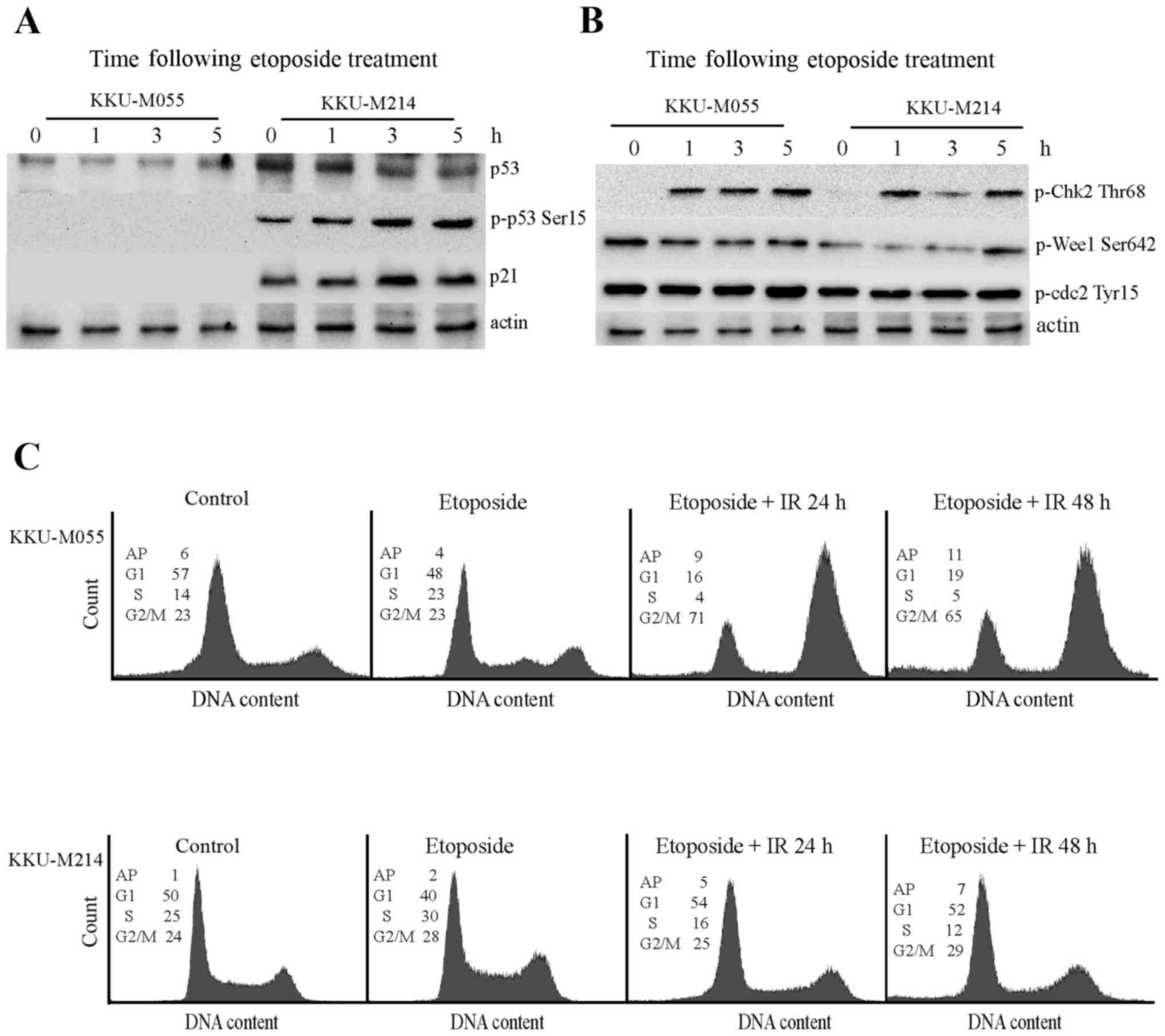 | Figure 4.Effects of etoposide on the DNA
damage response pathways in KKU-M055 and KKU-M214 cells. The cells
were treated with 4 Gy X-rays or etoposide alone (0.05 µg/ml) or
pretreated with etoposide for 24 h prior to X-ray irradiation. The
cells were collected at different time points for protein
extraction and cell cycle analysis. (A) The levels of p53, p-p53
Ser15 and p21 in extracts of KKU-M055 and KKU-M214 cells, following
etoposide treatment were determined by western blot analysis. (B)
The levels of relevant proteins for G2 checkpoint signaling in
extracts of KKU-M055 and KKU-M214 cells following etoposide
treatment were determined by western blot analysis. The detection
of actin was used as a loading control. (C) Cell cycle distribution
profiles were analyzed by flow cytometry. The numbers in the
histograms indicate the percentages of the cells in each phase of
the cell cycle (G1, S and G2/M) or AP. IR,
irradiation; p-p53 Ser15, tumor protein p53 phosphorylated at
Ser15; p21, cyclin-dependent kinase inhibitor 1A; p-Chk Thr68,
checkpoint kinase 2 phosphorylated at Thr68; Wee1, Wee1-like
protein kinase; Cdc2, cyclin-dependent kinase 1; AP,
aneuploidy. |
The cell cycle distribution profiles demonstrate
that etoposide induced a S-phase delay in KKU-M055 cells. Combined
treatment with 0.05 µg/ml etoposide and 4 Gy X-rays induced the
accumulation of KKU-M055 cells at the G2/M phase as
determined at 24 and 48 h following irradiation. By contrast,
treatment of KKU-M214 cells with etoposide alone or in combination
with radiation only had a minor impact on cell cycle distribution
(Fig. 4C).
Distinct modes of cell death are
induced by etoposide or radiation or a combination thereof in
KKU-M055 and KKU-M214 cells
The effect of etoposide on radiation-induced cell
death in KKU-M055 and KKU-M214 cells was investigated further. The
cells were exposed to X-rays, etoposide, or a combination of X-ray
irradiation and etoposide. The mode of cell death was evaluated
according to the nuclear morphological characteristics as described
in materials and methods. The results revealed that apoptosis was
the dominant mode of cell death in KKU-M055 cells (Fig. 5A and B), whereas mitotic catastrophe
was the dominant mode of cell death in KKU-M214 cells (Fig. 5A and B). Poly (ADP-ribose) polymerase
cleavage was clearly observed in KKU-M055 and KKU-M214 cells
following treatment with either X-rays or etoposide, or the
combined treatment of X-ray irradiation and etoposide (Fig. 5C). No reduction in full-length caspase
3 was observed, which fits well with the absence of cleaved caspase
3 (Fig. 5C). This result indicates
that cell death induced by etoposide or X-rays, or a combination
thereof, is caspase-independent.
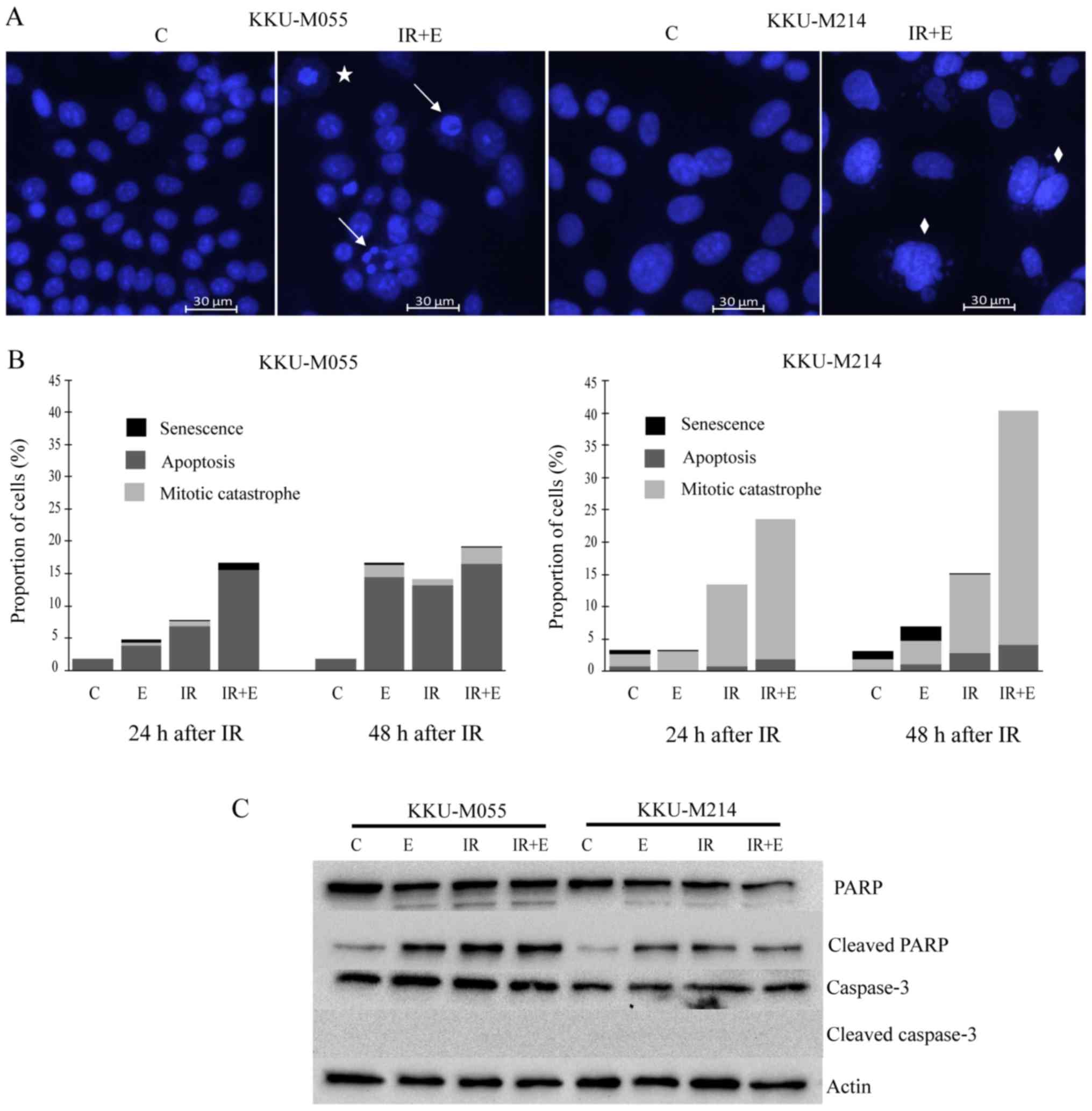 | Figure 5.Distinct modes of cell death induced
by etoposide or radiation or a combination thereof in KKU-M055 and
KKU-M214 cells. The cells were pretreated with 0.05 µg/ml etoposide
for 24 h. Subsequently, the cells were irradiated with X-rays (4
Gy) or left without irradiation. The control cells were neither
treated with etoposide nor irradiated. After 24 and 48 h, the cells
were stained with Hoechst 33342. Apoptosis, mitotic catastrophe or
senescence was identified as described in the Materials and
methods. (A) Representative images of nuclear staining with Hoechst
33342 indicates apoptotic (arrows), mitotic catastrophic (diamonds)
and senescent cells (star). (B) The frequencies of apoptosis,
mitotic catastrophe and senescence were quantified by fluorescence
microscopy of Hoechst 33342 nuclear-stained cells. (C) Levels of
key apoptotic proteins were determined by western blot analysis.
The detection of actin was used as a loading control C, control
cells; E, cells treated with etoposide; IR, irradiation; PARP, poly
(ADP-ribose) polymerase; IR+ET, cells treated with a combination of
X-rays and etoposide. |
Discussion
Resistance of cancer cells to radiation is the most
important reason for treatment failures of radiation therapy
(1,2,24,25). The synthetic-lethal targeting of
defective cell cycle checkpoints could enhance the sensitivity of
cancer patients to radiation (9,11,12). However, identification of individual
patient's tissue checkpoint defects is not practical in the clinic.
The present study demonstrated that etoposide radiosensitizes
p53-defective CCA cell lines, independent of their G2
checkpoint competencies.
The p53 protein serves a notable role in the
regulation of cell cycle checkpoints and cell death pathways
(25–27). p53 is crucial for G1
checkpoint induction but less essential for G2 checkpoint
regulation (28–30). It was previously reported that
KKU-M055 and KKU-M214 cell lines expressing a truncated p53 protein
are defective in G1 checkpoint control (10). Expression of a truncated p53 protein
was also indicated in the present study by western blot analyses.
Extracts of immortalized normal human cholangiocyte cells (MMNK1)
and human cervical carcinoma cells (SiHa) were used as reference
material to display full-length p53 bands in western blot
experiments (16,17). A limitation of the present study is
that there was no data of control experiments with CCA cell line
expressing full-length p53.
In the present study, it was revealed that KKU-M055
cells possess an effective G2 checkpoint. Effectiveness
of the G2 checkpoint of KKU-M055 cells was depicted by
the marked accumulation of cells at the G2/M phase,
together with the induction of Chk2, Wee1 and Cdc2 phosphorylation
following irradiation. However, the activation of the p53-p21 axis
in response to radiation in KKU-M055 cells could not be detected.
This finding indicates that the G2 checkpoint of
KKU-M055 cells is activated independent of the p53-p21 axis. By
contrast, the presence of a defective G2 checkpoint was
clearly demonstrated in KKU-M214 cells. These cells failed to halt
the cell cycle at the G2/M phase following irradiation.
The induction of p53 phosphorylation was observed following
radiation. However, p53 phosphorylation did not contribute to the
induction of cell cycle arrest in KKU-M214 cells at the
G2/M phase. The present study demonstrated a
radio-sensitizing effect of etoposide in CCA cell lines, which are
p53- and G1 checkpoint-defective. Evidence indicates
that different p53 defects exhibit distinct p53 activities
(26,31). The association between the
radiosensitizing effects of etoposide and different functional p53
proteins requires clarification.
Etoposide is a highly active inducer of DNA double
strand breaks and G2 arrest in mammalian cells (14,15). No
G2/M arrest was observed following the exposure of the
two CCA cell lines to etoposide alone. Incubation of KKU-M055 cells
(which have an intact G2 checkpoint) with etoposide
prior to irradiation markedly increased the percentage of the cells
in the G2/M phase compared with irradiation alone.
However, the increased induction of G2/M arrest by
etoposide was not observed in KKU-M214 cells (defective
G2 checkpoint). Etoposide and radiation had a
synergistic effect on the survival of the two CCA cell lines,
regardless of their G2 checkpoint functionalities. Thus,
it can be postulated that the G2/M arrest is not the
determinant mechanism for the radiosensitization activity of
etoposide.
The mechanism by which etoposide radiosensitizes the
two CCA cell lines may be associated with the promotion of cell
death. Etoposide may promote cell death by generating DNA double
strand breaks within the cells prior to irradiation. The existing
DNA double strand breaks would render the cell more vulnerable to
the killing effects of radiation. To investigate this hypothesis,
DNA damage could be quantified by analyzing well-defined markers of
DNA damage such as γ-histone H2AX or p53-binding protein 1 in
future research projects. In the present study, it was indicated
that the treatment of the cells of the two cell lines with
etoposide prior to irradiation increased cell death without
altering the dominant modes of cell death. Apoptosis and mitotic
catastrophe are the principal modes of cell death in
radiation-damaged cells (2).
Following irradiation, cancer cells with G2 checkpoint
defects cannot complete the repair of DNA damage before entering
into mitosis. The cells subsequently attempt to divide and
subsequently die through mitotic catastrophe (32). The findings from previous (10) and present studies indicate that p53
and G1 checkpoint functions are impaired in the two CCA
cell lines. Therefore, it is likely that G2 checkpoint
functionality determines the modes of cell death of these cells
following irradiation. Apoptosis was found to be the dominant mode
of cell death in KKU-M055 cells with an intact G2
checkpoint, whereas mitotic catastrophe was the dominant mode of
cell death in KKU-M214 cells with a defect in the G2
checkpoint.
It was previously reported that
etoposide/5-fluorouracil/leucovorin combination treatment improved
the overall survival of patients with advanced pancreatic or
biliary cancer (33,34). However, the success of this treatment
regime is limited due to its toxicity. The present study used very
low concentrations of etoposide (0.025 and 0.05 µg/ml; <0.1 µM)
and revealed clear radiosensitization effects of etoposide on the
two cell lines. This finding indicates that low concentrations of
etoposide could be used as a low cytotoxic radiosensitizer for the
treatment of CCA patients.
According to data from the SIB Bioinformatics
Resource Portal ExPASy, the KKU-M214 cell line is a KKU-M213 cell
line derivative (35). A
contamination of the KKU-M214 cell line with KKU-M213 cells cannot
be excluded in the present study. The current study investigated
radio-sensitizing effects of etoposide on two cell populations
differing in their G2 checkpoint status and clearly
indicated that the two cell populations differ in this status. The
two cell populations were sensitized by etoposide to undergo
radiation-induced cell death. Thus, interpretation of the study
results would not differ if the KKU-M214 cell line were in fact a
mixed intrahepatic CCA type.
In conclusion, the intrinsic radioresistance of
cancer cells is a core obstacle to the success of radiation
treatment. The efficiency of radiotherapy can be improved by
enhancing the radiosensitivity of cancer cells in vivo. The
present study demonstrated the radiosensitizing effect of etoposide
on two p53-defective CCA cell lines with either an intact or a
defective G2 checkpoint. This provides good evidence
that etoposide can be used as a radiosensitizer for tumors,
independent of the functionalities of their G2
checkpoints.
Acknowledgements
The present study was supported by Naresuan
University research fund (grant no. R2558C122).
References
|
1
|
Delaney G, Jacob S, Featherstone C and
Barton M: The role of radiotherapy in cancer treatment: Estimating
optimal utilization from a review of evidence-based clinical
guidelines. Cancer. 104:1129–1137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maier P, Hartmann L, Wenz F and Herskind
C: Cellular pathways in response to ionizing radiation and their
targetability for tumor radiosensitization. Int J Mol Sci.
17:E1022016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malik A, Sultana M, Qazi A, Qazi MH,
Parveen G, Waquar S, Ashraf AB and Rasool M: Role of natural
radiosensitizers and cancer cell radioresistance: An update. Anal
Cell Pathol (Amst). 2016:61465952016.PubMed/NCBI
|
|
4
|
Wang H, Zhang X, Teng L and Legerski RJ:
DNA damage checkpoint recovery and cancer development. Exp Cell
Res. 334:350–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morgan MA and Lawrence TS: Molecular
pathways: Overcoming radiation resistance by targeting DNA damage
response pathways. Clin Cancer Res. 21:2898–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Visconti R, Della Monica R and Grieco D:
Cell cycle checkpoint in cancer: A therapeutically targetable
double-edged sword. J Exp Clin Cancer Res. 35:1532016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Deckbar D, Jeggo PA and Löbrich M:
Understanding the limitations of radiation-induced cell cycle
checkpoints. Crit Rev Biochem Mol Biol. 46:271–283. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schmitt CA: Senescence, apoptosis and
therapy-cutting the lifelines of cancer. Nat Rev Cancer. 3:286–295.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gabrielli B, Brooks K and Pavey S:
Defective cell cycle checkpoints as targets for anti-cancer
therapies. Front Pharmacol. 3:92012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hematulin A, Sagan D, Sawanyawisuth K,
Seubwai W and Wongkham S: Association between cellular
radiosensitivity and G1/G2 checkpoint proficiencies in human
cholangiocarcinoma cell lines. Int J Oncol. 45:1159–1166. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Koniaras K, Cuddihy AR, Christopoulos H,
Hogg A and O'Connell MJ: Inhibition of Chk1-dependent G2 DNA damage
checkpoint radiosensitizes p53 mutant human cells. Oncogene.
20:7453–7463. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dillon MT, Good JS and Harrington KJ:
Selective targeting of the G2/M cell cycle checkpoint to improve
the therapeutic index of radiotherapy. Clin Oncol (R Coll Radiol).
26:257–265. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JM and Bernstein A: p53 mutations
increase resistance to ionizing radiation. Proc Natl Acad Sci USA.
90:pp. 5742–5746. 1993; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thakur DS: Topoisomerase II inhibitors in
cancer Treatment. Int J Pharma Sci Nanotechnol. 3:1173–1181.
2011.
|
|
15
|
Schonn I, Hennesen J and Dartsch DC:
Cellular responses to etoposide: Cell death despite cell cycle
arrest and repair of DNA damage. Apoptosis. 15:162–172. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maruyama M, Kobayashi N, Westerman KA,
Sakaguchi M, Allain JE, Totsugawa T, Okitsu T, Fukazawa T, Weber A,
Stolz DB, et al: Establishment of a highly differentiated
immortalized human cholangiocyte cell line with SV40T and hTERT.
Transplantation. 77:446–451. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee YS, Bae SM, Kwak SY, Park DC, Kim YW,
Hur SY, Park EK, Han BD, Lee YJ, Kim CK, et al: Cell cycle
regulatory protein expression profiles by adenovirus p53 infection
in human papilloma virus-associated cervical cancer cells. Cancer
Res Treat. 38:168–177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hematulin A, Sagan D, Eckardt-Schupp F and
Moertl S: NBS1 is required for IGF-1 induced cellular proliferation
through the Ras/Raf/MEK/ERK cascade. Cell Signal. 20:2276–2285.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Amornwichet N, Oike T, Shibata A, Ogiwara
H, Tsuchiya N, Yamauchi M, Saitoh Y, Sekine R, Isono M, Yoshida Y,
et al: Carbon-ion beam irradiation kills X-ray-resistant p53-null
cancer cells by inducing mitotic catastrophe. PLoS One.
9:e1151212014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vakifahmetoglu H, Olsson M and Zhivotovsky
B: Death through a tragedy: Mitotic catastrophe. Cell Death Differ.
15:1153–1162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fei P and El-Deiry WS: P53 and radiation
responses. Oncogene. 22:5774–5783. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Obchoei S, Weakley SM, Wongkham S,
Wongkham C, Sawanyawisuth K, Yao Q and Chen C: Cyclophilin A
enhances cell proliferation and tumor growth of liver
fluke-associated cholangiocarcinoma. Mol Cancer. 10:1022011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wattanawongdon W, Hahnvajanawong C, Namwat
N, Kanchanawat S, Boonmars T, Jearanaikoon P, Leelayuwat C,
Techasen A and Seubwai W: Establishment and characterization of
gemcitabine-resistant human cholangiocarcinoma cell lines with
multidrug resistance and enhanced invasiveness. Int J Oncol.
47:398–410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hematulin A, Meethang S, Ingkaninan K and
Sagan D: Derris scandens Benth extract potentiates radioresistance
of Hep-2 laryngeal cancer cells. Asian Pac J Cancer Prev.
13:1289–1295. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morrison R, Schleicher SM, Sun Y, Niermann
KJ, Kim S, Spratt DE, Chung CH and Lu B: Targeting the mechanisms
of resistance to chemotherapy and radiotherapy with the cancer stem
cell hypothesis. J Oncol. 2011:9418762011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mirzayans R, Andrais B, Scott A and Murray
D: New insights into p53 signaling and cancer cell response to DNA
damage: Implications for cancer therapy. J Biomed Biotechnol.
2012:1703252012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Speidel D: The role of DNA damage
responses in p53 biology. Arch Toxicol. 89:501–517. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chung JH and Bunz F: Cdk2 is required for
p53-independent G2/M checkpoint control. PLoS Genet.
6:e10008632010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Passalaris TM, Benanti JA, Gewin L, Kiyono
T and Galloway DA: The G(2) checkpoint is maintained by redundant
pathways. Mol Cell Biol. 19:5872–5881. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wasylishen AR and Lozano G: Attenuating
the p53 pathway in human cancers: Many means to the same end. Cold
Spring Harb Perspect Med. 6:a0262112016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim BM and Hong Y, Lee S, Liu P, Lim JH,
Lee YH, Lee TH, Chang KT and Hong Y: Therapeutic implications for
overcoming radiation resistance in cancer therapy. Int J Mol Sci.
16:26880–26913. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rao S, Cunningham D, Hawkins RE, Hill ME,
Smith D, Daniel F, Ross PJ, Oates J and Norman AR: Phase III study
of 5FU, etoposide and leucovorin (FELV) compared to epirubicin,
cisplatin and 5FU (ECF) in previously untreated patients with
advanced biliary cancer. Br J Cancer. 92:1650–1654. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Glimelius B, Hoffman K, Sjödén PO,
Jacobsson G, Sellström H, Enander LK, Linné T and Svensson C:
Chemotherapy improves survival and quality of life in advanced
pancreatic and biliary cancer. Ann Oncol. 7:593–600. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
SIB Bioinformatics Resource Portal
(ExPASy), . Cellosaurus KKU-M214 (CVCL_M264). http://web.expasy.org/cellosaurus/CVCL_M264August
9–2017
|