Introduction
Lung cancer is one of the most frequently diagnosed
cancers, and was the leading cause of cancer-associated mortality
globally in 2012 (1). Non-small-cell
lung cancer (NSCLC), represents ~85% of all newly diagnosed lung
cancer, and includes adenocarcinoma (gland-forming), squamous cell
carcinoma and large-cell carcinoma histological subtypes (2). Clinically, the majority of patients are
not suitable for treatment by surgical resection due to distant
metastases and advanced stage; therefore chemotherapeutic agents
hold promise for the treatment of NSCLC (3). Cis-diamminedichloroplatinum (cisplatin;
CDDP) is one of the most effective chemotherapeutic drugs,
exhibiting a wide spectrum of activities against various human
cancers, including NSCLC. It produces DNA intra-strand crosslinks
between adjacent purines by forming bivalent adducts with
nucleophilic sites on purines, thereby exerting its antitumor
effects (4).
However, the efficacy of CDDP in cancer treatment is
often restricted due to resistance, either intrinsic, as observed
in patients with lung, colorectal and prostate cancer, or acquired
following CDDP chemotherapy, as often observed in patients with
ovarian cancer (5). The course of
CDDP resistance appears to be multifactorial, including changes in
drug transport resulting in decreased drug accumulation, enhanced
drug detoxification, alterations in DNA repair and damage bypass
and/or changes in the apoptotic cell death pathways, such as the
phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt)/nuclear
factor-κB (NF-κB) signaling pathway (6,7). Besides,
patients with esophageal cancer that treated with CDDP were
reported to experience therapeutic failure or tumor recurrence
(8). Accordingly, agents that may
elevate the sensitivity to CDDP in human NSCLC are of therapeutic
interest, including heat shock protein family A member 12B
(HSPA12B).
HSPA12B is a distant member of the mammalian heat
shock protein 70 (HSP70) family, because it contains a HSP70 ATPase
domain (9). Previous research has
indicated the essential role of HSP70 in oncogenesis and
chemotherapy resistance (10). For
example, a previous study suggested that HSP70 served functional
roles in the progression of uterine cervical squamous cell
carcinoma (SCC), and that HSP70 knockdown enhanced chemosensitivity
to CDDP in cervical SCC cells (11).
Notably, several in vitro studies have examined the role of
HSPA12B in carcinogenesis and lung cancer progression, and have
identified it as a potential therapeutic target (12,13);
however, the potential role of HSPA12B in chemosensitivity has not
been described.
The present study aimed to investigate the
significance of HSPA12B overexpression in CDDP resistance in NSCLC
in vivo and in vitro, and to explore the molecular
mechanisms underlying the effect of HSPA12B expression.
Materials and methods
Cell lines
Human lung adenocarcinoma A549 cells were supplied
by the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China) and were cultured at 37°C in a humidified
atmosphere containing 5% CO2 in Ham's F12 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
sodium bicarbonate (2.2% w/v), L-glutamine (0.03% w/v), penicillin
(100 U/ml), streptomycin (100 g/ml), and 10% fetal calf serum
(Gibco; Thermo Fisher Scientific, Inc.).
pcDNA3.1-HSPA12B+/+
construction and transfection
Reverse transcription-polymerase chain reaction
(RT-PCR) of normal human fibroblast total RNA was conducted using
the following primers: Forward, 5′-ATCGCCACCTTCAAAAGGCAA-3′; and
reverse, 5′-CTGTGAGGACCACTTCACGA-3′. The cDNA was obtained and
cloned into pCR™ II (Invitrogen; Thermo Fisher
Scientific, Inc.), and full-length HSPA12B cDNA was then sub-cloned
into the pcDNA3.1 plasmid (Invitrogen; Thermo Fisher Scientific,
Inc.) to produce the pcDNA3.1-HSPA12B+/+ vector.
A549 cells were plated at a density of
5×105 in 6-cm dishes and transfected using Lipofectamine
2000® according to the manufacturer's instructions
(Invitrogen; Thermo Fisher Scientific, Inc.). A549 cells were
transfected with endotoxin-free preparations of
pcDNA3.1-HSPA12B+/+ or pcDNA3.1 (control) and harvested.
After 48 h transfection, the protein expression of HSPA12B was
assessed by western blotting to determine the
pcDNA3.1-HSPA12B+/+ transfection efficiency in A549
cells. A549/HSPA12B+/+ cells were then exposed to the
PI3K inhibitor LY294002 (24 µM; Cell Signaling Technology, Inc.,
Danvers, MA, USA), the Akt inhibitor Triciribine (30 µM,
calbiochem) and the NF-κB inhibitor caffeic acid phenethyl ester
(CAPE, 10 µM) (both from Sigma-Aldrich; Merck KGaA) for 1 h.
Subcutaneous implantation of tumor
cells
The present study was approved by the Ethics
Committee of Jinshan Hospital, Fudan University (Shanghai, China).
A549, A549/HSPA12B+/+ and A549/pcDNA3.1 cells were
harvested from sub-confluent cultures (50–70% confluence) following
a brief exposure to 0.25% trypsin (Sigma-Aldrich; Merck KGaA) and
0.2% EDTA. Trypsinization was halted by adding to the cell
suspension 100 ml fresh RPMl-1640 medium (Thermo Fisher Scientific,
Inc.) containing 10% FBS. The cells were washed once in serum-free
medium and resuspended in PBS. A total of 2×106 cells in
100 µl PBS were injected into the right flanks of 6-week-old BALB/c
male nude mice (weighing 18–20 g, n=12 in each group). Mice were
purchased from the Shanghai Laboratory Animal Center, Chinese
Academy of Sciences (Shanghai, China) and were housed in
polystyrene cages. Two mice were kept per cage with free access to
food and water, and a 12/12 h light/dark cycle, with an ambient
temperature of 20–25°C.
In vivo model
Tumors were established by subcutaneous injection of
2×106 A549 tumor cells (A549, A549/HSPA12B+/+
and A549/pcDNA3.1) into the right flank of the mice (n=12/group).
Tumor volumes were calculated as: π/6 × a2 × b (where a
is the short axis and b is the long axis). After 3 weeks, when
tumors reached ~100 mm3, the mice in each group (n=12)
were randomly divided into two subgroups: Control and CDDP
(n=6/subgroup). These mice received daily intravenous injections of
either CDDP (4 mg/kg body weight) or the same volume of PBS,
respectively. CDDP was administered daily (from 21 days after the
initial injection of tumor cells). The treatments lasted for 15
days, during which the size of the tumors was recorded. The mice
were euthanized 3 days after the last injection, and tumors were
excised. Euthanasia 3 days after the last injection was deemed a
humane end-point to minimize pain and distress of experimental mice
(14). The present study was approved
by the Ethics Committee of Jinshan Hospital, Fudan University,
Shanghai, China. All experimental procedures were performed in
strict accordance with the guidelines for the Care and Use of
Laboratory Animals published by the US National Institutes of
Health (NIH publication no. 85–23, revised 1996) (15).
In situ detection of apoptotic
cells
Apoptotic cells in the tumor tissues were detected
by TUNEL assay, according to the manufacturer's protocol for the
In Situ Cell Death Detection kit (cat. no. 11684817910;
Roche Diagnostics, GmbH, Mannheim, Germany). Resected tumors were
frozen, fixed in 10% formalin solution (pH 6.8–7.2; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for 18–20 h at room
temperature, embedded in paraffin and sectioned at 5 µm thickness.
The 5-µm sections were prepared by the Mayo Clinic Scottsdale
Histology Core Facility (Scottsdale, AZ, USA). A TUNEL assay for
apoptosis was conducted using an In Situ Cell Death
Detection kit (Roche Diagnostics, GmbH) according to the
manufacturer's instructions. Sections were deparaffinized in xylene
and then treated with a graded series of alcohol (100, 95, 90, 80
and 70% ethanol in double-distilled water) and rehydrated in PBS
(pH 7.5). Tissues were then treated with proteinase K solution (2
µg/ml; Roche Diagnostics) for 15 min at room temperature for
permeabilization. Endogenous peroxidase was inactivated by 3%
H2O2 (Sigma-Aldrich; Merck KGaA) in PBS for
30 min and sections were rinsed with PBS, immersed in citrate
buffer (pH=6.0) and then incubated with TdT and digoxigenin dUTP
(diluted 1:1) at 37°C for 60 min. Subsequently, the reaction was
ceased with 1× TdT stop buffer (17.4 g NaCl and 8.8 g tri-sodium
citrate
(Na3C6H5O7·2H2O)
in 1,000 ml double distilled water) at 37°C for 30 min and
anti-digoxigenin peroxidase conjugate was applied for an incubation
of 30 min at room temperature. The slides were developed using
0.05% diaminobenzidine substrate for 5 min at room temperature. For
the negative control, slides were incubated at 37°C for 60 min with
TdT buffer without TdT. As a positive control, slides were treated
with DNase (1 µg/ml; Sigma-Aldrich; Merck KGaA) at 37°C for 10 min.
Apoptotic cells were imaged under a fluorescence microscope (Nikon
Corporation, Tokyo, Japan). The TUNEL-positive cells were counted
in 10 randomly selected high-power fields at ×400 magnification.
The apoptosis index was calculated as previously described
(3).
Cell viability analysis
A Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) assay was used to
quantitatively evaluate cell viability. The cells (A549,
A549/HSPA12B+/+ and A549/pcDNA3.1) were seeded onto
96-well plates at a density of 1×104 cells/well for 24 h
at room temperature. Cells were exposed to in vitro
treatment with 1, 2, 4 and 8 µg/ml CDDP at room temperature for 24,
48 or 72 h. Subsequently, the culture medium was removed, and the
cells were washed with PBS; 100 µl Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) and 10 µl
CCK-8 solution were then added to each well, and incubated at 37°C
for 2.5 h. Following incubation, the optical density at 450 nm was
determined using a microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA). Finally, the CCK-8 readings of the treatment
group were divided by their corresponding control readings to
obtain the ratio of viable cells.
Western blot analysis
Protein was isolated from cells that were lysed in
radioimmunoprecipitation buffer (RIPA) containing protease
inhibitors at 4°C for 30 min. Cell lysates were prepared with a
RIPA lysis buffer kit (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), and the protein concentrations were quantified using a
Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). Proteins (30 µg/lane) were separated on SDS-PAGE (8% gel) and
transferred to polyvinylidene difluoride membranes (Amersham; GE
Healthcare, Chicago, IL, USA). The membranes were blocked in 5%
non-fat milk (Merck KGaA) overnight at 4°C. Membranes were then
probed with the following primary antibodies; anti-HSPA12B (cat.
no. ab116082; 1:500), anti-AKT (cat. no. ab8932; 1:200), anti-Bcl-2
(cat. no. ab37899; 1:200), anti-cleaved caspase-3 (cat. no.
ab13847; 1:500), anti-caspase-3 (cat. no. ab4051; 1:500),
anti-GAPDH (cat. no. ab9483; 1:200) and anti-β-actin (cat. no.
ab8227; 1:1,000) all purchased from Abcam (Cambridge, MA, USA) and
anti-p-AKT (cat. no. 9271S; 1:1,000), anti-IκBα (cat. no. 9242;
1:100), anti-p-IκBα (cat. no. 2859; 1:100) all purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA), and incubated
overnight at 4°C. Subsequently, protein bands were detected by
incubation with horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G (cat. no. A50-106P; 1:1,000; Origene Technologies,
Inc, Beijing, China) at room temperature for 1 h. Signals were
detected using an enhanced chemiluminescence kit (Wuhan Boster
Biotechnology Co., Ltd., Wuhan, China) and exposed to Kodak X-OMAT
film (Kodak, Rochester, NY, USA). Each experiment was performed at
least three times and the results were analyzed using Alpha View
Analysis Tools (AlphaViewSA software, version 3.2.2; ProteinSimple,
Santa Clara, CA, USA).
Statistical analysis
Data were expressed as mean ± standard deviation.
Statistical analyses were performed using SPSS statistical software
package standard version 16.0 (SPSS, Inc., Chicago, IL, USA).
Experiments were performed in triplicate. Statistical differences
among multiple independent groups were determined using a one-way
analysis of variance followed by a Dunnett's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
HSPA12B overexpression increased
resistance to CDDP in NSCLC cells in vitro
In order to examine whether HSPA12B expression
affected CDDP sensitivity in vivo, western blotting was
first performed to detect the pcDNA3.1-HSPA12B+/+
transfection efficiency in A549 cells. In the
A549/HSPA12B+/+ cells, the protein levels of HSPA12B
were significantly higher compared with the HSPA12B levels in the
untransfected A549 cells and the control A549/pcDNA3.1 cells
(Fig. 1A).
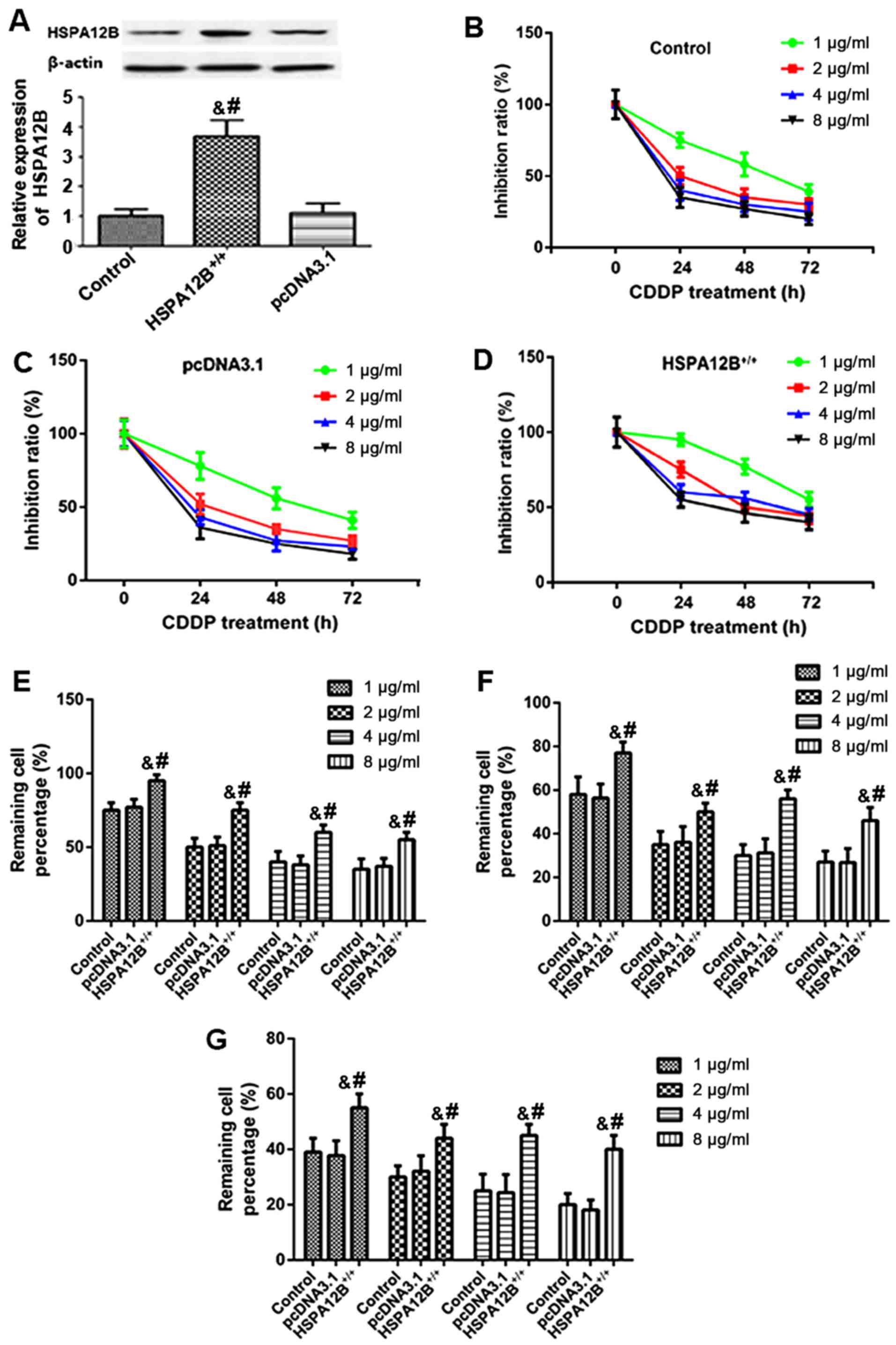 | Figure 1.Increased resistance to chemotherapy
in HSPA12B-overexpressing non-small cell lung cancer cells in
vitro. (A) Western blot analyses demonstrated that in the
pcDNA3.1-HSPA12B+/+-transfected A549 cells, the protein
expression of HSPA12B was significantly higher compared with the
endogenous HSPA12B levels in untransfected A549 cells and the
control pcDNA3.1-transfected A549 cells. (B) Untransfected A549
cells (control), (C) A549 cells transfected with the empty pcDNA3.1
vector and (D) A549 cells overexpressing HSPA12B were exposed to
in vitro treatment with 1, 2, 4 or 8 µg/ml CDDP at 24, 48,
and 72 h. The remaining cell percentage in (E) untransfected A549
cells (control), (F) A549 cells transfected with the empty pcDNA3.1
vector and (G) A549 cells overexpressing HSPA12B following exposure
to in vitro treatment with 1, 2, 4 or 8 µg/ml CDDP at 24,
48, and 72 h was shown. A CCK-8 assay was performed to evaluate
cell viability. Data are presented as the mean ± standard
deviation. &P<0.05 vs. untransfected control;
#P<0.05 vs. empty pcDNA3.1-transfected cells. CDDP,
cisplatin; HSPA12B, heat shock protein family A member 12B. |
Subsequently, A549/HSPA12B+/+ and
A549/pcDNA3.1 cells were exposed to in vitro treatment with
1, 2, 4, and 8 µg/ml CDDP. At 24, 48 and 72 h following treatment,
a CCK-8 assay was employed to evaluate cell viability. As
demonstrated in Fig. 1B-G, the
responses of HSPA12B-overexpressing cells to CDDP treatment were
inhibited in a dose- and time-dependent manner after 24, 48 and 72
h. The results also indicated that HSPA12B overexpression led to
significant increases in the cell viability of A549 cells in
response to CDDP treatment compared with the cells transfected with
the empty pcDNA3.1 vector (Fig.
1B-G), suggesting an increased resistance to CDDP in A549 cells
overexpressing HSPA12B.
HSPA12B overexpression increased the
resistance to CDDP in NSCLC cells in vivo
According to the in vitro experiments of
HSPA12B in CDDP resistance, whether HSPA12B affects CDDP resistance
in vivo was additionally examined. A549,
A549/HSPA12B+/+ and A549/pcDNA3.1 cells were injected
subcutaneously into the right flanks of nude mice. As indicated in
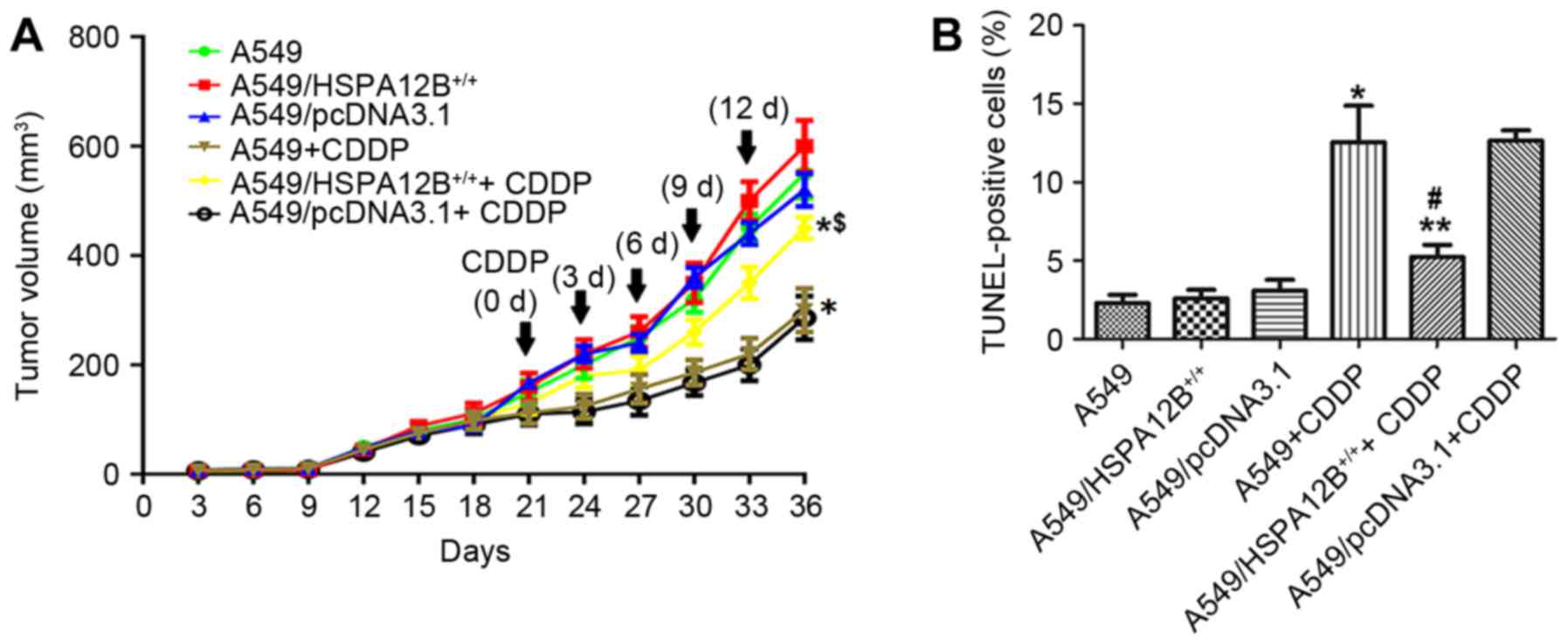
Fig. 2A, it was initially revealed
that CDDP treatment significantly inhibited tumor growth in mice
injected with A549 as compared with control mice. CDDP treatment
significantly inhibited tumor growth in mice injected with
A549/pcDNA3.1 cells and untransfected A549 cells compared with mice
injected with A549/HSPA12B+/+ cells. In mice treated
with CDDP, the A549 cell tumors reached a volume of 300±40
mm3 at 36 days post-treatment, which was significantly
smaller compared with the A549/HSPA12B+/+ cell tumors
(450±20 mm3) at 36 days after the initial injection of
tumor cells. In mice that did not receive CDDP treatment, no
significant difference in growth inhibition was observed in the
A549/HSPA12B+/+ tumors compared with the control
groups.
Tumor sections prepared from the groups were stained
with the TUNEL agent to detect apoptotic cells. The results in
Fig. 2B demonstrated that there were
more apoptotic cells in the control group tumors (A549 and
A549/pcDNA3.1) treated with CDDP compared with the control tumors
without CDDP treatment. However, fewer apoptotic cells were
observed in the A549/HSPA12B+/+ tumors treated with CDDP
compared with the A549+CDDP and A549/pcDNA3.1+CDDP control tumors.
In the groups that did not receive CDDP treatment, no significant
differences in the rate of cell apoptosis were observed among the
HSPA12B-overexpression and the control groups (A549,
A549/pcDNA3.1). Together, these data indicated that HSPA12B
overexpression attenuated CDDP-induced apoptosis in NSCLC
cells.
Increased HSPA12B induced
chemoresistance in NSCLC cells by modulating the PI3K/Akt/NF-κB
signaling pathway and apoptosis-associated proteins
The mechanism responsible for CDDP resistance is
associated with the inhibition of the propagation of the DNA damage
signal to the apoptotic machinery, including the activation of the
PI3K/Akt and its downstream NF-κB pathways, and the overexpression
of anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) and
interference in caspase activation (5,16). In
order to additionally explore the mechanism underlying the
inhibitory role of HSPA12B in CDDP-induced apoptosis, associated
proteins were investigated by western blotting (Fig. 3A). In the CDDP-treated
A549/HSPA12B+/+ solid tumors, the levels of
phosphorylated (p-)NF-κB inhibitor α (IκBα), p-Akt and Bcl-2 were
all significantly increased, whereas the levels of cleaved
caspase-3 were significantly decreased compared with CDDP-treated
A549 solid tumors, indicating that HSPA12B overexpression
diminished the effect of CDDP on A549 cells. No notable changes in
the levels of IκBα, Akt and caspase-3 were observed (Fig. 3A).
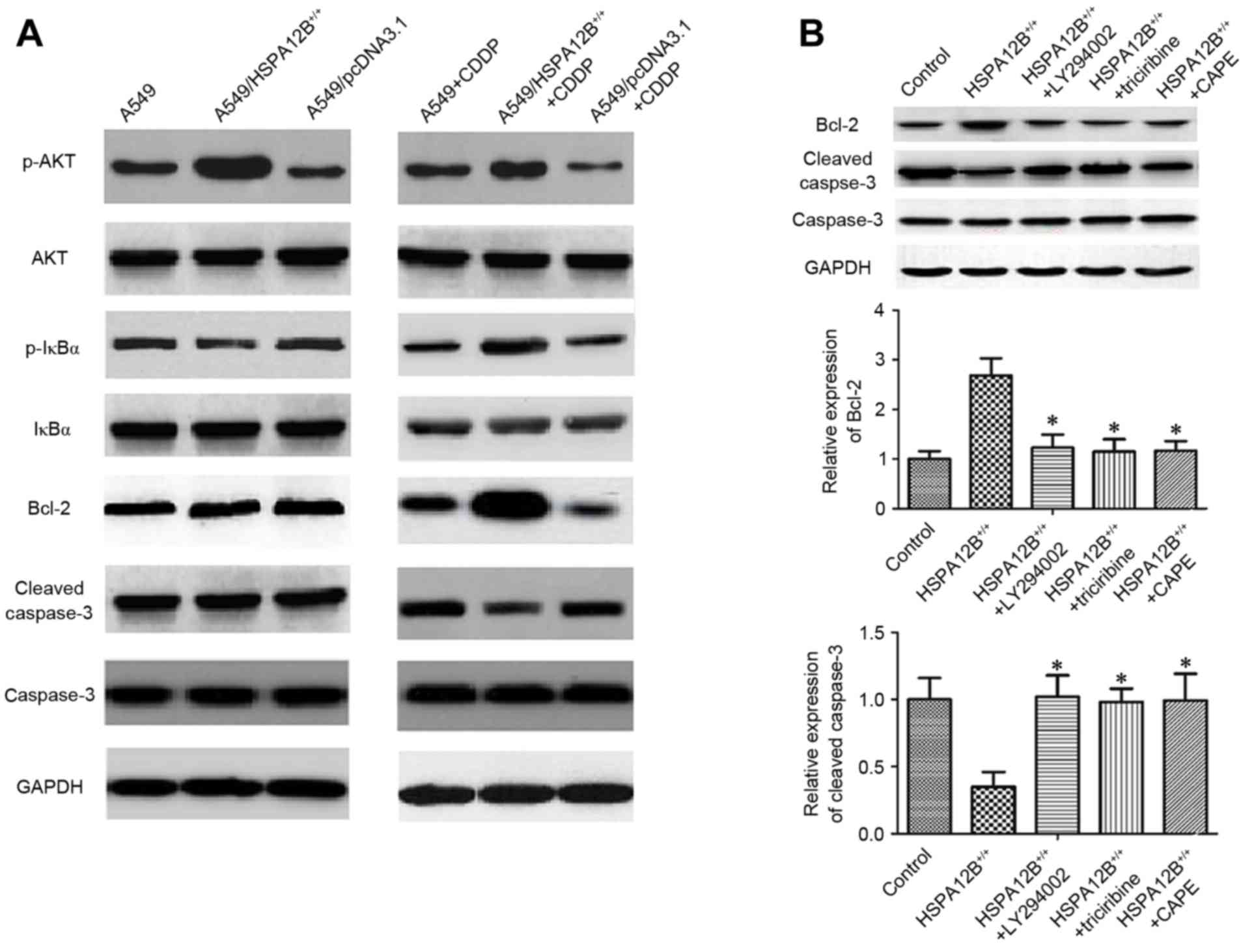 | Figure 3.Increased HSPA12B induces
chemoresistance in NSCLC cells by modulating PI3K/Akt/NF-κB
signaling pathway and apoptosis-associated proteins. (A) In the
CDDP-treated A549/HSPA12B+/+ solid tumors, the
expression of p-IκBα, p-Akt and Bcl-2 were significantly increased
while cleaved caspase-3 expression was significantly decreased
compared with CDDP-treated A549 tumors. (B) The PI3K inhibitor
LY294002, Akt inhibitor triciribine and NF-κB inhibitor CAPE
significantly increased cleaved caspase-3 expression and inhibited
the Bcl-2 level in HSPA12B+/+ cells compared with the
control groups. Data are presented as the mean ± standard
deviation. *P<0.05 vs. HSPA12B+/+. CDDP, cisplatin;
HSPA12B, heat shock protein family A member 12B; PI3K,
phosphoinositide 3-kinase; Akt, protein kinase B; NF-κB nuclear
factor-κB; p-, phosphorylated; IκBz, nuclear factor of κ light
polypeptide gene enhancer in B-cells inhibitor, α; CAPE, caffeic
acid phenethyl ester; Bcl-2, B-cell lymphoma 2. |
Finally, to verify the mechanism of HSPA12B-induced
CDDP resistance, A549/HSPA12B+/+ cells were exposed to
the PI3K inhibitor LY294002, the Akt inhibitor Triciribine and the
NF-κB inhibitor caffeic acid phenethyl ester (CAPE), and the
expression levels of Bcl-2, caspase-3 and cleaved caspase-3 were
analyzed. As demonstrated in Fig. 3B,
the levels of caspase-3 did not differ significantly amongst all
the groups, but LY94002, triciribine and CAPE significantly
increased the levels of cleaved caspase-3 and decreased the levels
of Bcl-2. These data implicate the PI3K/Akt/NF-κB signaling pathway
and apoptosis-associated proteins in HSPA12B-induced CDDP
resistance in NSCLC cells.
Discussion
Despite significant advances in oncology over
previous decades, lung cancer still has a high mortality rate
(1). CDDP is a platinum
chemotherapeutic agent and is widely used for the treatment of lung
cancer (17). DNA is the primary
target of CDDP; CDDP-induced DNA damage results in characteristic
cellular changes, including the inhibition of DNA synthesis,
suppression of RNA transcription, effects on the cell cycle, and
the therapeutically beneficial process of apoptosis (16–18).
Drug resistance to CDDP is a critical problem in the
context of cancer treatment, and the mechanism appears to be
complex. Cancer cells can develop CDDP resistance through
alterations in drug transport systems that lead to decreased
intracellular CDDP accumulation; through increased drug
detoxification activity due to the elevated levels of intracellular
scavengers such as glutathione and/or metallothioneins; through
alterations in DNA repair involving increased nucleotide excision
repair, inter-strand crosslink repair or loss of mismatch repair;
through alterations in DNA damage tolerance mechanisms; and through
changes in the apoptotic cell death pathways (6,7).
Therefore, novel methods or molecules that may enhance
chemosensitivity to CDDP and enable development of novel
therapeutic methods to treat NSCLC are required.
The present study investigated the significance of
HSPA12B overexpression in CDDP chemosensitivity in the A549 cell
line, and the molecular mechanisms underlying the effects of
HSPA12B overexpression. Firstly, HSPA12B overexpression was
suggested to contribute to CDDP resistance in vitro. CDDP
treatment alone significantly inhibited cell growth, whereas CDDP
treatment of HSPA12B-overexpressing resulted in significantly
attenuated growth inhibition.
Following this, mice with A549/HSPA12B+/+
tumors received doses of CDDP as described. The tumor volumes were
monitored during the study period at least twice a week.
HSPA12B-overexpression alone was not associated with any
significant changes in tumor volume or apoptotic cell numbers
compared with the control tumors. Furthermore, significantly
decreased tumor volumes and increased levels of apoptotic cells
were identified following CDDP treatment. However, HSPA12B
overexpression led to a significant attenuation of CDDP-induced
inhibition of tumor growth inhibition and CDDP-induced
apoptosis.
To the best of our knowledge, the data from the
present study represent the first evidence that HSPA12B serves an
essential role in CDDP resistance. HSPA12B belongs to the HSP70
family, which is involved in modulating chemosensitivity in various
types of cancer cells (10,11). Further investigation in the present
study found that HSPA12B overexpression contributes to CDDP
resistance via p-IκBα, p-Akt and Bcl-2 upregulation and cleaved
caspase-3 downregulation. Consistent with this observation,
increased levels of cleaved caspase-3, and decreased Bcl-2 levels
were identified in the present study following treatment with
PI3K/Akt/NF-κB signaling pathway inhibitors in HSPA12B-expressing
cells.
The PI3K/Akt/NF-κB signaling pathway is known to be
involved in promoting tumor cell survival, invasive behavior, and
chemosensitivity in various malignancies (19). A previous study has demonstrated that
CDDP activates p-Akt in A549 cells, and that blockage of the
PI3K/Akt pathway with chemical inhibitors moderately sensitizes
A549 cells to CDDP-induced apoptosis, and reduces cell viability
(20). An additional study indicated
that NF-κB was a downstream target of the PI3K/Akt pathway in
triptolide-induced apoptosis in MM.1 cells, and that PI3K/Akt may
serve a central role in the effect of triptolide on
dexamethasone-resistant and -sensitive multiple myeloma cell lines
(21). In addition, the
anti-apoptotic protein Bcl-2 is well-known to be transcriptionally
regulated by NF-κB, and to regulate mitochondria-mediated apoptosis
(22). Therefore, it raises the
possibility that CDDP primarily modulates the PI3K/Akt pathway,
which regulates the NF-κB pathway, which affects Bcl-2 expression,
and subsequently the expression of the apoptotic protein cleaved
caspase-3 is altered. These alterations thereby regulate NSCLC cell
growth and apoptosis.
Strategies for overcoming CDDP resistance include
combined treatment with CDDP plus drugs that specifically target
cancer cells, combinations of CDDP with compounds that target
effectors involved in CDDP resistance, and the development of novel
platinating drugs (5). HSPA12B siRNA
may be effective in modulating the PI3K/Akt/NF-κB signaling pathway
involved in CDDP resistance, providing a potential treatment for
NSCLC. However, future clinical trials are required to confirm this
conclusion.
To conclude, these data demonstrate that HSPA12B
overexpression enhances CDDP resistance through the regulation of
p-Akt and p-IκBα in the PI3K/Akt/NF-κB signaling pathway in NSCLC
cells. These experimental data support the development of targeted
strategies employing HSPA12B siRNA complementary to conventional
cytotoxic therapies for NSCLC, which contributes to the formulation
of potential therapeutics for improving the current treatment
modalities for patients with NSCLC.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Travis WD, Brambilla E and Riely GJ: New
pathologic classification of lung cancer: Relevance for clinical
practice and clinical trials. J Clin Oncol. 31:992–1001. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma G, Cai H, Gao L, Wang M and Wang H:
sCLU regulates cisplatin chemosensitivity of lung cancer cells in
vivo. World J Surg Oncol. 13:802015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan JM, Li XD, Liu ZY, Hou GQ, Kang JH,
Huang DY and Du SX: Cisplatin induces apoptosis via upregulating
Wrap53 in U-2OS osteosarcoma cells. Asian Pac J Cancer Prev.
12:3465–3469. 2011.PubMed/NCBI
|
|
5
|
Koberle B, Tomicic MT, Usanova S and Kaina
B: Cisplatin resistance: Preclinical findings and clinical
implications. Biochim Biophys Acta. 1806:172–182. 2010.PubMed/NCBI
|
|
6
|
Kartalou M and Essigmann JM: Mechanisms of
resistance to cisplatin. Mutat Res. 478:23–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rabik CA and Dolan ME: Molecular
mechanisms of resistance and toxicity associated with platinating
agents. Cancer Treat Rev. 33:9–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Law S and Wong J: The current management
of esophageal cancer. Adv Surg. 41:93–119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Han Z, Truong QA, Park S and Breslow JL:
Two Hsp70 family members expressed in atherosclerotic lesions. Proc
Natl Acad Sci USA. 100:pp. 1256–1261. 2003; View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ren A, Yan G, You B and Sun J:
Down-regulation of mammalian sterile 20-like kinase 1 by heat shock
protein 70 mediates cisplatin resistance in prostate cancer cells.
Cancer Res. 68:2266–2274. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoshidomi K, Murakami A, Yakabe K, Sueoka
K, Nawata S and Sugino N: Heat shock protein 70 is involved in
malignant behaviors and chemosensitivities to cisplatin in cervical
squamous cell carcinoma cells. J Obstet Gynaecol Res. 40:1188–1196.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Steagall RJ, Hua F, Thirunazukarasu M,
Zhan L, Li C, Maulik N and Han Z: Abstract 3600: HspA12B promotes
angiogenesis through suppressing AKAP12 and up-regulating VEGF
pathway. Circulation. 118 Suppl 18:S4492008.
|
|
13
|
Ma H, Lu T, Zhang X, Li C, Xiong J, Huang
L, Liu P, Li Y, Liu L and Ding Z: HSPA12B: A novel facilitator of
lung tumor growth. Oncotarget. 6:9924–9936. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang B, Liu ZM, Hao FG and Wang M:
siRNA-directed clusterin silencing promotes cisplatin antitumor
activity in human non-small cell lung cancer xenografts in
immunodeficient mice. Eur Rev Med Pharmacol Sci. 18:1595–1601.
2014.PubMed/NCBI
|
|
15
|
Bayne K: Revised guide for the care and
use of laboratory animals available. American physiological
society. Physiologist. 39:199–208, 111. 1996.PubMed/NCBI
|
|
16
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Su J, Wu S, Tang W, Qian H, Zhou H and Guo
T: Reduced SLC27A2 induces cisplatin resistance in lung cancer stem
cells by negatively regulating Bmi1-ABCG2 signaling. Mol Carcinog.
55:1822–1832. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lippard SJ: Platinum, Gold, and Other
Metal Chemotherapeutic Agents. 209. American Chemical Society;
Washington, DC: 1983, doi: 10.1021/bk-1983-0209. View Article : Google Scholar
|
|
19
|
Azijli K, Weyhenmeyer B, Peters GJ, de
Jong S and Kruyt FA: Non-canonical kinase signaling by the death
ligand TRAIL in cancer cells: discord in the death receptor family.
Cell Death Differ. 20:858–868. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang M, Liu ZM, Li XC, Yao YT and Yin ZX:
Activation of ERK1/2 and Akt is associated with cisplatin
resistance in human lung cancer cells. J Chemother. 25:162–169.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang M, Huang J, Pan HZ and Jin J:
Triptolide overcomes dexamethasone resistance and enhanced
PS-341-induced apoptosis via PI3k/Akt/NF-kappaB pathways in human
multiple myeloma cells. Int J Mol Med. 22:489–496. 2008.PubMed/NCBI
|
|
22
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor kappa B and its significance
in prostate cancer. Oncogene. 20:7342–7351. 2001. View Article : Google Scholar : PubMed/NCBI
|