Introduction
Colorectal cancer (CRC) is one of the principal
causes of cancer worldwide (1).
Unfortunately, even if a 5-year survival prognosis can be given to
90% of patients in the early stages, disappointing treatment
outcomes are recorded in subjects with extensive local invasion or
distant metastases. Generally, their 5-year survival rate is less
than 15% (2) and, even if suitable
for receiving adjuvant chemotherapy, disease-related deaths remain
stubbornly high.
A recent steady stream of important breakthroughs
has dramatically improved our understanding of CRC. One such
discovery identified the BRAF mutation as common in metastatic CRC
patients, especially those with a right-side colon cancer and a
poorly differentiated tumor (3).
Vemurafenib is a potent and selective inhibitor of mutated BRAF.
Chapman et al (4) revealed
that vemurafenib achieved 40% response rates in melanoma with a
BRAF mutation. However, unlike melanoma, the effect of vemurafenib
in CRC patients with a BRAF mutation is often negligible, resulting
in a clinical response in only 5% of patients (5). This discrepancy of outcomes suggests
that different cancer types may present important variations even
if they share the same BRAFV600E mutation.
Growing evidence has revealed that resistance to
BRAFV600E inhibition depends in part on altered c-Met
signaling in cancers. Blocking c-Met signaling may therefore help
reverse resistance to vemurafenib in BRAFV600E-targeted
therapy (6,7). Byeon et al (6) have reported that dual inhibition of
BRAFV600E and c-Met leads to a reversal of the
epithelial-to-mesenchymal transition, resulting in decreased
resistance to chemotherapy and a positive therapeutic response in
thyroid cancer. To date, only a few studies have focused on the
effects of c-Met inhibitors and anti-BRAF agents as a combined
treatment option for CRCs. We therefore investigated the combined
effect of vemurafenib and PHA-665752, a c-Met inhibitor, on in
vitro and in vivo growth of human CRC cells, with the
goal of identifying suitable clinical combinations.
Materials and methods
Animals
Fifty-six female BALB/c nu/nu nude mice (age, 4–5
weeks; weight, 20 g each) were purchased from HFK Bioscience Co.,
Ltd. (Beijing, China) and raised under specific pathogen-free
conditions at the Animal Experimental Center of the Fourth Hospital
of Hebei Medical University (Shijiazhuang, China). The animals were
allowed to adapt to the housing conditions for 5 days before being
used in experiments. The experimental protocol pertaining to the
animal study was approved by the Institutional Animal Care and the
Ethics Committee of the Fourth Hospital of Hebei Medical University
In addition, animal studies were conducted in accordance with the
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals.
Cell lines and antibodies
Human CRC cell lines RKO and HT-29
(BRAFV600E mutant) were acquired from the Cell Bank of
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). HT-29 cells were cultured in McCoy's 5A medium
and RKO cells were cultured in MEM (both from Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Cells were maintained in the
presence of 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C
in an atmosphere containing 5% CO2. The medium was
supplemented with 10% fetal calf serum (FBS; PAN-Biotech GmbH,
Aidenbach, Germany).
The following rabbit polyclonal antibodies were
purchased from Bioworld Technology (Louis Park, MN, USA):
Anti-phosphorylated (p)-c-Met (BS4752), anti-AKT (BS1007),
anti-p-AKT (BS4007), and anti-p-extracellular signal-regulated
kinase (anti-p-ERK) (BS5016). An anti-hepatocyte growth factor
(HGF) rabbit polyclonal antibody (BS1025R) was purchased from Bioss
Inc. (Woburn, MA, USA). Vemurafenib was purchased from Cayman
Chemical Co. (Ann Arbor, MI, USA); PHA-665752 was purchased from
Selleck Chemicals LLC (Houston, TX, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT)
assay
Cells were plated on 96-well microtiter plates at a
density of 3×103 cells/well, and cultured overnight to
allow for cell attachment. Cells were treated with PHA-665752 (1,
1.5, 2, 2.5, 3, 3.5, or 4 µmol/l) and/or vemurafenib (0.01, 0.1,
0.5, 1, 3, 6, or 9 µmol/l). Each microtiter plate was incubated for
4 h at 37°C. The half-maximal inhibitory concentration
(IC50) of PHA-665752 was calculated as follows:
Percent inhibition=[1–Mean absorbance of
exeperimental wells - Mean absorbance of blank wellsMean absorbance
of control wells - Mean absorbance of blank wells]x100%
The IC50 was calculated when it was equal
to 50%. In single-drug treatment experiments, a suitable
IC50 value for vemurafenib could not be obtained because
of low response to the drug. Thus, in the combined treatment
experiment, we used a fixed concentration of PHA-665752 and a
varying concentration of vemurafenib. The MTT assay determined the
viability of CRC cells at 24, 48, and 72 h. A Thermo Plate
microplate reader (Thermo Fisher Scientific, Inc.) was used to
measure sample absorbance at 490 nm.
Flow cytometry
For cell cycle detection, cells were seeded in
6-well plates at a density of 12×104 cells/well for
HT-29 and 9×104 cells/well for RKO cells. Cells were
washed three times with cold phosphate-buffered saline (PBS) and
then stained with 50 µl propidium oxide (Sigma-Aldrich, St. Louis,
MO, USA) for 30 min. Each sample was harvested at log phase and
fixed in 70% ethanol at 4°C overnight in the dark. Quantification
of cell cycle distribution was performed with a FACScan system
(Beckman Coulter, Inc., Brea, CA, USA). The percentage of cells in
G0/G1, S, and G2-M phases was calculated and compared.
Mouse xenograft studies
Cancer cells (5.5×107) were
subcutaneously implanted into the left abdominal region of mice.
Both PHA-665752 and vemurafenib were prepared by dilution in
dimethyl sulfoxide (DMSO). Treatments were administered when tumor
length reached ~0.5–0.7 cm. Mice were divided into four groups
(n=10/group): vemurafenib group [75 mg/kg peroral (p.o.) twice a
day, interval time ≥8 h], PHA-665752 group [25 mg/kg
intraperitoneal (i.p.) every other day], combined group
(vemurafenib 75 mg/kg p.o. twice a day, interval time ≥8 h;
PHA-665752 25 mg/kg i.p. every other day), and DMSO control group
(2.5% DMSO 200 µl i.p. every other day). Tumor volume was measured
every 3 days, and calculated using the following formula:
Tumor volume=[large diameter of tumor
x(small diameter of tumor)2]/2
The treatment lasted for 3 weeks. Then, mice were
sacrificed under anesthesia, and tumor tissue was obtained and
fixed in 10% formaldehyde solution prior to immunohistochemical
analysis.
Immunohistochemical analysis
Samples were fixed by formalin, embedded in
paraffin, and cut into sections (4–6-µm thick). Immunochemical
staining (IHC) was performed using the Envision plus detection
system (Dako, Glostrup, Denmark) according to the manufacturer's
instructions. Sections were incubated with methanol/hydroperoxide
(9:1) for 20 min at room temperature to block endogenous peroxidase
activity, and washed with PBS. Sections were incubated at 4°C
overnight with primary antibodies against HGF (1:100), p-c-Met
(1:100), p-Akt (1:200), and p-ERK (1:200). Secondary antibodies
(Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China)
were added, sections were incubated at 37°C for 45 min, and washed
with PBS. DAB staining was used for coloring sections and nuclei
were counterstained with hematoxylin. Yellow, yellowish-brown, or
darker immunohistochemical staining was considered as positive. The
density of positive staining was measured using a computerized
image system composed of a charge-coupled device camera (DFC420;
Leica Microsystems Imaging Solution, Ltd., Cambridge, UK) connected
to a microscope (DM IRE2; Leica). The mean number of immunopositive
cells was determined in five fields-of-view at ×400 magnification
using Leica Qwin Plus V3 software (Leica Microsystems GmbH,
Wetzlar, Germany).
Statistical analysis
Data for continuous variables are shown as mean ±
standard deviation (SD). Differences among groups were compared
using Student's t-test, one-way analysis of variance (ANOVA).
Statistical significance was considered at P-values <0.05. All
data were analyzed using SPSS software (version 21.0; SPSS, Inc.,
Chicago, IL, USA).
Results
Growth suppression by PHA-665752 and
resistance to vemurafenib in CRC cells in vitro
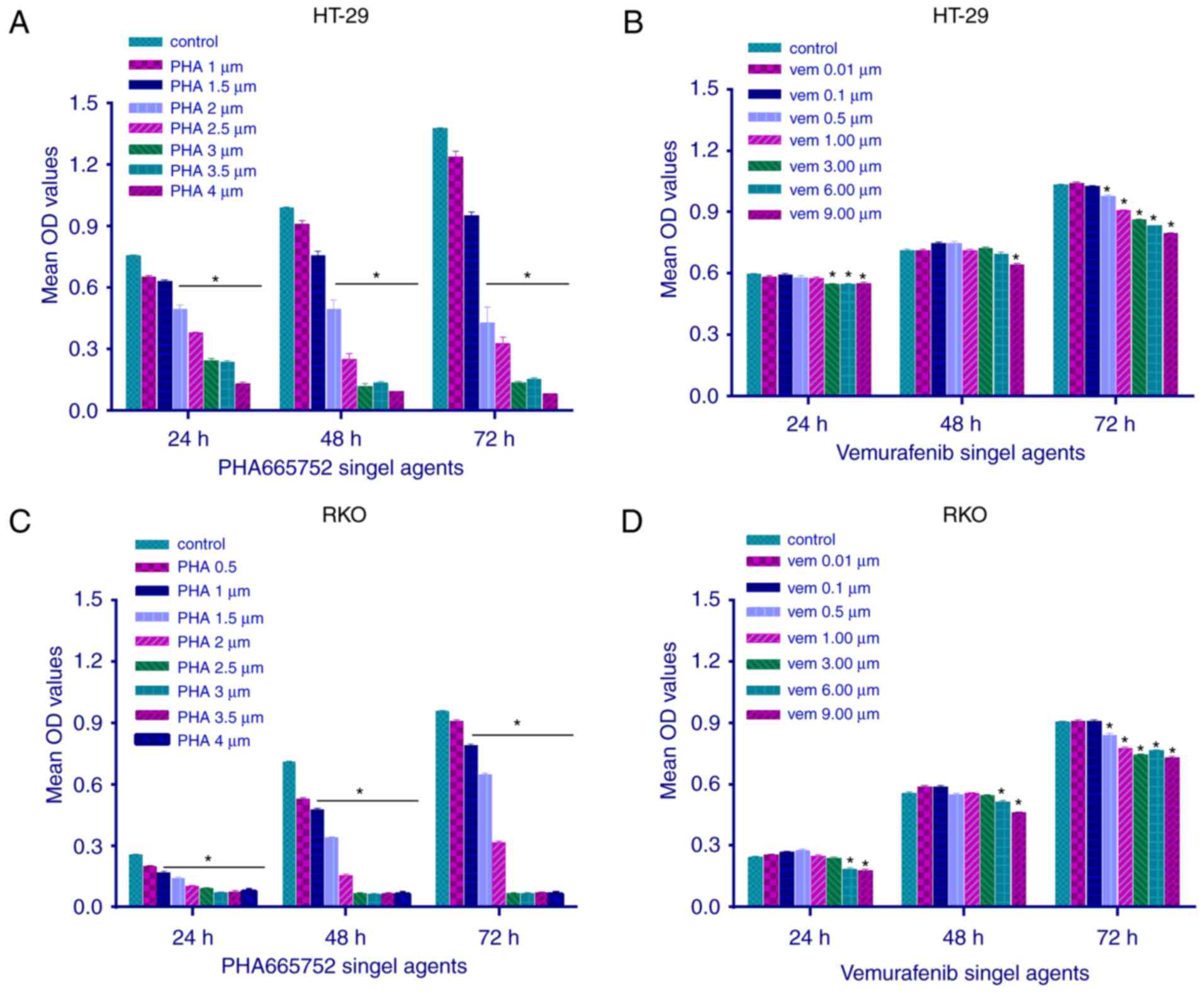
The effect of PHA-665752 on the viability of RKO and
HT-29 cells was evaluated using the MTT assay at 24, 48, and 72 h
(Fig. 1). PHA-665752 significantly
inhibited the proliferation of CRC cells with a
BRAFV600E mutation in a time- and dose-dependent manner
(P<0.05). In addition, IC50 values of PHA-665752
against HT-29 and RKO cells were 2 and 1 µmol/l, respectively
(Fig. 1A and C). Vemurafenib slightly
inhibited cell growth only at a sufficiently high concentration;
consequently, we could not calculate its IC50 value
(Fig. 1B and D; P<0.05). These
findings suggested that CRC cells with a BRAFV600E
mutation were resistant to vemurafenib therapy, whereas the
c-Met-related pathway could mediate CRC cell growth and
proliferation in vitro.
Combined PHA-665752 and vemurafenib
treatment is more effective than either agent alone in inhibiting
CRC cell proliferation
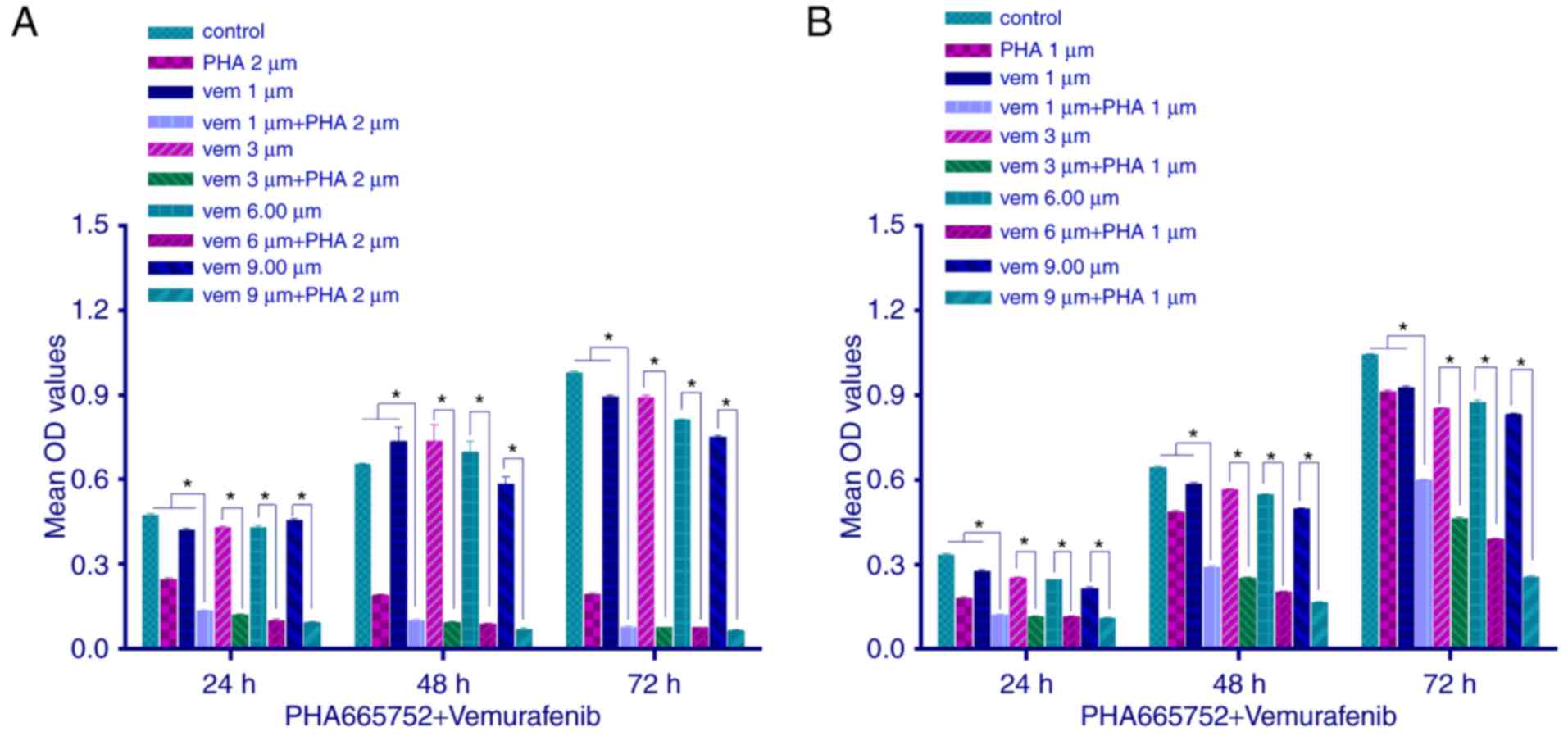
We explored the proliferation of CRC cells by MTT
assay, following a combined treatment with PHA-665752 (2 µmol/l for
HT-29 cells and 1 µmol/l for RKO cells) and vemurafenib at variable
doses. Cell proliferation was inhibited (P<0.05) to a greater
extent with the combined treatment than with any of the two drugs
alone. Moreover, as the concentration of vemurafenib but not
PHA-665752 increased, the inhibitory effect, too, increased
(Fig. 2; P<0.05). These results
indicated that in vitro combined therapy using PHA-665752
plus vemurafenib could significantly decrease tumor cell growth,
with PHA-665752 reversing vemurafenib drug resistance.
Effect of PHA-665752, vemurafenib, and
a combination of both drugs on cell cycle progression
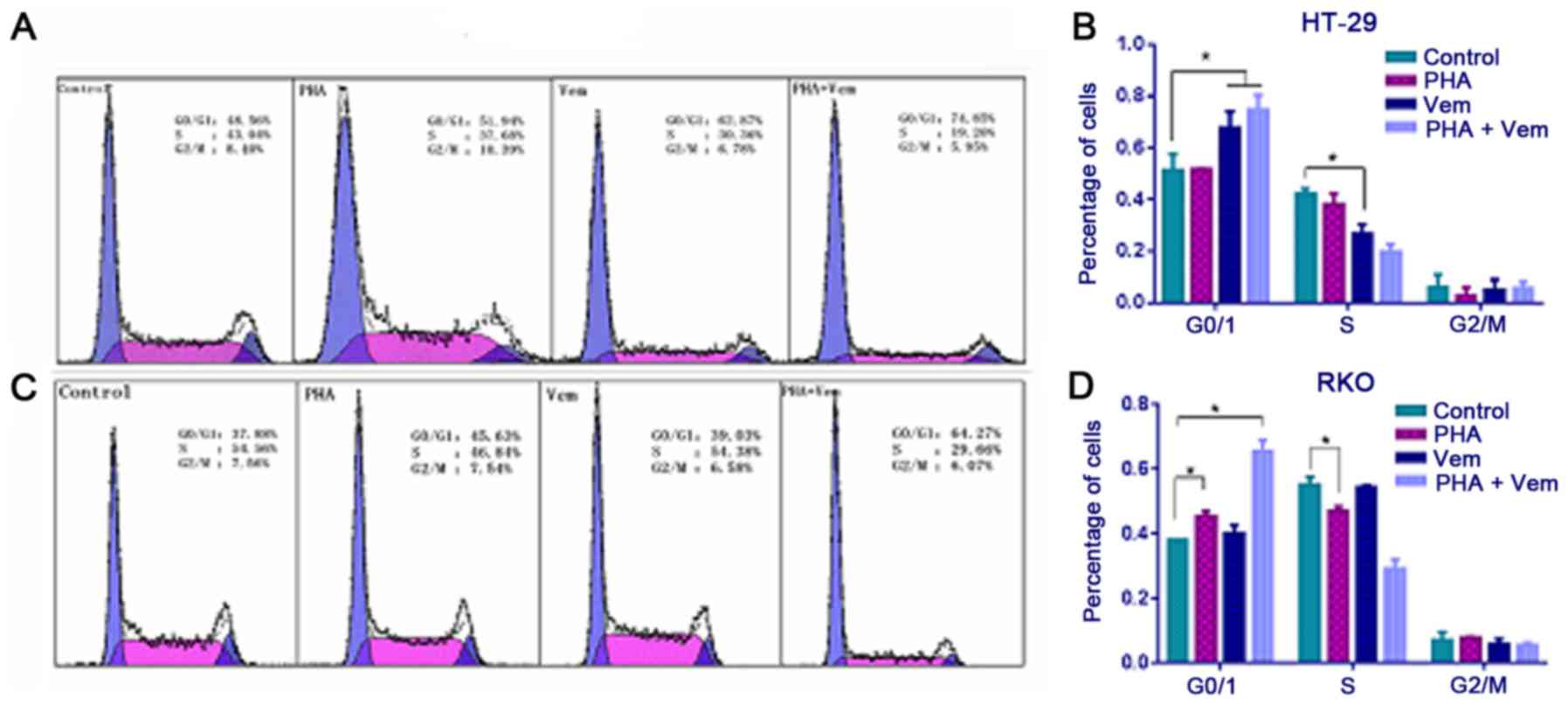
Flow cytometry revealed that treatment with
vemurafenib caused a marked increase with G0/G1 and decrease with S
phase frequency in HT-29 cells compared with the control group
(Figs. 3A and B; P<0.05). In RKO
cells, the proportion of cells in G0/G1 increased following
PHA-665752 treatment (P<0.05), whereas that in S phase decreased
relative to the control (Fig. 3B;
P<0.05). A combination of PHA-665752 and vemurafenib had a
marked effect on G0/G1 phase frequency compared with the control in
both CRC cell lines (Fig. 3C and D;
P<0.05). This finding implied that HT-29 cells in G0/G1 phase
were killed by vemurafemib, whereas RKO cells in G0/G1 were merely
blocked by PHA-665752; their effect was significantly potentiated
when the drugs were combined.
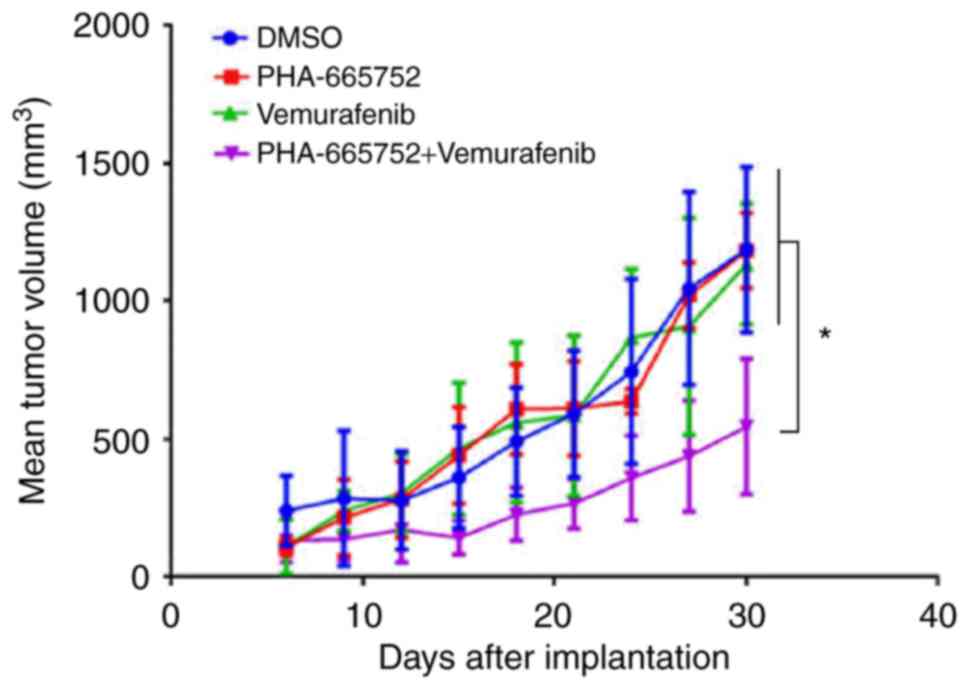
Combined PHA-665752 and vemurafenib treatment is
more effective than either agent alone in suppressing growth of
mouse xenografts bearing CRC cells. Thirty days after being
subcutaneously implanted in the left abdominal region, tumor
volumes were measured. Tumors that received combined treatment,
PHA-665752 alone, vemurafenib alone, or DMSO were: 549.22±240.93,
1207.25±129.53, 1137.31±220.21, 1213.85±295.34 mm3,
respectively (Fig. 4). Thus, tumor
volume was significantly smaller in mice subjected to combined
treatment (P<0.05). These findings suggested that combined
treatment was more effective in controlling tumor growth in mice
than either PHA-665752 or vemurafenib alone.
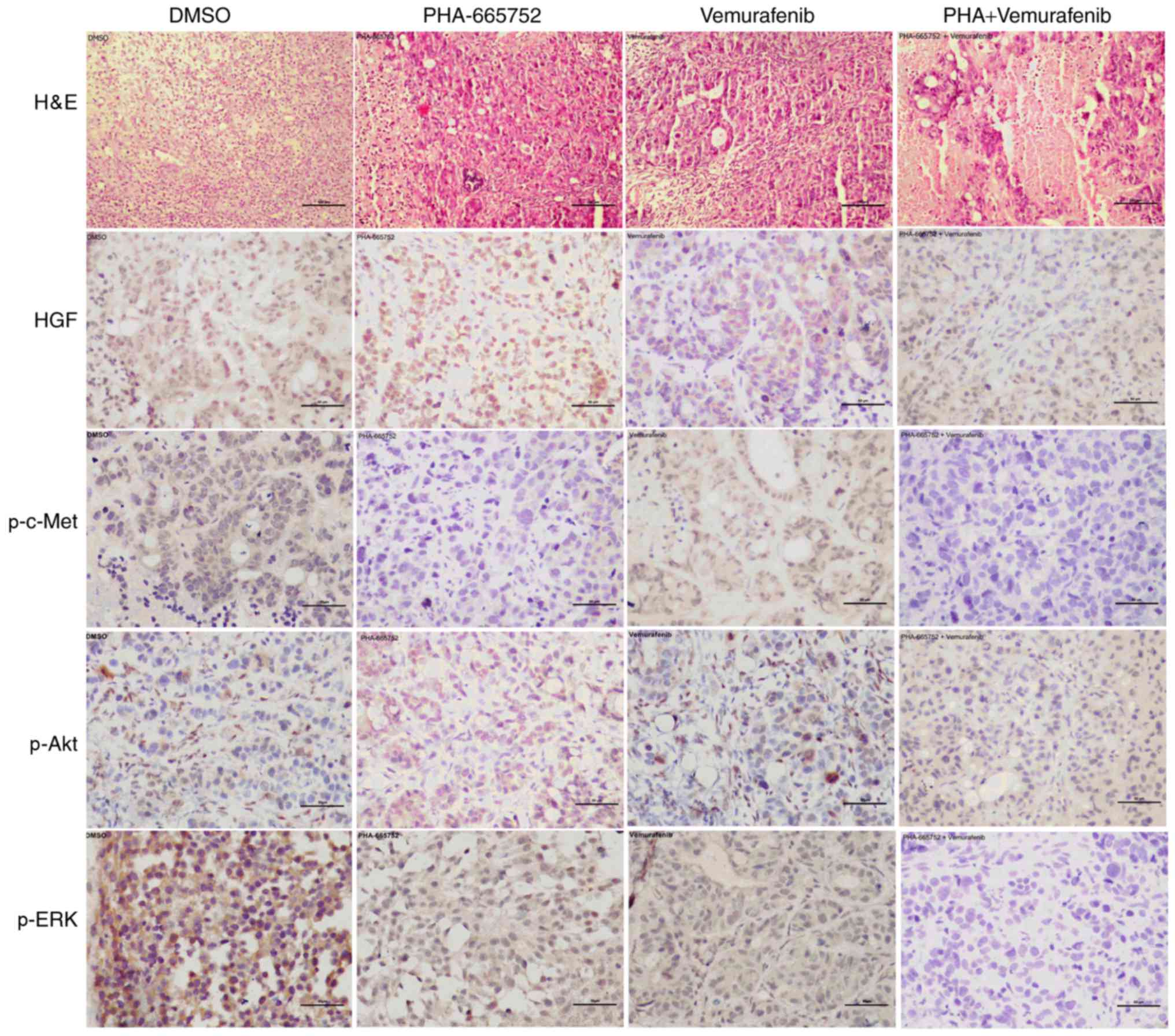
Combined therapy with PHA-665752 and vemurafenib
indicates involvement of the PI3K/AKT and MEK/ERK signaling pathway
in vivo. Finally, we used immunohistochemistry to evaluate
the expression of p-c-Met, p-ERK, and p-AKT following inhibition
with PHA-665752 alone, vemurafenib alone, or a combination of the
two. Except for HGF, the expression of the other target proteins
was significantly lower in mice treated with PHA-665752 plus
vemurafenib than DMSO (Fig. 5). These
findings suggest that effectiveness of the combined anticancer
treatment may depend on a shift of the phosphorylation status of
ERK and AKT.
Discussion
BRAF is a protein directly downstream from RAS in
the classical MAPK cascade (8). RAS
activates RAF by recruiting RAF and simulating its dimerization. In
CRC, BRAF mutations occur most commonly at the V600 site,
particularly in patients with right-side colon cancer, who are more
likely to present with poorly differentiated tumors (9,10). Cancers
with a BRAFV600E mutation have been shown to lead to a
poor prognosis (11). Therefore,
anti-BRAFV600E therapy is considered effective in
oncotherapy. Vemurafenib, a selective inhibitor of
BRAFV600E, has achieved a high objective response rate
in ~50% of melanomas, improving overall survival compared with
traditional chemotherapy (4). In
contrast to the high response rates seen in melanomas, therapeutic
efficacy in CRC has been poorly characterized. In our study, we
report that the c-Met inhibitor PHA-665752 could suppress CRC cell
survival when used alone, whereas the BRAFV600E
inhibitor vemurafenib had no significant effect on reducing CRC
cell viability. Interestingly, dual inhibition of c-Met and
BRAFV600E resulted in a significantly stronger
inhibitory effect on cancer cells than either agent alone.
Therefore, inhibition of BRAF signaling together with
c-Met-targeted therapy may offer a new effective approach for CRC
treatment in CRC patients with a BRAFV600E mutation.
Along with tumor initiation, progression, and even
metastases, tumor cells' viability, proliferation, and death
represent important parameters. Here, cell cycle analysis indicates
that BRAFV600E and c-Met inhibitors alone can only block
cell growth of RKO and HT-29 cells by inducing G0/G1 arrest.
However, vemurafenib plus PHA-665752 inhibit cell proliferation of
both cell lines, indicating that the antitumor effect of the
combined treatment may be achieved by interrupting normal cell
cycling profiles. The finding is in line with previous studies on
inhibition of the cell cycle in gastric cancer and non-small cell
lung cancer (12,13).
In vivo, cancer develops around a solid tumor
in the context of a tumor microenvironment (14,15). This
specific microenvironment is not only a cause but also the
consequence of tumorigenesis. Tumor, mesenchymal, and nominal cells
co-evolve dynamically through direct and/or indirect cellular
interactions to elicit various specific biological programs
(16,17). The tumor microenvironment is important
for tumor initiation, maintenance, and even metastasis (18); however, how and why the tumor
microenvironment can contribute to cancer remains unclear (19). In our study, PHA-665752 showed
significant antitumor ability in vitro, but poor inhibition
of tumor volume growth in vivo. This may be due to the
complex microenvironment network composed of fibroblasts,
multipotent stromal cells, blood vessels, immune cells, and
secreted factors such as cytokines. Therefore, further studies
involving manipulation of the tumor microenvironment could offer an
approach to prevent and treat cancer.
Activation of the PI3K/AKT and MEK/ERK pathways is
common in different types of cancer (20–23) and,
in solid tumors, it is often associated with poor prognosis
(24). AKT and ERK play crucial roles
in cell proliferation, metastasis, angiogenesis, and drug response.
The phosphorylated state of both AKT and ERK is indicative of the
activity of tumor signaling (25,26).
Moreover, a high expression of both proteins coincides with higher
tumor cell viability and proliferation, and lower death in basic
experiments, but a poorer prognosis in clinical practice (27). Finally, the PI3K/AKT pathway plays an
important role in progression of CRC cells with a BRAF mutation,
which can accelerate invasion, migration, and infiltration
(28). Here, we treated CRC cells
bearing the BRAFV600E mutation with vemurafenib and a
c-Met inhibitor, individually or in combination.
Immunohistochemistry revealed no significant decrease in expression
of p-AKT and p-ERK after treatment with vemurafenib alone. However,
the c-Met inhibitor synergized with vemurafenib to result in low
expression of P-AKT and P-ERK. These findings constitute an
indirect proof of the impact the combined therapy has in
suppressing tumorigenesis and development. Furthermore, it suggests
a possible mechanism for cancer cell resistance to vemurafenib and
the BRAF inhibitor. Additional in-depth studies are warranted to
explore the exact underlying mechanism.
In conclusion, combined treatment with PHA-665752
and vemurafenib suppresses in vitro and in vivo CRC
cell growth more effectively than treatment with either agent
alone. The targeting of c-Met combined with vemurafenib may
represent an effective approach in the management of CRC patients
with a BRAFV600E mutation.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 81172332).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin PC, Yang YF, Tyan YC, Hsiao ES, Chu
PC, Lee CT, Lee JC, Chen YM and Liao PC: Identification of
Phosphorylated Cyclin-Dependent Kinase 1 Associated with Colorectal
Cancer Survival Using Label-Free Quantitative Analyses. PLoS One.
11:e01588442016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clarke CN and Kopetz ES: BRAF mutant
colorectal cancer as a distinct subset of colorectal cancer:
Clinical characteristics, clinical behavior, and response to
targeted therapies. J Gastrointest Oncol. 6:660–667.
2015.PubMed/NCBI
|
|
4
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cohen R, Cervera P, Svrcek M, Pellat A,
Dreyer C, de Gramont A and André T: BRAF-mutated colorectal cancer:
What is the optimal strategy for treatment. Curr Treat Options
Oncol. 18:92017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Byeon HK, Na HJ, Yang YJ, Ko S, Yoon SO,
Ku M, Yang J, Kim JW, Ban MJ, Kim JH, et al: Acquired resistance to
BRAF inhibition induces epithelial-to-mesenchymal transition in
BRAF (V600E) mutant thyroid cancer by c-Met-mediated AKT
activation. Oncotarget. 8:596–609. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Filitis DC, Rauh J and Mahalingam M: The
HGF-cMET signaling pathway in conferring stromal-induced
BRAF-inhibitor resistance in melanoma. Melanoma Res. 25:470–478.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gnad F, Doll S, Song K, Stokes MP, Moffat
J, Liu B, Arnott D, Wallin J, Friedman LS, Hatzivassiliou G and
Belvin M: Phosphoproteome analysis of the MAPK pathway reveals
previously undetected feedback mechanisms. Proteomics.
16:1998–2004. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barresi V, Bonetti LR and Bettelli S:
KRAS, NRAS, BRAF mutations and high counts of poorly differentiated
clusters of neoplastic cells in colorectal cancer: Observational
analysis of 175 cases. Pathology. 47:551–556. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao J, Sun ZW, Li YY and Shen L: Mutations
of KRAS and BRAF in Chinese patients with colorectal carcinoma:
Analyses of 966 cases. Zhonghua Bing Li Xue Za Zhi. 41:579–583.
2012.(In Chinese). PubMed/NCBI
|
|
11
|
Jang S, Hong M, Shin MK, Kim BC, Shin HS,
Yu E, Hong SM, Kim J, Chun SM, Kim TI, et al: KRAS and PIK3CA
mutations in colorectal adenocarcinomas correlate with aggressive
histological features and behavior. Hum Pathol. 65:21–30. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen CT, Kim H, Liska D, Gao S,
Christensen JG and Weiser MR: MET activation mediates resistance to
lapatinib inhibition of HER2-amplified gastric cancer cells. Mol
Cancer Ther. 11:660–669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Joshi M, Rice SJ, Liu X, Miller B and
Belani CP: Trametinib with or without vemurafenib in BRAF mutated
non-small cell lung cancer. PLoS One. 10:e01182102015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sikkandhar MG, Nedumaran AM, Ravichandar
R, Singh S, Santhakumar I, Goh ZC, Mishra S, Archunan G, Gulyás B
and Padmanabhan P: Theranostic probes for targeting tumor
microenvironment: An overview. Int J Mol Sci. 18:pii: E10362017.
View Article : Google Scholar
|
|
15
|
Munn DH, Sharma MD, Johnson TS and
Rodriguez P: IDO, PTEN-expressing Tregs and control of
antigen-presentation in the murine tumor microenvironment. Cancer
Immunol Immunother. 66:1049–1058. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Casey SC, Amedei A, Aquilano K, Azmi AS,
Benencia F, Bhakta D, Bilsland AE, Boosani CS, Chen S, Ciriolo MR,
et al: Cancer prevention and therapy through the modulation of the
tumor microenvironment. Semin Cancer Biol. 35 Suppl:S199–S223.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Whitfield JR and Soucek L: Tumo r
microenvironment: Becoming sick of Myc. Cell Mol Life Sci.
69:931–934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rakhra K, Bachireddy P, Zabuawala T,
Zeiser R, Xu L, Kopelman A, Fan AC, Yang Q, Braunstein L, Crosby E,
et al: CD4(+) T cells contribute to the remodeling of the
microenvironment required for sustained tumor regression upon
oncogene inactivation. Cancer Cell. 18:485–498. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kenny PA, Lee GY and Bissell MJ: Targeting
the tumor microenvironment. Front Biosci. 12:3468–3474. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kuo YH, Chiang EI, Chao CY, Rodriguez RL,
Chou PY, Tsai SY, Pai MH and Tang FY: Dual inhibition of key
proliferation signaling pathways in triple-negative breast cancer
cells by a novel derivative of taiwanin A. Mol Cancer Ther.
16:480–493. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mann KM, Ying H, Juan J, Jenkins NA and
Copeland NG: KRAS-related proteins in pancreatic cancer. Pharmacol
Ther. 168:29–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bresin A, D'Abundo L, Narducci MG,
Fiorenza MT, Croce CM, Negrini M and Russo G: TCL1 transgenic mouse
model as a tool for the study of therapeutic targets and
microenvironment in human B-cell chronic lymphocytic leukemia. Cell
Death Dis. 7:e20712016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krpina K, Babarović E, Španjol J, Đorđević
G, Maurer T and Jonjić N: Correlation of tumor-associated
macrophages and NK cells with bladder cancer size and T stage in
patients with solitary low-grade urothelial carcinoma. Wien Klin
Wochenschr. 128:248–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ocana A, Vera-Badillo F, Al-Mubarak M,
Templeton AJ, Corrales-Sanchez V, Diez-Gonzalez L, Cuenca-Lopez MD,
Seruga B, Pandiella A and Amir E: Activation of the PI3K/mTOR/AKT
pathway and survival in solid tumors: Systematic review and
meta-analysis. PLoS One. 9:e952192014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baba Y, Tamura T, Satoh Y, Gotou M, Sawada
H, Ebara S, Shibuya K, Soeda J and Nakamura K: Panitumumab
interaction with TAS-102 leads to combinational anticancer effects
via blocking of EGFR-mediated tumor response to trifluridine. Mol
Oncol. 11:1065–1077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu L, Liao JZ, He XX and Li PY: The role
of autophagy in hepatocellular carcinoma: Friend or foe.
Oncotarget. 8:57707–57722. 2017.PubMed/NCBI
|
|
27
|
Wang X, Shi W, Shi H, Lu S, Wang K, Sun C,
He J, Jin W, Lv X, Zou H and Shu Y: TRIM11 overexpression promotes
proliferation, migration and invasion of lung cancer cells. J Exp
Clin Cancer Res. 35:1002016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
He K, Chen D, Ruan H, Li X, Tong J, Xu X,
Zhang L and Yu J: BRAFV600E-dependent Mcl-1 stabilization leads to
everolimus resistance in colon cancer cells. Oncotarget.
7:47699–47710. 2016.PubMed/NCBI
|