Introduction
Cervical cancer is the most common type of malignant
neoplasm in the female reproductive system and ranks as the second
most common cause of gynecological cancer-associated mortality,
with 47,130 new cases and 8,010 mortalities reported in the United
States in 2014 (1). Despite decreases
in the incidence and mortality rates of cervical cancer, which are
largely due to progress in surgical treatment (2) radiotherapy (3) and chemotherapy (4), cervical cancer remains a substantial
threat to women's health globally (5). It has been hypothesized that the
carcinogenesis and development of cervical cancer are associated
with sexual behavior, childbirth, persistent human papillomavirus
(HPV) infection and smoking (6).
Extensive efforts have been made to clarify the genetic and
epigenetic mechanisms that underlie the growth and invasion of
cervical cancer. However, the pathogenesis remains understood.
Pelvic lymph node metastasis may serve as an independent prognostic
factor (7); however, the lack of
specific tumor markers for the prediction of cervical cancer
mortality and invasion has yet to be resolved.
Long non-coding RNA (lncRNA) is defined as RNA of
>200 nucleotides without protein-coding function. It has been
reported that the portion of the human genome that encodes proteins
may be <2% (8). A growing number
of lncRNAs have been recognized as key regulators, rather than
simply transcriptional noise. They are associated with various
biological processes, including genomic imprinting, X chromosome
inactivation, chromatin modification, transcription interference,
transcription activation and nuclear transport (8–10). lncRNAs
are one of the most highly expressed classes of non-coding RNAs in
human cervical tissue, and their relevance to cervical cancer is
steadily becoming more apparent (11). A previous study demonstrated that
lncRNA expression levels differ, with statistical significance,
between three cervical intraepithelial neoplasia grades, indicating
that these transcripts may be involved in the development and
progression of pre-cancerous lesions (12). Studies on this topic have revealed
that aberrant expression of certain lncRNAs, including HOX
transcript antisense RNA (HOTAIR), metastasis-associated
lung adenocarcinoma transcript 1, cervical carcinoma-expressed PCNA
regulatory lncRNA, lncRNA-EBIC (also known as thymopoietin
pseudogene 2), growth arrest-specific 5, and lncRNA-LET (also known
as NPTN intronic transcript 1), serves critical roles in cervical
cancer development, invasion and metastasis (13–18).
However, none of these lncRNAs has been applied as biomarker for
clinical diagnosis. Furthermore, the precise function of the
majority of lncRNAs remains unknown despite extensive research
efforts. Thus, there is an urgent requirement to identify the
mechanisms of interaction between lncRNAs and coding genes.
In the present study, a high-throughput microarray
was employed to analyze lncRNA and mRNA expression profiles in
samples of cancerous and normal cervical tissue. Gene Ontology (GO)
and pathway enrichment analyses investigated enriched functions of
the predicted targets, allowing the establishment of an lncRNA-mRNA
correlation network. The results illustrated that differences in
the expression levels of lncRNAs, in addition to mRNAs, may be
associated with cervical cancer pathogenesis; the comprehensive
analysis of lncRNAs and mRNA may lay a foundation for further
investigation in the diagnosis and treatment of cervical
cancer.
Materials and methods
Patient specimens and RNA
extraction
A total of 6 cervical carcinoma samples and 6 normal
cervical tissues were collected for microarray. The 6 cervical
carcinoma samples were labeled C1 and C3-7, C2 was diagnosed
cervical carcinoma in situ and was not suitable for
microarray. For validation, another 20 normal and 30 cancerous
cervical tissue samples were also selected. The number of cervical
cancer tissues was subsequently increased to 60 for the study of
the association between ENST00000551152 expression and
clinicopathological parameters. Patient characteristics are
summarized in Table I. All the tissue
samples were obtained between January 2014 to January 2015 from the
Department of Gynecology, First Affiliated Hospital of Sun Yat-sen
University (Guangzhou, China). Samples were selected based on the
diagnosis of cervical cancer, determined by at least two
pathologists and no patients had received chemotherapy or
radiotherapy prior to surgery. All provided written, informed
consent. The present study was approved by the Medical Ethics
Committees at the First Affiliated Hospital of Sun Yat-sen
University. All samples were stored at −80°C until RNA
extraction.
 | Table I.Correlation between ENST00000551152
expression and clinicopathological characteristics in early-stage
squamous cervical cancer. |
Table I.
Correlation between ENST00000551152
expression and clinicopathological characteristics in early-stage
squamous cervical cancer.
|
|
| ENST00000551152
expression, n (%) |
|
|---|
|
|
|
|
|
|---|
| Characteristic | All patients, n | Low | High | P-value |
|---|
| Total | 60 | 14 (23.3) | 46 (76.7) |
|
| Age, years |
|
|
| 0.297 |
| ≤42 | 33 | 6 (18.2) | 27 (81.8) |
|
|
>42 | 27 | 8 (29.6) | 19 (70.4) |
|
| FIGO stage |
|
|
| 0.017a |
| IB1 | 29 | 12 (41.4) | 17 (58.6) |
|
| IB2 | 8 | 1 (12.5) | 7 (87.5) |
|
| IIA1 | 17 | 1 (5.9) | 16 (94.1) |
|
| IIA2 | 6 | 0 (0.0) | 6 (100.0) |
|
| Tumor size, cm |
|
|
| 0.022a |
| ≤4 | 39 | 13 (33.3) | 26 (66.7) |
|
|
>4 | 21 | 1 (4.8) | 20 (95.2) |
|
| Differentiation
grade |
|
|
| 0.056 |
|
Well-differentiated | 7 | 4 (57.1) | 3 (42.9) |
|
|
Moderately differentiated | 25 | 6 (24.0) | 19 (76.0) |
|
| Poorly
differentiated | 28 | 4 (14.3) | 24 (85.7) |
|
| Extent of stromal
invasion |
|
|
| 0.001 |
|
<1/2 | 29 | 12 (41.4) | 17 (58.6) |
|
| ≥1/2 | 31 | 2 (6.5) | 29 (93.5) |
|
| LVSI |
|
|
| 0.053a |
|
Yes | 11 | 0 (0.0) | 11 (100.0) |
|
| No | 49 | 14 (28.6) | 35 (71.4) |
|
| PLNM |
|
|
| 0.026a |
|
Yes | 14 | 0 (0.0) | 14 (100.0) |
|
| No | 46 | 14 (30.4) | 32 (69.6) |
|
| SCC antigen,
ng/ml |
|
|
| 0.004 |
|
<1.5 | 27 | 11 (40.7) | 16 (59.3) |
|
|
≥1.5 | 33 | 3 (9.1) | 30 (90.9) |
|
Extraction of total RNA from fresh tissues was
performed using RNAiso Plus reagent (Takara Biotechnology Co.,
Ltd., Dalian, China) and the total RNA content of each sample was
quantified using a NanoDrop ND-1000 (Thermo Fisher Scientific,
Inc., Wilmington, DE, USA). Standard denaturing agarose gel
electrophoresis was performed to evaluate the RNA integrity.
RNA labeling and array
hybridization
Following the Agilent One-Color Microarray-Based
Gene Expression Analysis protocol (Agilent Technologies, Inc.,
Santa Clara, CA, USA), sample labeling and array hybridization were
implemented in the 6 cancer tissues and 6 normal cervical tissues.
Ribosomal RNA was removed to leave purified mRNA using an
mRNA-ONLY™ Eukaryotic mRNA Isolation Kit (Epicentre; Illumina,
Inc., San Diego, CA, USA). The entire lengths of the transcripts
were transcribed into fluorescent complementary RNA (cRNA) without
3′ bias by using random priming. The labeled cRNA was then purified
with an RNeasy Mini Kit (Qiagen China Co., Ltd, Shanghai, China). A
mixture containing 5 µl 10X blocking agent, 1 µg 25X fragmentation
buffer and 1 µg of each labeled cRNA sample was fragmented and then
heated for 30 min at 60°C. To dilute the labeled cRNA, 25 µl 2X GE
Hybridization buffer (GE Healthcare Life Sciences, Logan, UT, USA)
was added. After dispensing into the gasket slide, 50 µl of
hybridization solution was applied to the lncRNA expression
microarray slide. Incubation of the slides was performed in an
Agilent hybridization oven at 65°C for 17 h (Agilent Technologies,
Inc.). Washing, fixing and scanning of the hybridized arrays were
completed with the Agilent DNA microarray scanner (part no.
G25005C).
Data analysis of lncRNA and mRNA
expression profiles
The general profiles of human lncRNAs and
protein-coding transcripts from the 6 cervical carcinoma samples
and 6 normal cervical tissues were detected using the Arraystar
Human lncRNA Microarray V3.0 (Arraystar, Inc., Rockville, MD, USA);
using this array, ~26,109 coding transcripts and 30,586 lncRNAs are
detectable. Array images were analyzed by Agilent Feature
Extraction software (version 11.0.1.1; Agilent Technologies, Inc.).
Following quantile normalization and data processing in GeneSpring
GX v11.5.1 software package (Agilent Technologies, Inc.), lncRNA
and mRNAs flagged as Present or Marginal (‘all Target Value’) in ≥6
out of 12 samples and were selected for further analysis. Volcano
plot filtering was employed to validate the significance of the
differences in the lncRNA and mRNA expression levels between the
cancer and normal cervical tissues. GeneSpring GX was used to
perform hierarchical clustering. GO and pathway analyses were
performed using standard enrichment computation.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Following total RNA extraction as described, cDNA
synthesis was performed for 15 min at 37°C and 5 sec at 85°C with
PrimeScript RT Master Mix (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. Primer sequences are
listed in Table II. SYBR Premix Ex
Taq (Takara Biotechnology Co., Ltd.) was used with 2 µl template
cDNA in each reaction for qPCR with a 7500 Fast Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.). In the
40 cycles of PCR, pre-denaturation was performed for 30 sec at
95°C, while the parameters for denaturation and annealing were set
at 95°C for 5 sec and 60°C for 34 sec, respectively. The
2−ΔΔCq value [ΔCq=Cq(RNA)-Cq(GAPDH)] (19) was used to determine an average cycle
threshold value from triplicate reactions and calculate the level
of genomic expression (20). Single
product amplification was ensured by the generated melting
curves.
| Table II.Primers used for quantitative
polymerase chain reaction analysis of long non-coding RNA
levels. |
Table II.
Primers used for quantitative
polymerase chain reaction analysis of long non-coding RNA
levels.
| Sequence ID | Primer sequence (5′
to 3′) |
|---|
| TCONS_00001368 | F:
CACACAAGGACTGGAGCAAA |
|
| R:
CACCTAACCCACCACATTCA |
| NR_033746 | F:
GCAGCTCAGGTTCTCCAAAT |
|
| R:
CCCTCTTTAGCCTGTTGGTC |
| NR_027122 | F:
CTGTCCTCCTGCTCTTTGC |
|
| R:
GAGTTTTGGGTTCACGGATA |
|
ENST00000421943 | F:
GGGACCAGGAATGTGAACTT |
|
| R:
TGCCCTCAGATGTGAAACTC |
|
ENST00000439076 | F:
ACAGGCGGCAGAGAAGAAG |
|
| R:
GACACACGCAGTCATTCAGG |
|
ENST00000448991 | F:
GCAGACTTGACCTCTTGGC |
|
| R:
ATAGTGGGTATCGGGGGTG |
| uc001iot.1 | F:
GAGAAGAGGCGAACGAGG |
|
| R:
GTGGGACAGCCAATACATAAT |
|
ENST00000414085 | F:
CGCAGAACTTTGCTGGAGA |
|
| R:
GAAATACAGAGTCAGAGAGCGTG |
|
ENST00000421498 | F:
GACCATGCTGTTGAAACCAC |
|
| R:
TCAAGGAGAGCACAAGGAACT |
|
ENST00000428667 | F:
TTTCCATACCCAGCCAACTT |
|
| R:
CTTCCTGCACTGCCAACCT |
|
ENST00000443523 | F:
CCTGGCTGGAGATGCTTACT |
|
| R:
GGTTCCTGTTGGGACTTTAGA |
|
ENST00000551152 | F:
GCAAGAACTGAGACCTGACG |
|
| R:
TAAGCACACCACTCCACTGC |
| GAPDH | F:
GGGAAACTGTGGCGTGAT |
|
| R:
GAGTGGGTGTCGCTGTTGA |
GO and pathway analysis
GO is a functional analysis that annotates genes and
attributes associated with their expression using ontological
categories, including ‘biological process’, ‘cellular component’
and ‘molecular function’. GO categories (http://www.geneontology.org) were applied to the
differentially expressed lncRNAs (21). To analyze how the target genes
function in the cellular pathways, the Kyoto Encyclopedia of Genes
and Genomes (KEGG; http://www.kegg.jp) (22) database was used. The P-value, which
represents the importance of the pathway, was used with a cut-off
0.5, with lower values indicating greater significance.
lncRNA-mRNA correlation network
The lncRNA-mRNA correlation network was established
according to lncRNA target predictions along with differentially
expressed lncRNA and mRNA profiles using Cytoscape (http://www.cytoscape.org) (23). The lncRNAs and mRNAs that were
selected to create the network were those with Pearson correlation
coefficients ≥0.99.
Cell culture
The cervical carcinoma cell lines HeLa, SiHa, MS751
and C33A were purchased from the American Type Culture Collection
(Manassas, VA, USA) and cultured in Dulbecco's Modified Eagle's
medium (Gibco; Thermo Fisher Scientific, Inc.) or RPMI-1640 medium
(BRI, Rockville, MD, USA), supplemented with 10% fetal bovine serum
(Hyclone; GE Healthcare Life Sciences), penicillin (100 U/ml), and
streptomycin (100 µg/ml). The conditions during cell culture were
5% CO2, 95% humidified air, and 37°C.
Statistical analysis
Statistical analyses were performed using SPSS
software (version 13.0; SPSS, Inc., Chicago, IL, USA). All data are
presented as the mean ± standard deviation. The Student's t-test
was used for evaluating the statistical significance of differences
in the means between two groups. The association between lncRNA
expression and clinicopathological features was assessed using the
χ2 test and Fisher's exact test. P<0.05 was
considered to indicate a statistically significant difference.
Receiver operating characteristic (ROC) curve analysis was employed
to define the cutoff value for high expression of lncRNA by the
(0,1)-criterion, and the area under the curve was calculated as
previously described (24).
Results
Aberrant lncRNA and mRNA expression in
cervical cancer
The profiles of lncRNAs and mRNAs in paired cervical
cancer and normal cervical tissue samples were produced using
microarray technology to investigate the potential biological
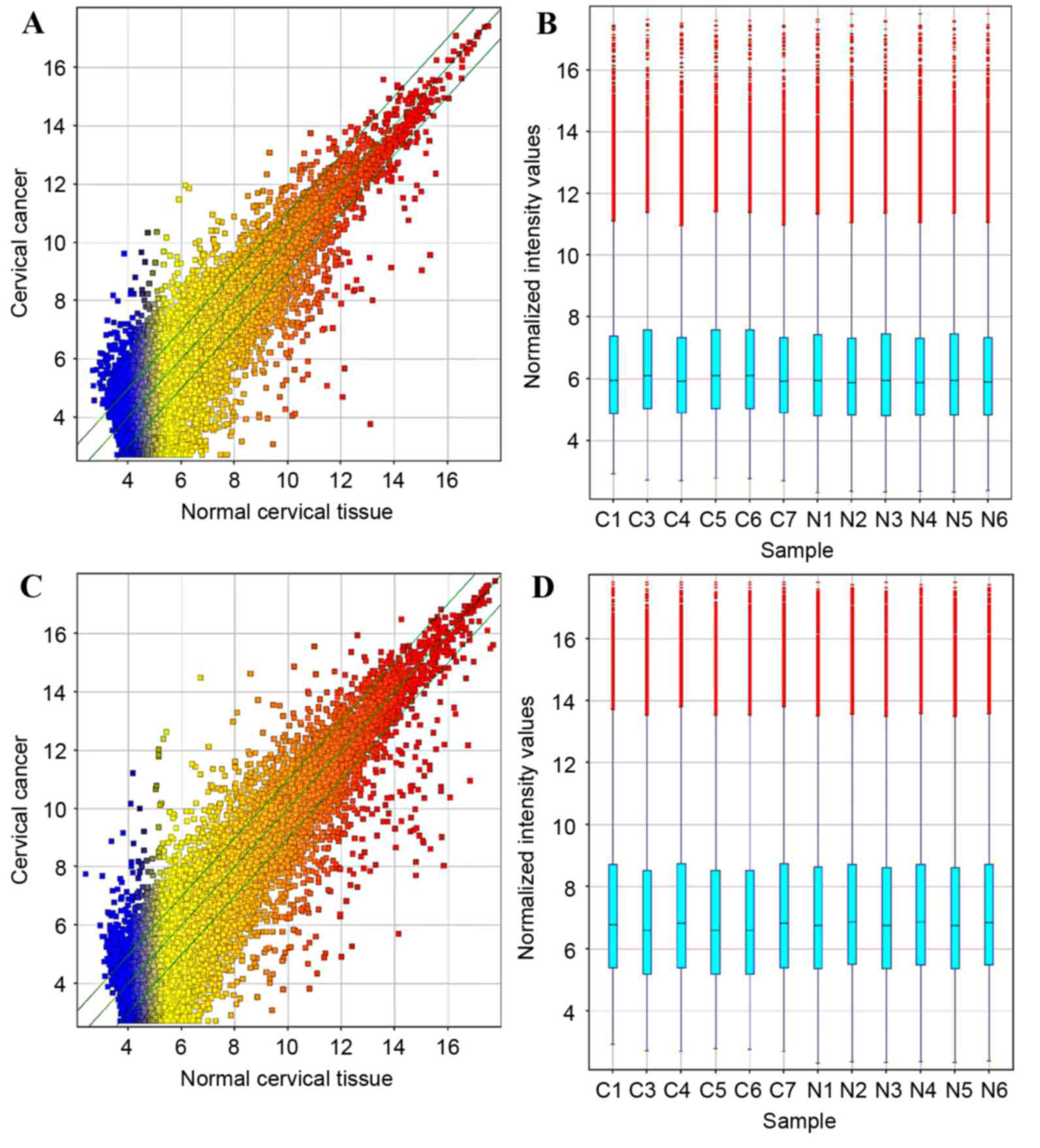
function of lncRNAs in cervical cancer (Fig. 1A and B). Relative to normal tissue,
5,844 (19.1%) lncRNAs presented differential expression in cancer
tissue (fold-change, ≥2) among the 30,586 detected lncRNA
transcripts. This included 2,574 upregulated 3,270 downregulated
lncRNAs. The most prominently upregulated and downregulated lncRNAs
were uc002jcf.3 (fold-change, 57.214) and NR_027122 (fold-change,
620.995), respectively.
In the mRNA expression profile data, 1,538
upregulated mRNAs and 2,898 downregulated mRNAs were detected
(Fig. 1C and D). Among these mRNAs,
the most prominently upregulated and downregulated mRNAs were Kelch
domain-containing 7B (fold-change, 224.009) and keratin 1
(fold-change, 339.108), respectively.
Ontological and pathway analysis of
target genes of differentially expressed lncRNAs
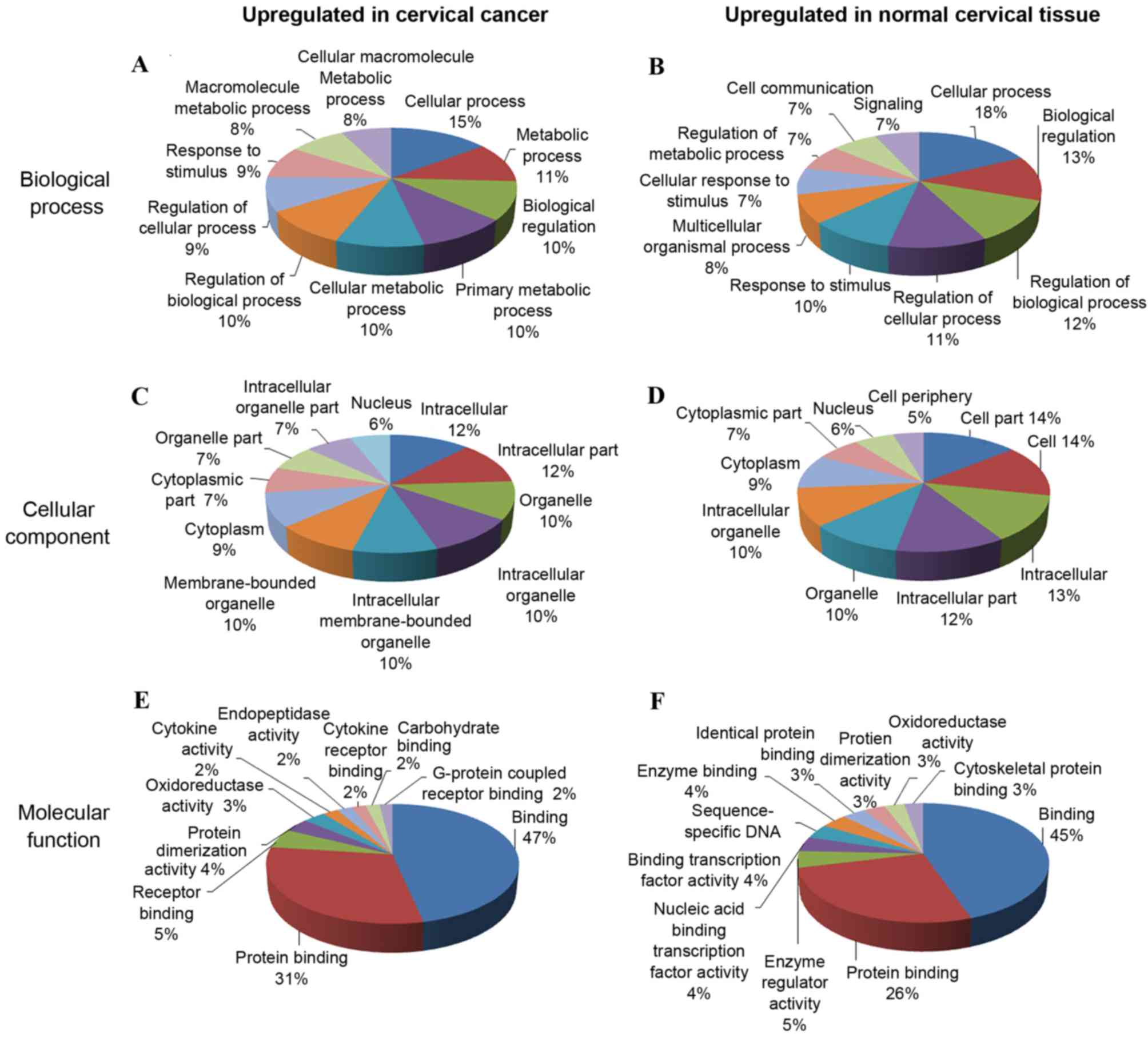
To investigate a possible correlation between
functional grouping and the differentially expressed lncRNAs, GO
annotation categories including ‘biological process’ (Fig. 2A and B), ‘cellular component’
(Fig. 2C and D) and ‘molecular
function’ (Fig. 2E and F) were
explored for the differentially expressed mRNAs of the samples. In
the GO ‘biological process’ classification, the majority of the
upregulated and downregulated GO annotations were associated with
‘cellular process’ and ‘regulation of cellular process’. The
‘cellular component’ classification search revealed that a large
proportion of genes that were upregulated in cervical cancer were
annotated as GO categories ‘intracellular’, ‘intracellular part’,
‘organelle’ and ‘intracellular organelle’; ‘cell’ and ‘cell part’
accounted for the greatest numbers of downregulated genes. The most
common ‘molecular function’ GO annotations for all differentially
expressed genes were associated with ‘binding’ and ‘protein
binding’.
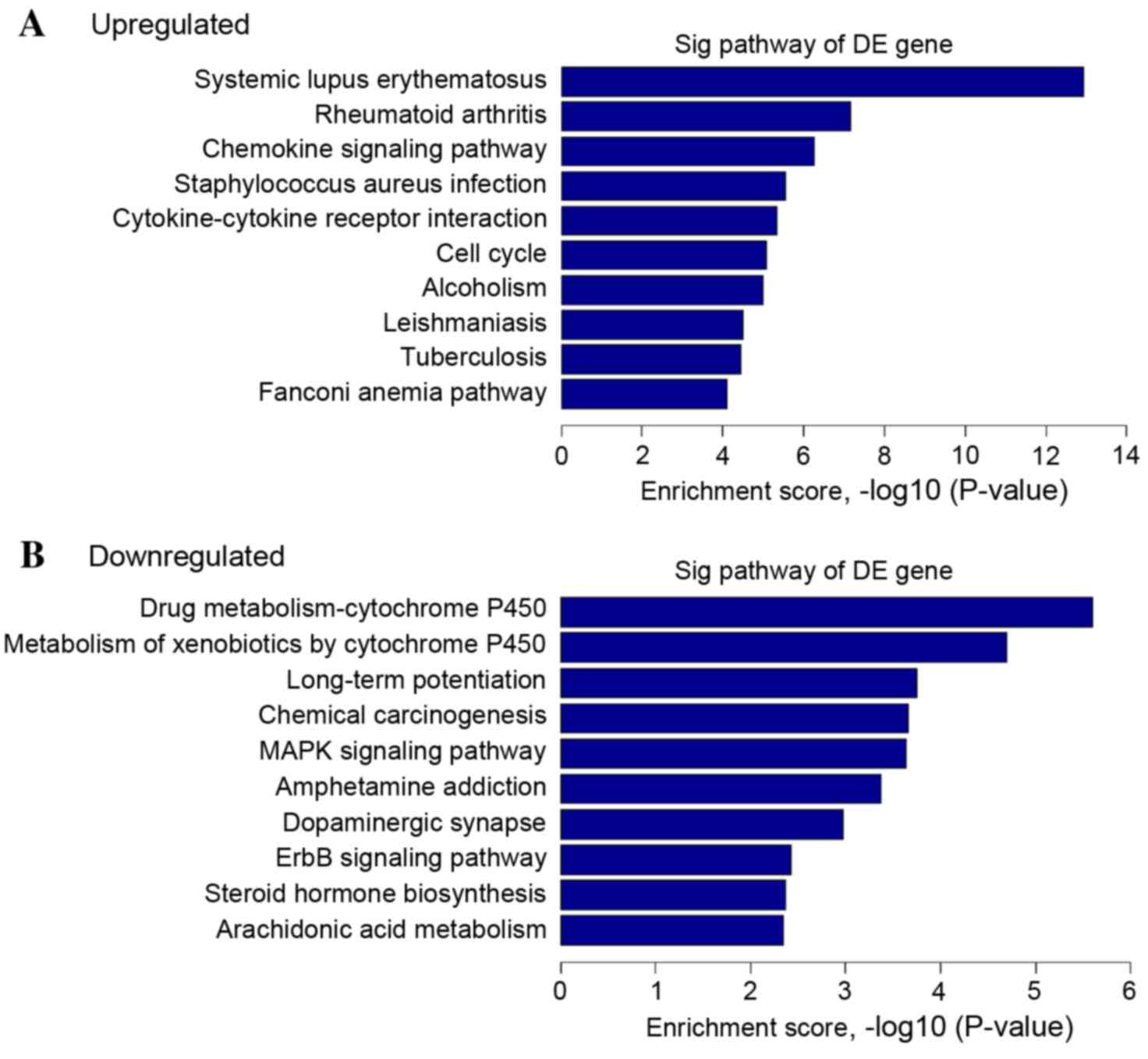
Additionally, pathway analysis was accomplished with
the KEGG database (22) to
investigate the biological pathways associated with the mRNAs with
the most pronounced differential expression in cervical cancer. A
total of 40 upregulated pathways were identified in the cervical
cancer tissue, which were correlated with the KEGG pathway
categories ‘cytokine-cytokine receptor interaction’, ‘chemokine
signaling pathway’, ‘transcriptional misregulation in cancer’ and
‘cell cycle’. A total of 37 downregulated pathways were identified,
which were associated with the categories ‘MAPK signaling pathway’,
‘pathways in cancer’ and ‘Wnt signal pathway’; all have been
previously reported to be associated with cervical cancer (Fig. 3) (25,26).
Construction of lncRNA-mRNA
correlation network
In total, 592 network nodes and 934 associations
between 12 lncRNAs and 580 coding genes were included in the CNC
network, within which there were 500 positive correlation pairs,
and 434 negative correlation pairs. The co-expression network
demonstrated that one lncRNA could act on 141 coding genes
maximally, and that one coding gene corresponded to a maximum of 5
lncRNAs. The mutual regulation between lncRNAs and mRNAs in
cervical cancer was also indicated in the CNC network.
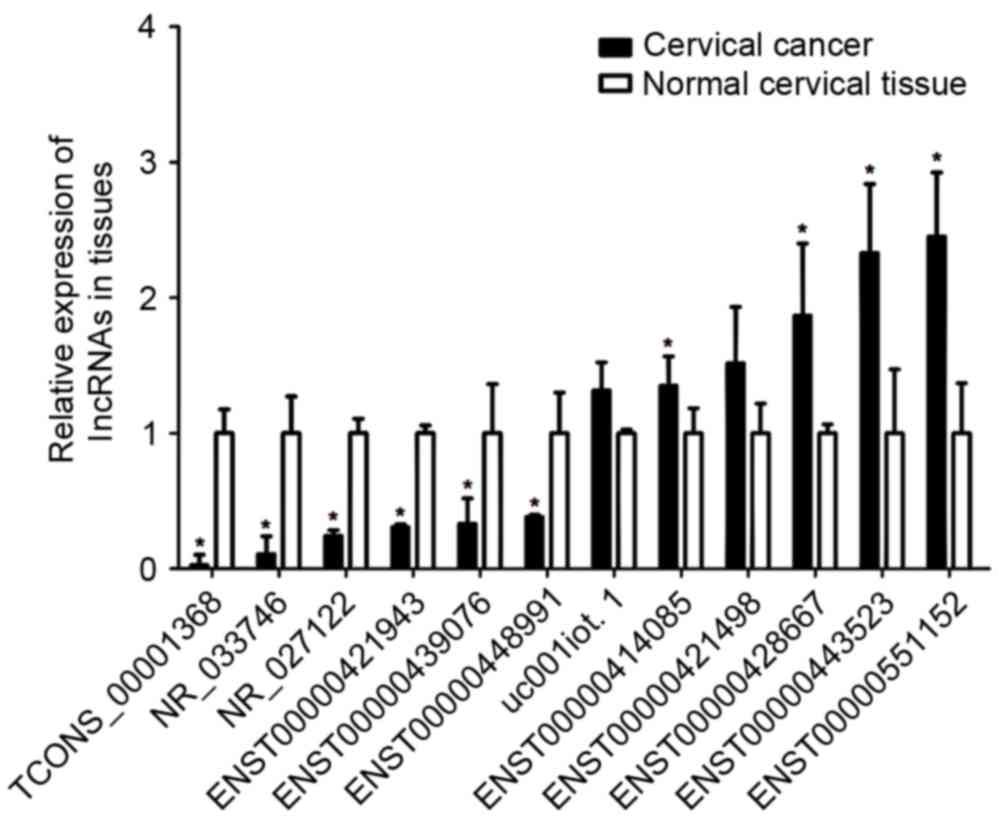
RT-qPCR validation
To confirm the microarray results, 12 lncRNAs were
selected at random and validated by RT-qPCR. The expression levels
of these lncRNAs were detected in 30 cervical carcinoma tissues and
20 normal cervical tissues. The analysis revealed that the lncRNAs
ENST00000414085, ENST00000428667, ENST00000551152 and
ENST00000443523 were upregulated, and that ENST00000421943,
NR_027122, ENST00000448991, ENST00000439076, TCONS_00001368 and
NR_033746 were downregulated in the cervical carcinoma tissues
relative to the normal cervical tissues (P<0.05; Fig. 4), in good consistency with the
microarray results. However, the lncRNAs ENST00000421498 and
uc001iot.1 showed no statistically significant differences in
expression between cancerous tissue and normal tissues. In summary,
the vast majority of the lncRNAs assessed by RT-qPCR were in line
with the trends observed by lncRNA microarray.
For further study on the potential function of
lncRNAs, the upregulated lncRNA ENST00000551152 and downregulated
lncRNA TCONS_00001368 were investigated in cervical carcinoma cell
lines. It was observed that ENST00000551152 was overexpressed in
SiHa, HeLa, C33A and MS751 cervical cancer cell lines compared with
normal cervical cancer tissues, whereas the expression of
TCONS_00001368 was decreased in these cervical cancer cell lines
(P<0.05; Fig. 5).
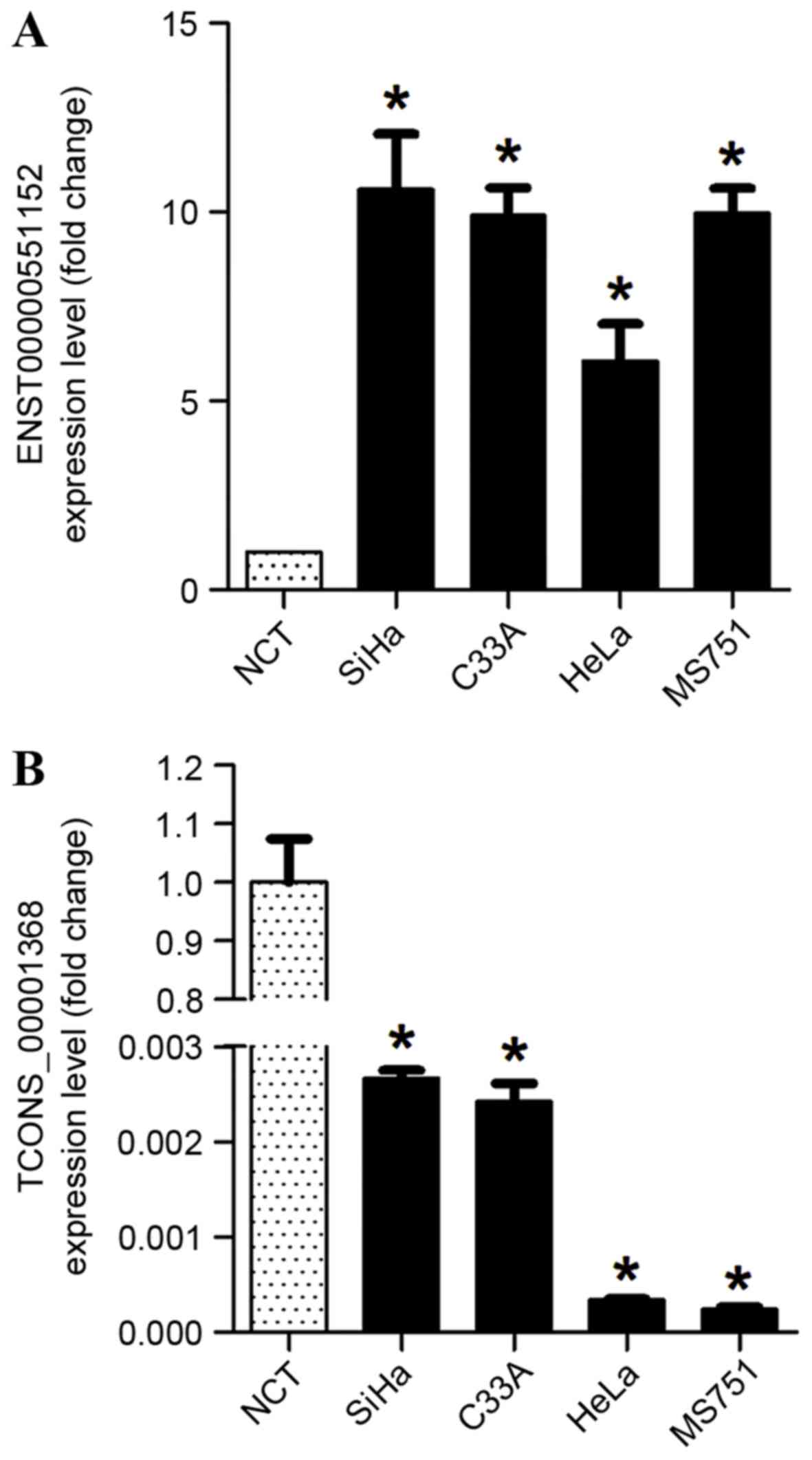 | Figure 5.Relative expression levels of two
lncRNAs in cervical cancer cell lines. (A) ENST00000551152 lncRNA,
which was upregulated in cervical cancer tissue samples, was
confirmed to be significantly overexpressed in SiHa, HeLa, C33A and
MS751 cervical cancer cell lines when compared with NCT. (B)
TCONS_00001368 lncRNA, which was significantly downregulated in
cervical cancer tissue samples, was also confirmed to be
downregulated in SiHa, HeLa, C33A and MS751 cervical cancer cell
lines when compared with NCT. *P<0.05 vs. NCT. lncRNA, long
non-coding RNA; NCT, normal cervical tissues. |
Correlation of ENST00000551152
expression and clinicopathological parameters
As ENST00000551152 had the highest level of
overexpression among the 12 lncRNAs selected for validation, the
association between ENST00000551152 expression level and
clinicopathological characteristics in cervical cancer patients was
examined to reveal the potential role of this lncRNA in cervical
cancer pathogenesis. Additionally, 20 normal and 60 cancerous
cervical tissue samples were selected for ENST00000551152 level
quantification. The data indicated that the expression of
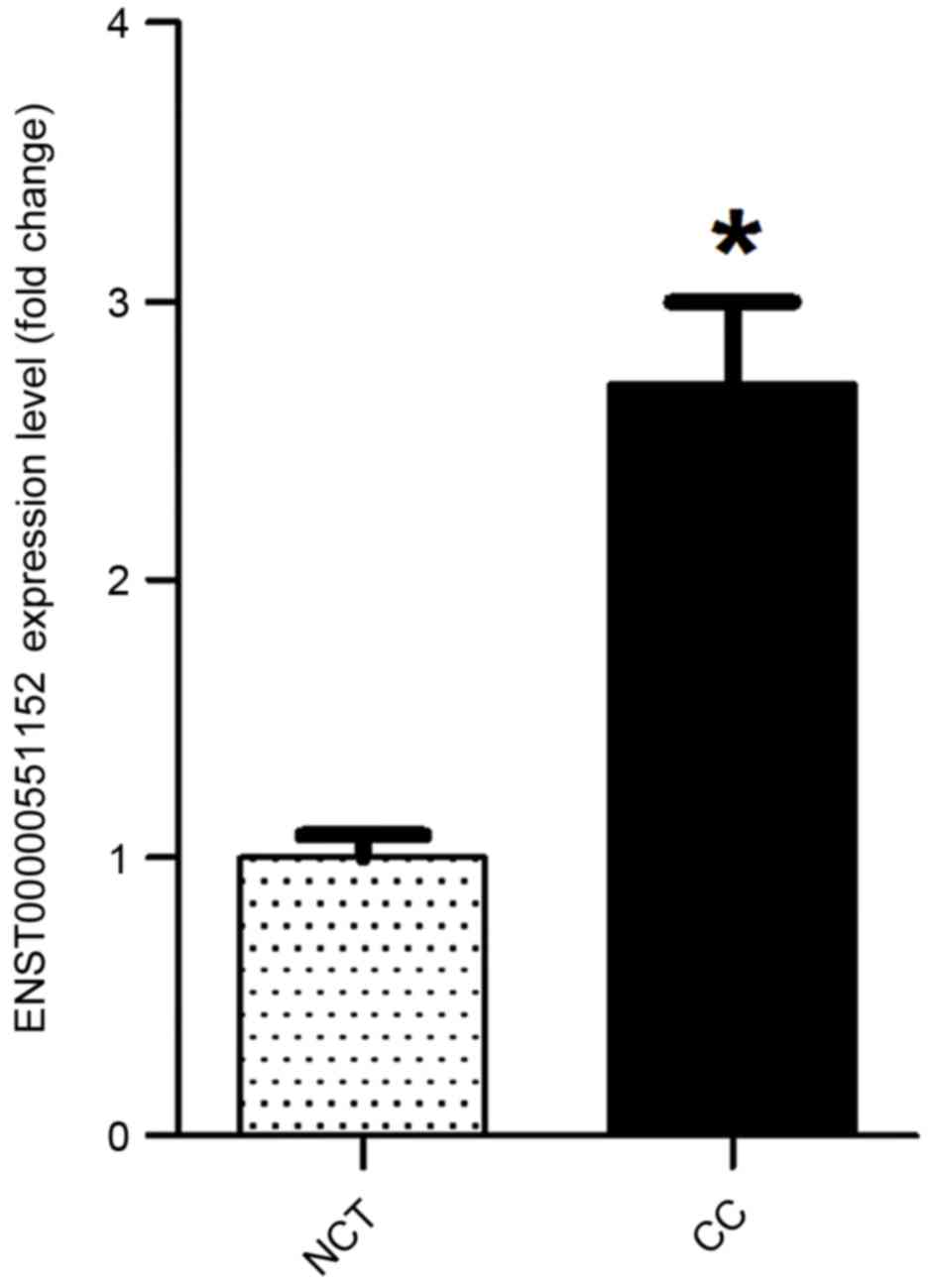
ENST00000551152 in cancer tissues was significantly increased by
2.45 mean fold-change compared with that in normal cervical tissues
(P<0.05; Fig. 6).
ROC curve analysis was employed to determine the
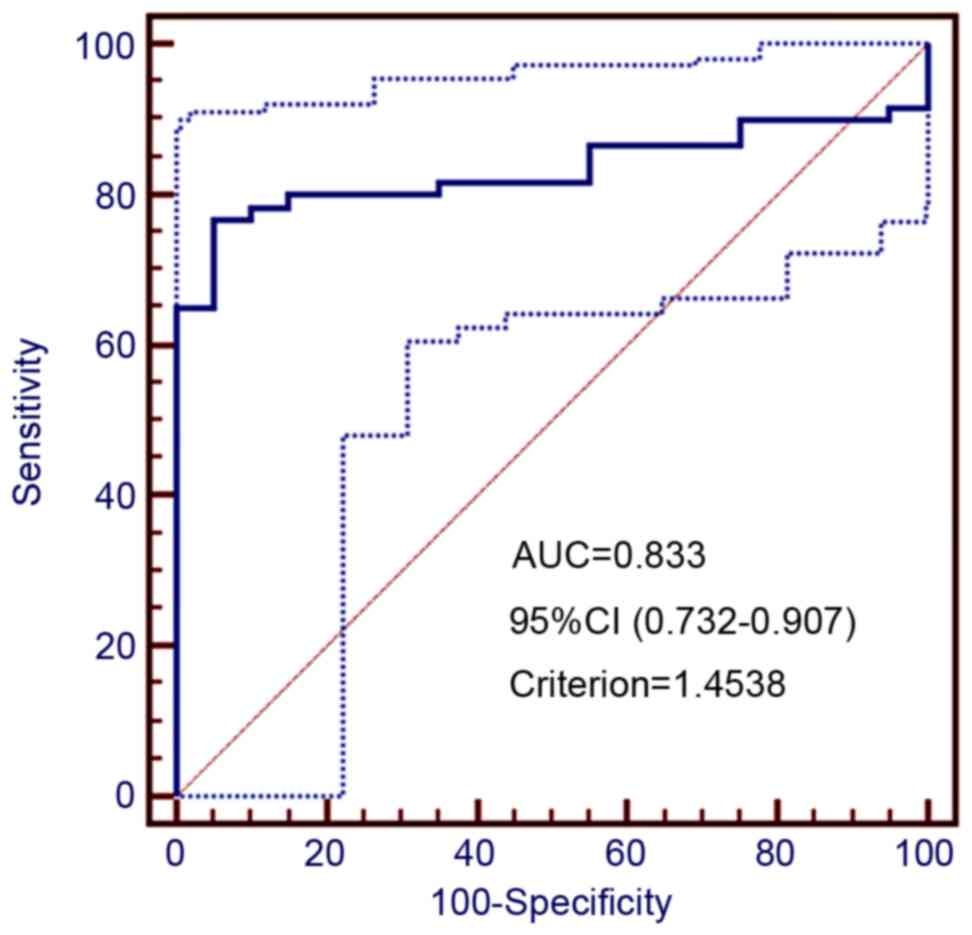
cutoff value for high ENST00000551152 expression (Fig. 7). Based on this analysis, a total of
46 cancer tissue samples had ≥1.45-fold increased ENST00000551152
levels and were defined as having high level of expression, whereas
14 cases had <1.45-fold expression and were considered as having
low level expression. Significant correlations were identified
between the ENST00000551152 expression level and several prognostic
risk factors, including the International Federation of Gynecology
and Obstetrics (FIGO) stage (P=0.017), tumor size (P=0.022), extent
of stromal invasion (P=0.001), pelvic lymph node metastasis
(P=0.026) and SCC expression level (P=0.004). No significant
difference was found between the expression of ENST00000551152 and
other variables, including age, differentiation grade and
lymphovascular space invasion (Table
I). Taken together, the data indicate that the lncRNA
ENST00000551152 may serve a critical role in cervical cancer
pathogenesis.
Discussion
The significance of lncRNA in cancer pathogenesis
and the correlations between various types of human cancers and the
aberrant expression of lncRNAs have been elaborated in numerous
studies in the past decade (27,28).
Certain lncRNAs behave like oncogenes or tumor-suppressors,
performing important functions in cancer initiation, progression,
metastasis or recurrence. For example, the lncRNA HOTAIR
promotes cellular proliferation, cell cycle progression, migration,
and invasion via inhibiting p21 in cervical cancer, and thus
functions as an oncogene (17). By
contrast, lncRNA-LET serves as a tumor suppressor; the overall
survival of patients with cervical cancer with downregulated
lncRNA-LET is markedly poorer than in those with lncRNA-LET
upregulation (14). Thus, it is
hypothesized that lncRNAs may be key regulators in cervical cancer
pathogenesis. However, the exact pathogenesis-associated functions
of lncRNAs in cervical cancer remain unclear. Thus, it is necessary
to identify the expression patterns of lncRNAs on a large scale,
which is likely to aid in the identification of novel biomarkers
and provide a potential therapeutic target for further
research.
In the present study, the differential expression
patterns of lncRNAs and mRNAs were profiled by comparing cervical
cancer and normal cervical tissue samples to identify the
pathogenesis-associated functions of lncRNAs. A total of 5,844
lncRNAs and 4,436 mRNAs were differentially expressed (with
fold-change, ≥2) according to the microarray results. Among the
5,844 lncRNAs, 2,574 were upregulated and 3,270 were downregulated,
a large portion of which have not yet been functionally
characterized. To confirm the consistency of the microarray, 12
lncRNAs were randomly selected for quantification by RT-qPCR in 30
cervical cancer and 20 normal cervical tissue samples. The RT-qPCR
results were largely consistent with the microarray data,
demonstrating that the high-throughput microarray was able to
reflect the actual expression patterns of lncRNAs in cervical
cancer tissue samples. In order to gain a better understanding of
the biological function of lncRNAs, the expression levels of
ENST00000551152 (upregulated) and TCONS_00001368 (downregulated)
were assessed in cervical cancer cell lines. Furthermore, the
association between the expression of ENST00000551152 and
clinicopathological variables were analyzed in tissue samples. It
was identified that ENST00000551152 expression levels were closely
associated with FIGO stage, tumor size, stromal invasion, pelvic
lymph node metastasis and SCC antigen expression level. Thus,
ENST00000551152 shows great potential as a biomarker in cervical
cancer pathogenesis. Due to the limits of the present study,
further study should be conducted to investigate the mechanism by
which lncRNAs affect the biology of cervical cancer cells.
Analysis of differentially expressed mRNAs, which
were potential targets of the differentially expressed lncRNAs, was
completed utilizing the KEGG pathway annotation database in order
to increase understanding of the possible functional roles of the
lncRNAs. Relative to normal cervical tissues, cervical cancer
tissue exhibited upregulated mRNAs that were associated with 40
KEGG pathways. The pathways included ‘cytokine-cytokine receptor
interaction’, ‘chemokine signaling pathway’, and ‘cell cycle’, all
of which have previously been implicated by a number of studies
(29–31). The downregulated mRNAs were enriched
for 37 pathways, of which ‘MAPK signaling pathway’ was the most
frequently reported. The CNC network suggested that the regulatory
interaction between lncRNAs and mRNAs is complex in cervical
cancer. Although elaboration of the exact mechanisms of those
genes' involvement in cervical cancer was not achieved, lncRNAs
with differential expression may be participants in cervical cancer
by regulating these coding genes.
In conclusion, the present study has demonstrated
the comprehensive expression profile of lncRNAs and mRNAs in
cervical cancer via microarray technology. The differential
expression of lncRNAs and mRNAs were observed in cervical cancer
samples relative to paired normal cervical tissues. The potential
correlation between lncRNAs and protein-coding genes and the roles
of lncRNAs in cervical cancer were investigated via bioinformatics
analyses, including a CNC network, KEGG pathway annotation and GO
category classification. Detailed regulatory mechanisms remain to
be further elaborated. Furthermore, each lncRNA and associated mRNA
interaction could be a candidate diagnostic marker or therapeutic
target for cervical cancer, and further investigation is
required.
Acknowledgements
The present study was supported by National Natural
Science Foundation of China (grant no. 8167100337), the Natural
Science Foundation of Guangdong Province (grant no. 2015A030313073)
and Science and Technology Program of Guangzhou (grant no.
201510010289).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar
|
|
2
|
Kucukmetin A, Biliatis I, Ratnavelu N,
Patel A, Cameron I, Ralte A and Naik R: Laparoscopic radical
trachelectomy is an alternative to laparotomy with improved
perioperative outcomes in patients with early-stage cervical
cancer. Int J Gynecol Cancer. 24:135–140. 2014. View Article : Google Scholar
|
|
3
|
Rijkmans EC, Nout RA, Rutten IH, Ketelaars
M, Neelis KJ, Laman MS, Coen VL, Gaarenstroom KN, Kroep JR and
Creutzberg CL: Improved survival of patients with cervical cancer
treated with image-guided brachytherapy compared with conventional
brachytherapy. Gynecol Oncol. 135:231–238. 2014. View Article : Google Scholar
|
|
4
|
Tewari KS, Sill MW, Long HJ III, Penson
RT, Huang H, Ramondetta LM, Landrum LM, Oaknin A, Reid TJ, Leitao
MM, et al: Improved survival with bevacizumab in advanced cervical
cancer. N Engl J Med. 370:734–743. 2014. View Article : Google Scholar
|
|
5
|
Weinmann S, Williams AE, Kamineni A, Buist
DS, Masterson EE, Stout NK, Stark A, Ross TR, Owens CL, Field TS
and Doubeni CA: Cervical cancer screening and follow-up in 4
geographically diverse US health care systems, 1998 through 2007.
Cancer-Am Cancer Soc. 121:2976–2983. 2015.
|
|
6
|
Koskas M, Rodier JM, Bretel JJ, Bonneau C,
Luton D, Touboul C and Rouzier R: Life after uterine cancer. Rev
Prat. 64:816–820. 2014.
|
|
7
|
Gien LT and Covens A: Lymph node
assessment in cervical cancer: Prognostic and therapeutic
implications. J Surg Oncol. 99:242–247. 2009. View Article : Google Scholar
|
|
8
|
Caley DP, Pink RC, Trujillano D and Carter
DR: Long noncoding RNAs, chromatin and development.
ScientificWorldJournal. 10:90–102. 2010. View Article : Google Scholar
|
|
9
|
Lee JT, Davidow LS and Warshawsky D: Tsix,
a gene antisense to Xist at the X-inactivation centre. Nat Genet.
21:400–404. 1999. View
Article : Google Scholar
|
|
10
|
Hung T and Chang HY: Long noncoding RNA in
genome regulation: Prospects and mechanisms. RNA Biol. 7:582–585.
2010. View Article : Google Scholar
|
|
11
|
Ravasi T, Suzuki H, Pang KC, Katayama S,
Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, et al:
Experimental validation of the regulated expression of large
numbers of non-coding RNAs from the mouse genome. Genome Res.
16:11–19. 2006. View Article : Google Scholar
|
|
12
|
Gibb EA, Becker-Santos DD, Enfield KS,
Guillaud M, Niekerk Dv, Matisic JP, Macaulay CE and Lam WL:
Aberrant expression of long noncoding RNAs in cervical
intraepithelial neoplasia. Int J Gynecol Cancer. 22:1557–1563.
2012. View Article : Google Scholar
|
|
13
|
Cao S, Liu W, Li F, Zhao W and Qin C:
Decreased expression of lncRNA GAS5 predicts a poor prognosis in
cervical cancer. Int J Clin Exp Pathol. 7:6776–6783. 2014.
|
|
14
|
Jiang S, Wang HL and Yang J: Low
expression of long non-coding RNA LET inhibits carcinogenesis of
cervical cancer. Int J Clin Exp Pathol. 8:806–811. 2015.
|
|
15
|
Jiang Y, Li Y, Fang S, Jiang B, Qin C, Xie
P, Zhou G and Li G: The role of MALAT1 correlates with HPV in
cervical cancer. Oncol Lett. 7:2135–2141. 2014. View Article : Google Scholar
|
|
16
|
Sun NX, Ye C, Zhao Q, Zhang Q, Xu C, Wang
SB, Jin ZJ, Sun SH, Wang F and Li W: Long noncoding RNA-EBIC
promotes tumor cell invasion by binding to EZH2 and repressing
E-cadherin in cervical cancer. PLoS One. 9:e1003402014. View Article : Google Scholar
|
|
17
|
Jing L, Yuan W, Ruofan D, Jinjin Y and
Haifeng Q: HOTAIR enhanced aggressive biological behaviors and
induced radio-resistance via inhibiting p21 in cervical cancer.
Tumour Biol. 36:3611–3619. 2015. View Article : Google Scholar
|
|
18
|
Yang M, Zhai X, Xia B, Wang Y and Lou G:
Long noncoding RNA CCHE1 promotes cervical cancer cell
proliferation via upregulating PCNA. Tumour Biol. 36:7615–7622.
2015. View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Sui W, Lin H, Peng W, Huang Y, Chen J,
Zhang Y and Dai Y: Molecular dysfunctions in acute rejection after
renal transplantation revealed by integrated analysis of
transcription factor, microRNA and long noncoding RNA. Genomics.
102:310–322. 2013. View Article : Google Scholar
|
|
21
|
Kanehisa M, Goto S, Sato Y, Furumichi M
and Tanabe M: Tanabe, KEGG for integration and interpretation of
large-scale molecular data sets. Nucleic Acids Res. 40:D109–D114.
2012. View Article : Google Scholar
|
|
22
|
Gene Ontology Consortium, . Gene ontology,
gene ontology consortium: Going forward. Nucleic Acids Res.
43:D1049–D1056. 2015. View Article : Google Scholar
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar
|
|
24
|
Luo R, Zhang M, Liu L, Lu S, Zhang CZ and
Yun J: Decrease of fibulin-3 in hepatocellular carcinoma indicates
poor prognosis. PLoS One. 8:e705112013. View Article : Google Scholar
|
|
25
|
Su PH, Lin YW, Huang RL, Liao YP, Lee HY,
Wang HC, Chao TK, Chen CK, Chan MW, Chu TY, et al: Epigenetic
silencing of PTPRR activates MAPK signaling, promotes metastasis
and serves as a biomarker of invasive cervical cancer. Oncogene.
32:15–26. 2013. View Article : Google Scholar
|
|
26
|
Chen Q, Cao HZ and Zheng PS: LGR5 promotes
the proliferation and tumor formation of cervical cancer cells
through the Wnt/β-catenin signaling pathway. Oncotarget.
5:9092–9105. 2014.
|
|
27
|
QI P and Du X: The long non-coding RNAs, a
new cancer diagnostic and therapeutic gold mine. Mod Pathol.
26:155–165. 2013. View Article : Google Scholar
|
|
28
|
Qiu MT, Hu JW, Yin R and Xu L: Long
noncoding RNA: An emerging paradigm of cancer research. Tumour
Biol. 34:613–620. 2013. View Article : Google Scholar
|
|
29
|
Valle-Mendiola A, Weiss-Steider B,
Rocha-Zavaleta L and Soto-Cruz I: IL-2 enhances cervical cancer
cells proliferation and JAK3/STAT5 phosphorylation at low doses,
while at high doses IL-2 has opposite effects. Cancer Invest.
32:115–125. 2014. View Article : Google Scholar
|
|
30
|
Xiang T, Du L, Pham P, Zhu B and Jiang S:
Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell
cycle arrest in human cervical cancer cells via the ROS-dependent
mitochondrial pathway. Cancer Lett. 364:79–88. 2015. View Article : Google Scholar
|
|
31
|
Pahne-Zeppenfeld J, Schröer N,
Walch-Rückheim B, Oldak M, Gorter A, Hegde S and Smola S: Cervical
cancer cell-derived interleukin-6 impairs CCR7-dependent migration
of MMP-9-expressing dendritic cells. Int J Cancer. 134:2061–2073.
2014. View Article : Google Scholar
|