Introduction
The incidence of thyroid cancer has rapidly
increased over the past few years and thyroid cancer is now the
fifth most common cancer type diagnosed in women in the USA
(1,2).
Papillary thyroid cancer (PTC) accounts for >80% of thyroid
tumors and is associated with a relatively favorable prognosis
subsequent to surgical treatment. However, PTC tumors that are
difficult to resect and those that metastasize remain a challenge
to treat with long-term success (3–5). Examples
of such challenging cases include recurrent thyroid cancer and
anaplastic thyroid cancer (ATC) (6).
It has been reported that recurrence is experienced in 20–30% of
patients with thyroid cancer within the first 20 years post-surgery
(7). Radioactive iodine and
thyrotropin-suppressive therapies are used to treat these patients;
however, persistent metastasis and dedifferentiation despite these
treatments associate them with poor prognoses (4,5). The
10-year survival rate for patients with recurrent disease is only
10%, and this value has remained unchanged for numerous years due
to the limited progress in the available treatments for recurrent
thyroid cancer (8).
BRAF mutations have been identified in a variety of
human cancer types, including thyroid cancer, malignant melanoma,
ovarian tumors and colorectal cancer (CRC) (9,10). Over 40
BRAF mutations have been reported, the most commonly reported being
BRAF V600E, which results from a thymine-to-adenosine
transformation at position 1799 in exon 15, causing a
valine-to-glutamate substitution at residue 600 in the peptide
(11). The activity of the BRAF V600E
protein is 10-fold greater than that of the wild-type protein and
cannot be regulated appropriately (12). The resulting continuous activation of
BRAF V600E activates the mitogen assisted protein kinase (MAPK)
signaling pathway and promotes tumor progression (12).
The BRAF V600E mutation occurs in 29–83% cases of
PTC, the most common subtype of thyroid cancer, and 24% cases of
ATC, the most aggressive and lethal subtype of thyroid cancer
(13–15). In addition to acting as a biomarker
for diagnosis, the BRAF V600E expression is associated with
aggressive and iodine-resistant phenotypes of thyroid cancer
(14). It has been reported that
thyroid cancers harboring the BRAF V600E mutation tend to exhibit
other factors indicative of a poor prognosis, including
extra-thyroidal extension, lymph node metastasis, advanced stage,
iodine-131 resistance, recurrent disease and distant metastasis
(16–21). Even in papillary thyroid
microcarcinoma (with diameters <1 cm), the BRAF V600E mutation
is associated with extra-thyroidal extension and lymph node
metastasis (22,23).
The BRAF V600E protein has been investigated as a
therapeutic target in a number of studies and various BRAF V600E
inhibitors have been identified (24–26).
Sorafenib is a first-generation BRAF V600E inhibitor. However, the
mechanism by which the effect of sorafenib is mediated remains
unclear due to its ability to inhibit multiple kinases (27,28).
Vemurafenib and dabrafenib are selective BRAF V600E inhibitors,
which have demonstrated therapeutic activity in phase 1 and 2
clinical trials in patients with BRAF V600E-mutation-induced
metastatic melanoma, and they have been approved by the Food and
Drug Administration (29). However,
these inhibitors are unable to efficiently suppress the progression
of other types of cancer and resistance to the inhibitors developed
within 6–7 months, even in melanoma (30,31). In
CRC, endothelial growth factor receptor (EGFR)-mediated
re-proliferation serves an essential role in the resistance to BRAF
inhibition (32). It has been
reported that overexpression of EGFR results in constitutive
activation of the MAPK signaling pathway and promotes cancer cell
proliferation, even during treatment with selective BRAF V600E
inhibitors (32). In the present
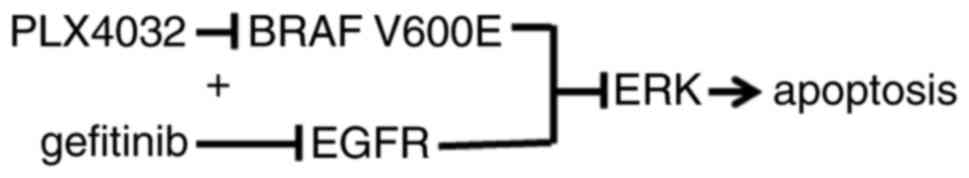
study, BRAF V600E was inhibited and downregulation of EGFR was
induced in PTC cell lines to determine whether this combined
strategy was able to efficiently block cancer cell proliferation,
and if it may be a potential novel treatment for thyroid
cancer.
Materials and methods
Cell lines and reagents
The K1 and BCPAP cell lines were purchased from
Sigma-Aldrich (Merck KGaA Darmstadt, Germany). K1 and BCPAP are
papillary thyroid cancer cell lines, and K1 cells are a derivative
of the GLAG-66 cell line (33). Both
cell lines were passaged for <3 months from stocks generated
from the first or second passage of the original cells. The
mutation status of these cell lines is reported in Table I. BCPAP cells were maintained in
Dulbecco's modified Eagle medium with 10% fetal bovine serum (FBS),
and K1 cells were maintained in RPMI-1640 supplemented with 10%
FBS. The cells were maintained at 37°C in a humidified incubator
with 5% carbon dioxide. The BRAF V600E-selective inhibitor PLX4032
was obtained from Plexxikon, Inc. (Berkeley, CA, USA) and the
EGFR-selective inhibitor, gefitinib, was obtained from Roche
Diagnostics (Basel, Switzerland). Inhibitors were dissolved in DMSO
and the stock solutions were stored at −20°C.
 | Table I.Basic information and genetic
profiles of the cell lines. |
Table I.
Basic information and genetic
profiles of the cell lines.
| Cell line | Subtype | BRAF variation | EGFR variation |
|---|
| K1 | PTC | GTG-GAG | Wild-type |
| BCPAP | PTC | GTG-GAG | CAG-CAA |
Cellular proliferation assays
Proliferation of K1 and BCPAP cells was evaluated
using the MTT assay (Sigma-Aldrich; Merck KGaA). Cells were plated
in 96-well microtiter plates at a density of 3×103
cells/well in a volume of 180 µl DMEM with FBS. PLX4032 with or
without gefitinib was diluted in media containing 1% DMSO at 10X
the final assay concentrations (PLX4032: 0.01, 0.03, 0.1, 0.3, 1
and 3 µM; gefitinib: 0.125 µM). After 24 h of culture, 20 µl each
drug dilution was added in triplicate to separate wells. Cells were
assayed for proliferation at 24, 48, 72, 96 and 120-h time points.
The percentage of inhibition was calculated using the following
formula: 100-(mean absorbance of experimental wells/mean absorbance
of control wells) ×100. The half maximal inhibitory concentration
(IC50) values were determined by calculating the
regression of plots produced from the logarithms of concentration
vs. percentage inhibition, using XLfit software (version 4.2; ID
Business Solutions Ltd., Guildford, UK).
Western blot analysis
Cells were seeded at 70–75% confluence in 6-well
plates, 1 day prior to drug treatment. Cells were cultured at the
aforementioned drug concentrations and times at 37°C with 5%
CO2, and were harvested and lysed in 1X cell lysis
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA). After a
20-min incubation on ice, the lysates were centrifuged at 12,000 ×
g for 15 min at 4°C to clear insoluble debris. The protein
concentrations of the lysates were then determined with a BCA kit
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
equal amounts of total protein from cell lysates and tumor lysates
(10 µg for each lane) were resolved on 4–12% NuPage gradient
polyacrylamide gels (Invitrogen; Thermo Fisher Scientific, Inc.)
before they were blotted onto polyvinylidene fluoride membrane (GE
Healthcare Life Sciences, Little Chalfont, UK). The membranes were
first blocked with 5% dry fat free milk for 1 h at room temperature
and washed twice with Tris-Buffered Saline containing 0.1% Tween-20
(Affymetrix, Inc.; Thermo Fisher Scientific, Inc.). The blocked
membranes were probed with rabbit polyclonal antibodies against
human phosphorylated-extracellular regulated kinase (p-ERK1/2)
(Cell Signaling Technology, Inc.; cat no. 4370S; dilution,
1:1,000), ERK1/2 (Sigma-Aldrich; Merck KGaA; cat no. M5670;
dilution, 1:1,000), β-actin (cat no. sc-130656; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; dilution, 1:2,000) and
incubated for 2 h at room temperature. Followed by incubation for 1
h at room temperature with horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (cat no. sc-2004; dilution,
1:5,000; Santa Cruz Biotechnology, Inc.). A chemiluminescent signal
was generated using Amersham ECL Plus Western Blotting Detection
reagents (GE Healthcare, Chicago, IL, USA) and detected with a
Fujifilm LAS-3000 imager (Fujifilm, Tokyo, Japan). The densitometry
was performed using Multi Gauge 3.0 software (Fujifilm).
Flow cytometry
Cells were seeded in 6-well plates at
4×105 cells/well and incubated for 24 h prior to
treatment with PLX4032 and/or gefitinib. A total of five treatment
groups were analyzed: PLX4032 monotreatment for 1 day; PLX4032
monotreatment for 5 days; PLX4032 combined with gefitinib for 1
day; and PLX4032 combined with gefitinib for 5 days. The
concentrations of PLX4032 and gefitinib were 0.206 and 0.125 µM,
respectively. Untreated cells were used as a control group.
Apoptosis was evaluated by measuring the exposure of
phosphatidylserine on cell membranes using Annexin V-FITC apoptosis
detection kits (Sigma-Aldrich; Merck KGaA). Following the
treatments, cell pellets were resuspended in a solution containing
5 µg/ml propidium iodide and 1 µg/ml Annexin V-fluorescein
isothiocyanate for 15 min at room temperature in darkness.
Subsequently, the cells were assessed by a flow cytometer equipped
with CellQuest 5.1 software (BD Biosciences, Franklin Lakes, NJ,
USA).
Statistical methods
GraphPad Prism 7.0 software (GraphPad Software,
Inc., La Jolla, CA, USA) was used to analyze all data and the
results are presented as the mean ± standard error. Significant
differences were determined by one-way analysis of variance
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Co-treated with gefitinib and PLX4032
had a stronger anti-proliferation efficacy
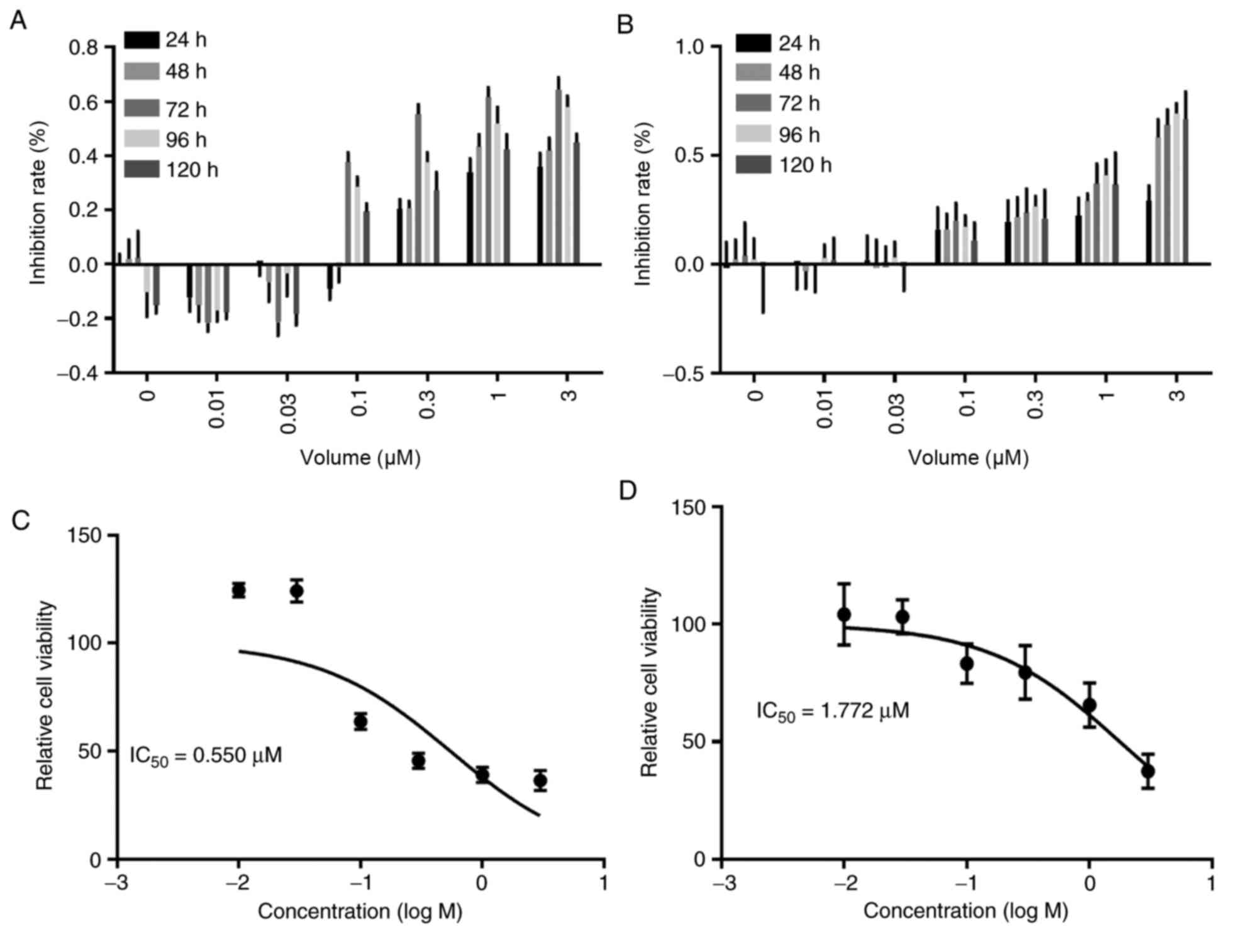
To evaluate the anti-proliferation efficacy of
PLX4032, the IC50 values of PLX4032 were obtained for K1
and BCPAP cells using an MTT assay at different time-points. Both
cell lines were most sensitive to PLX4032 at 72 h. The
IC50 in K1 cells (0.550 µM) was compared with BCPAP
cells (1.772 µM) (Fig. 1). It has
been reported that the IC50 of gefitinib is 0.125 µM for
other cancer cell lines (32). The
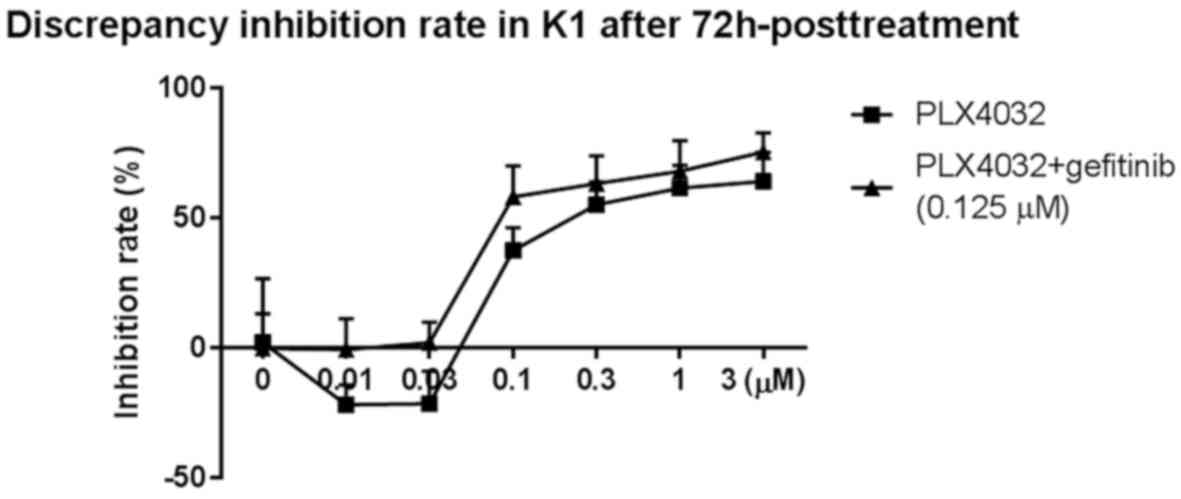
IC50 for PLX4032 in K1 cells co-treated with gefitinib
was determined at the 72-h time-point. The combination suppressed
proliferation more effectively than PLX4032 treatment alone,
resulting in a lower IC50 of 0.206 µM (Fig. 2).
Reactivation of the MAPK signaling
pathway was found in papillary thyroid cancer cell lines
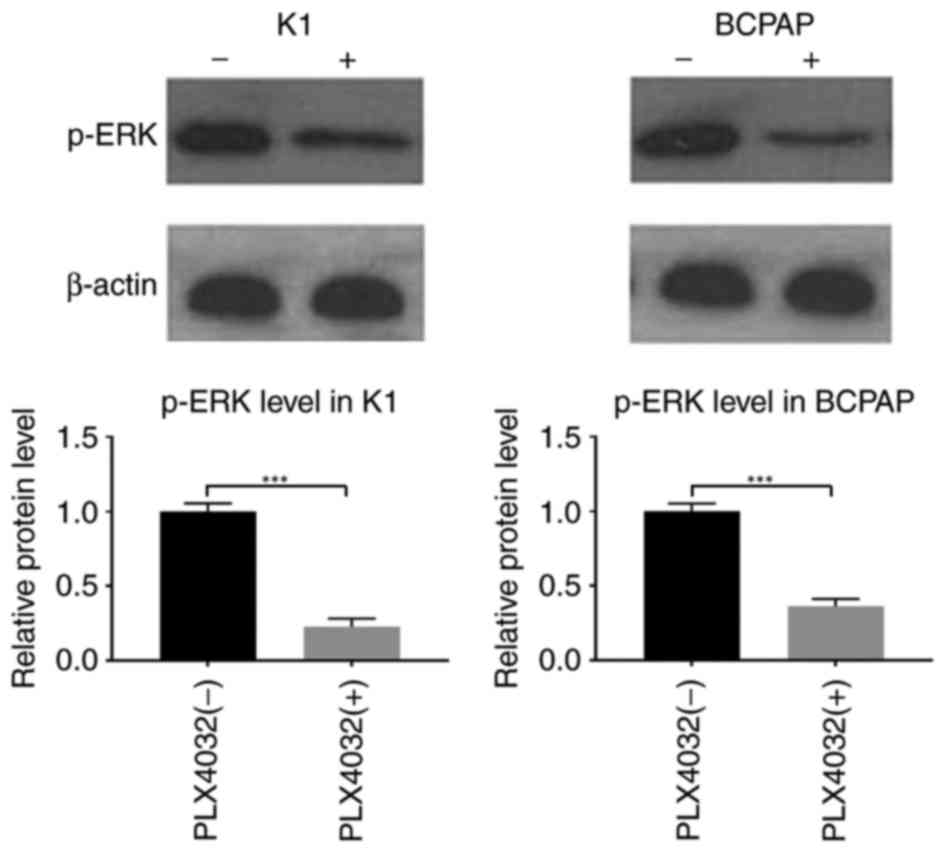
To investigate the mechanism of inhibitor
resistance, the activity of the MAPK signaling pathway was
examined. ERK is a key enzyme in this pathway; therefore, the
phosphorylation of ERK was measured in order to evaluate MAPK
pathway activity. K1 and BCPAP cells were treated with PLX4032 (K1:
0.2 µM; BCPAP: 1.772 µM) for 12 h. Western blot analyses indicated
that p-ERK levels were low in the PLX4032-treated groups compared
with the control groups, suggesting that MAPK activity was
suppressed by PLX4032 (Fig. 3). K1
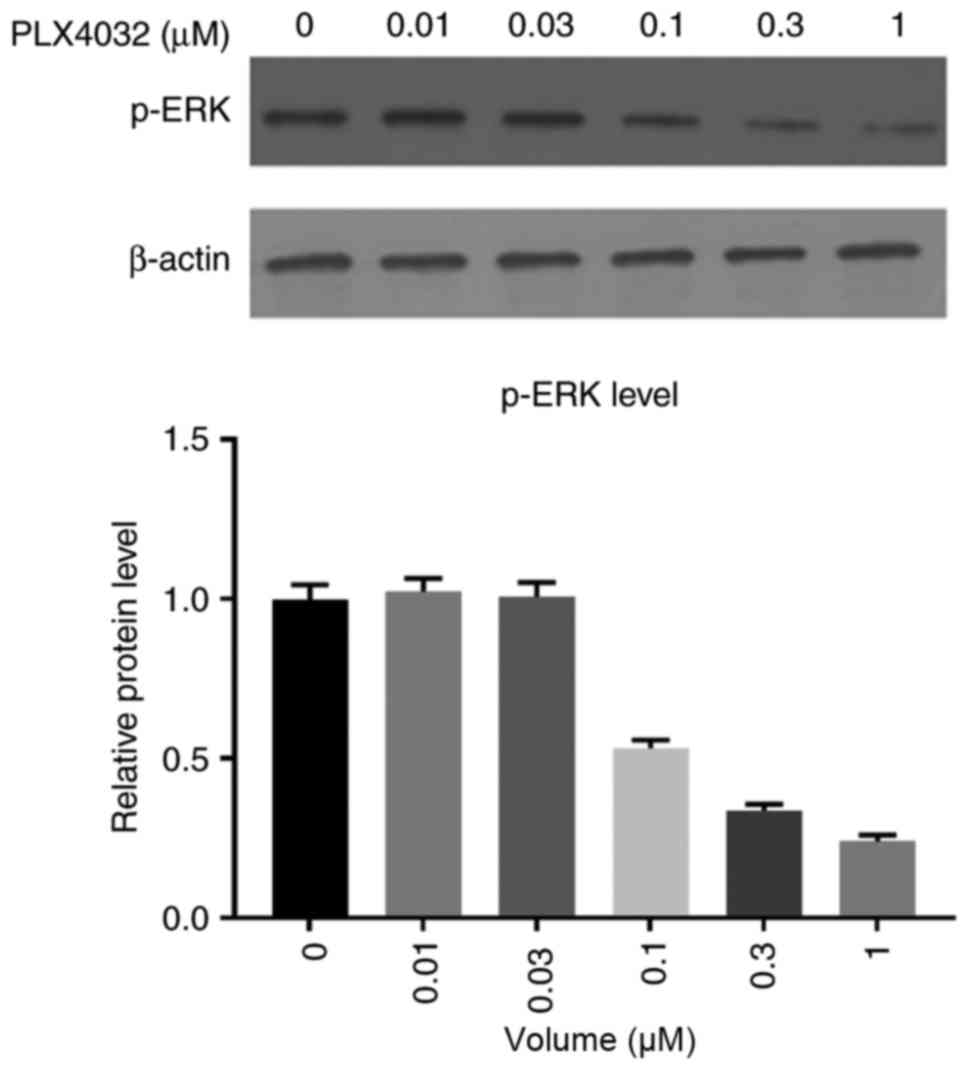
was more sensitive to PLX4032; therefore K1 was used in the
following experiments. Treatment of K1 cells with varying
concentrations (0, 0.01, 0.03, 0.1, 0.3 and 1 µM) of PLX4032
demonstrated that this inhibition of MAPK was dose-dependent
(Fig. 4). Subsequently, K1 cells were
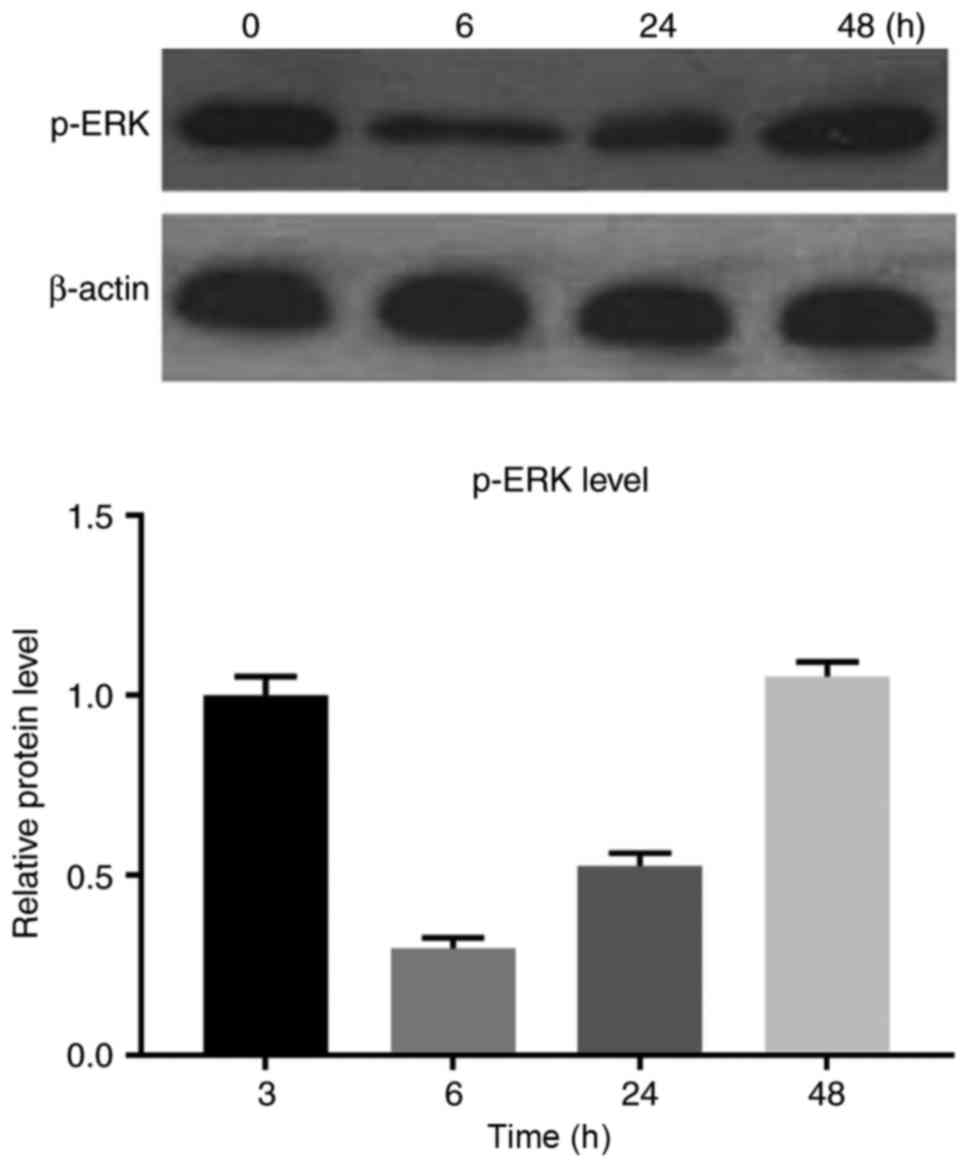
treated with 0.550 µM PLX4032 and the p-ERK levels were examined at
different time points (0, 6, 24 and 48 h). The lowest level of
p-ERK was detected at the 6 h time-point, after which the p-ERK
levels increased, indicating reactivation of the MAPK signaling
pathway (Fig. 5).
Co-treated with gefitinib and PLX4032
promoted apoptosis
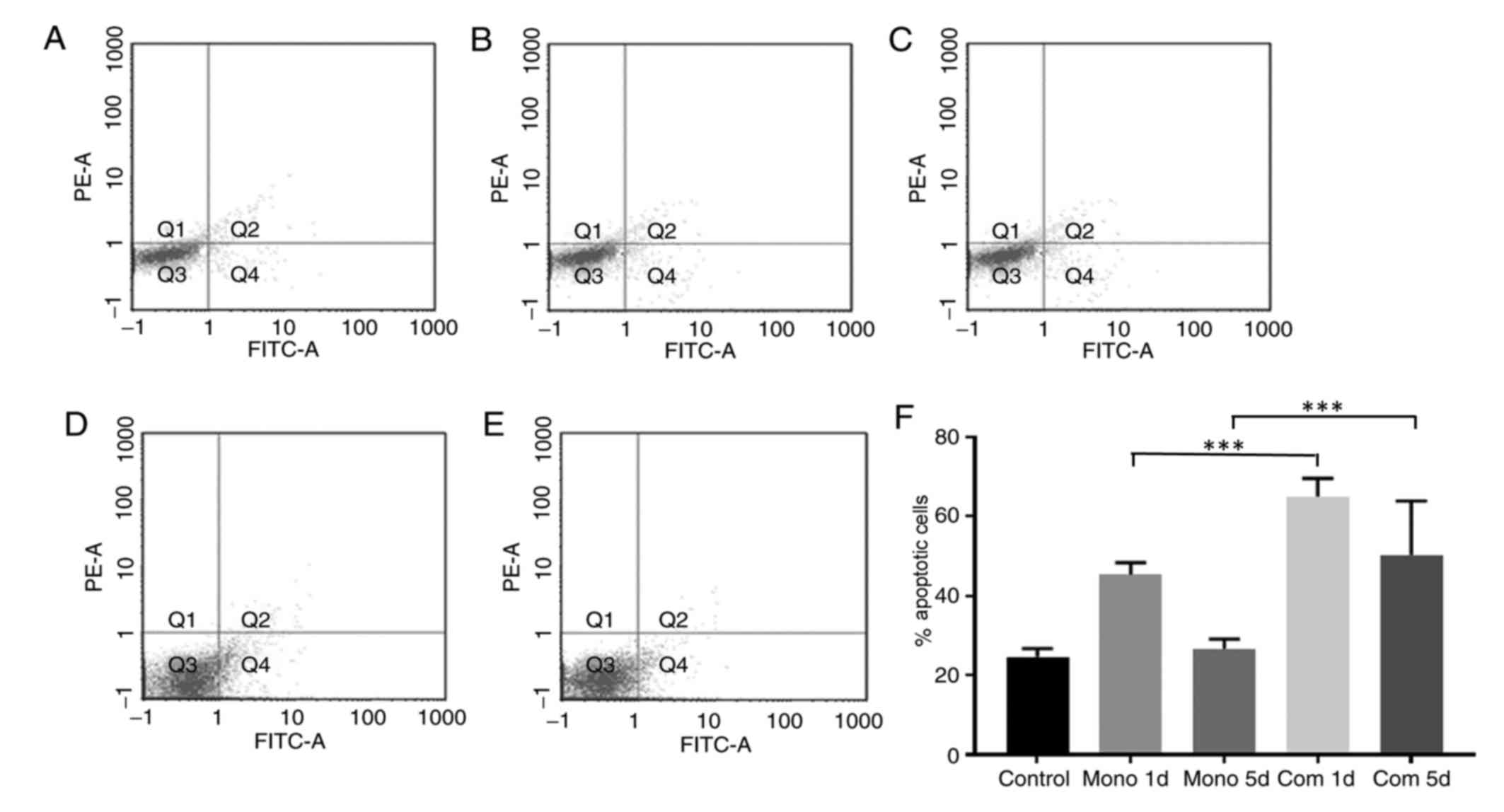
Flow cytometric analysis of apoptosis indicated that
apoptosis was elevated in cells cultured with PLX4032 for 1 day
compared with cells in the control group. However, apoptosis in the
treated group decreased from day 1 to 5, indicating a decrease in
the antitumor activity of PLX4032. Apoptosis of K1 cells treated
with PLX4032 and gefitinib was significantly elevated compared with
the control group and the PLX4032 monotreatment group (P<0.05).
In the co-treated group, there was no significant difference
between the level of apoptosis on days 1 and 5, which indicates
continued suppression over time (Fig.
6). Additionally, the combined treatment with an EGFR inhibitor
was able to increase the antitumor efficacy of BRAF V600E
inhibitors and suppress drug resistance in PTC cells (Fig. 7).
Discussion
The BRAF V600E mutation has been reported in
numerous types of cancer, including thyroid cancer, CRC and
melanoma (9,10). BRAF V600E is an oncogene, and serves
an important role in tumorigenesis and cancer progression. The
protein encoded by BRAF V600E is >10-fold more active compared
with the wild-type protein and is not regulated by normal feedback
mechanisms (12). BRAF V600E can
continuously activate the MAPK signaling pathway, thus promoting
tumor proliferation, invasion and metastasis (34). BRAF V600E mutations occur at different
rates in different cancer types, but the presence of the mutation
consistently indicates poor prognosis (14,15).
BRAF V600E is one of the most common genetic
alterations in thyroid cancer. It occurs in 29–83% of PTCs, which
comprise >80% of all thyroid tumors (13). PTCs harboring the BRAF V600E mutation
have more aggressive clinicopathological features, including
extra-thyroidal extension and lymph node metastasis (13–15).
Seeing as there are limited therapies for metastatic thyroid
cancer, novel treatments are essential to improve disease outcomes
for patients exhibiting this mutation. Inhibitors targeting the
BRAF V600E protein have been evaluated in clinical trials. However,
the inhibitor response depends on the type of cancer (24,25,30). The
selective inhibitor PLX4032 has exhibited beneficial therapeutic
effects on metastatic melanoma, but no effect on colon or thyroid
cancers (32). Numerous mechanisms to
account for the resistance of thyroid cancer to BRAF inhibition
have been suggested, including the following: Activation of the
RAF/MEK/ERK pathway caused by BRAF alternative splicing; activation
of the phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase
(AKT) pathway through hepatocyte growth factor receptor; autocrine
neuregulin 1-mediated human epidermal growth factor receptor 3
activation of the PI3K/AKT and RAS/RAF/MEK/ERK pathways; autocrine
interleukin-6-mediated activation of the janus kinase/signal
transducer and activator of transcription 3 and RAS/RAF/MEK/ERK
pathways, and increased autophagy (31,35–37).
In CRC, it has been reported that EGFR
overexpression promotes tumor proliferation (32). The present study determined whether
PLX4032 combined with the EGFR inhibitor, gefitinib, was able to
suppress thyroid cancer cell proliferation. Co-treatment with
gefitinib significantly increased the antitumor activity of
PLX4032. Frasca et al (38)
reported that co-treatment with PLX4032 was able to decrease the
IC50 of gefitinib. These data indicate that PLX4032
(vemurafenib) and gefitinib are able to produce synergistic
effects. Vemurafenib and gefitinib are safe for use in patients,
and therapeutic regimen combining BRAF and MAPK inhibitors may
result in greater efficacy and fewer side effects.
To further understand the molecular mechanism of
BRAF inhibitor resistance, activity of the MAPK pathway was
examined. In thyroid cancer cell lines, the MAPK pathway was
suppressed by PLX4032. This inhibition was the most effective after
6 h of treatment. MAPK activity gradually increased after 6 h,
indicating that the pathway was reactivated. Danysh et al
(39) reported that treatment of PTC
cell lines with a selective BRAF inhibitor for 5 months led to the
acquisition of resistance to the inhibitor through a spontaneous
KRAS G12D mutation. Thus, cancer cells can acquire short-term and
long-term resistance to BRAF inhibition. In the present study,
combined treatment with gefitinib and PLX4032 continuously
suppressed the MAPK pathway, indicating that EGFR serves an
important role in the resistance of cells to BRAF inhibition.
As MAPK signaling has an established association
with apoptosis, the survival of PLX4032-treated thyroid cancer
cells was quantified. Flow cytometry demonstrated that PLX4032
treatment initially increased apoptosis, but this effect declined
after 5 days of treatment, indicating that the effect of PLX4032 on
cell survival was short-term. Combined treatment with gefitinib
significantly increased apoptosis and prolonged this effect
(Fig. 7).
In conclusion, PTC cells harboring a BRAF V600E
mutation may become resistant to selective BRAF inhibition through
reactivation of the EGFR/MAPK pathway. Combined treatment with an
EGFR inhibitor is able to increase the antitumor efficacy of BRAF
V600E inhibitors and suppress drug resistance in PTC cells.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data that were generated or analyzed in this
study are included in this manuscript.
Authors' contributions
YJ and CZ conceived and designed the study. CH and
YY conducted the experiments. XZ and YL performed the statistical
analysis; MG interpreted the statistical analysis, reviewed and
made final approval of the version to be published. All authors
read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Davies L and Welch HG: Increasing
incidence of thyroid cancer in the United States, 1973–2002. JAMA.
295:2164–2167. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tuttle RM, Haddad RI, Ball DW, Byrd D,
Dickson P, Duh QY, Ehya H, Haymart M, Hoh C, Hunt JP, et al:
Thyroid carcinoma, version 2.2014. J Natl Compr Canc Netw.
12:1671–1680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antonelli A, Fallahi P, Ferrari SM, Carpi
A, Berti P, Materazzi G, Minuto M, Guastalli M and Miccoli P:
Dedifferentiated thyroid cancer: A therapeutic challenge. Biomed
Pharmacother. 62:559–563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schlumberger M, Brose M, Elisei R,
Leboulleux S, Luster M, Pitoia F and Pacini F: Definition and
management of radioactive iodine-refractory differentiated thyroid
cancer. Lancet Diabetes Endocrinol. 2:356–358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haddad RI, Lydiatt WM, Ball DW, Busaidy
NL, Byrd D, Callender G, Dickson P, Duh QY, Ehya H, Haymart M, et
al: Anaplastic thyroid carcinoma, version 2.2015. J Natl Compr Canc
Netw. 13:1140–1150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xing M, Westra WH, Tufano RP, Cohen Y,
Rosenbaum E, Rhoden KJ, Carson KA, Vasko V, Larin A, Tallini G, et
al: BRAF mutation predicts a poorer clinical prognosis for
papillary thyroid cancer. J Clin Endocrinol Metab. 90:6373–6379.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Durante C, Haddy N, Baudin E, Leboulleux
S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De
Vathaire F and Schlumberger M: Long-term outcome of 444 patients
with distant metastases from papillary and follicular thyroid
carcinoma: Benefits and limits of radioiodine therapy. J Clin
Endocrinol Metab. 91:2892–2899. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations in the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garnett MJ and Marais R: Guilty as
charged: B-RAF is a human oncogene. Cancer Cell. 6:313–319. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsai J, Lee JT, Wang W, Zhang J, Cho H,
Mamo S, Bremer R, Gillette S, Kong J, Haass NK, et al: Discovery of
a selective inhibitor of oncogenic B-Raf kinase with potent
antimelanoma activity. Proc Natl Acad Sci USA. 105:pp. 3041–3046.
2008; View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim KH, Kang DW, Kim SH, Seong IO and Kang
DY: Mutations of the BRAF gene in papillary thyroid carcinoma in a
Korean population. Yonsei Med J. 45:818–821. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Namba H, Nakashima M, Hayashi T, Hayashida
N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T and
Yamashita S: Clinical implication of hot spot BRAF mutation, V599E,
in papillary thyroid cancers. J Clin Endocrinol Metab.
88:4393–4397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing M: BRAF mutation in thyroid cancer.
Endocr Relat Cancer. 12:245–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sherman SI, Angelos P, Ball DW, Beenken
SW, Byrd D, Clark OH, Daniels GH, Dilawari RA, Ehya H, Farrar WB,
et al: Thyroid carcinoma. J Natl Compr Canc Netw. 3:404–457.
2005.PubMed/NCBI
|
|
17
|
DeGroot LJ, Kaplan EL, McCormick M and
Straus FH: Natural history, treatment, and course of papillary
thyroid carcinoma. J Clin Endocrinol Metab. 71:414–424. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mazzaferri EL and Kloos RT: Clinical
review 128: Current approaches to primary therapy for papillary and
follicular thyroid cancer. J Clin Endocrinol Metab. 86:1447–1463.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sherman SI, Brierley JD, Sperling M, Ain
KB, Bigos ST, Cooper DS, Haugen BR, Ho M, Klein I, Ladenson PW, et
al: Prospective multicenter study of thyroid carcinoma treatment:
Initial analysis of staging and outcome. National thyroid cancer
treatment cooperative study registry group. Cancer. 83:1012–1021.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tanaka K, Sonoo H, Hirono M, Ohkubo S,
Nomura T, Ikeda M, Nakajima K and Kurebayashi J: Retrospective
analysis of predictive factors for recurrence after curatively
resected papillary thyroid carcinoma. Surg Today. 35:714–719. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mazzaferri EL and Jhiang SM: Long-term
impact of initial surgical and medical therapy on papillary and
follicular thyroid cancer. Am J Med. 97:418–428. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin KL, Wang OC, Zhang XH, Dai XX, Hu XQ
and Qu JM: The BRAF mutation is predictive of aggressive
clinicopathological characteristics in papillary thyroid
microcarcinoma. Ann Surg Oncol. 17:3294–3300. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee X, Gao M, Ji Y, Yu Y, Feng Y, Li Y,
Zhang Y, Cheng W and Zhao W: Analysis of differential BRAF(V600E)
mutational status in high aggressive papillary thyroid
microcarcinoma. Ann Surg Oncol. 16:240–245. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Brose MS, Cabanillas ME, Cohen EEW, Wirth
L, Sherman SI, Riehl T, Yue H and Sherman E: An open label,
multi-center phase 2 study of the BRAF inhibitor vemurafenib in
patients with metastatic or unresectable papillary thyroid cancer
(PTC) positive for the BRAF V600 mutation and resistant to
radioactive iodine (NCT01286753, NO25530). Eur J Cancer. 49
Suppl:S132013.
|
|
25
|
Dadu R, Shah K, Busaidy NL, Waguespack SG,
Habra MA, Ying AK, Hu MI, Bassett R, Jimenez C, Sherman SI and
Cabanillas ME: Efficacy and tolerability of vemurafenib in patients
with BRAF(V600E)-positive papillary thyroid cancer: M.D. Anderson
cancer center off label experience. J Clin Endocrinol Metab.
100:E77–E81. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Falchook GS, Millward M, Hong D, Naing A,
Piha-Paul S, Waguespack SG, Cabanillas ME, Sherman SI, Ma B, Curtis
M, et al: BRAF inhibitor dabrafenib in patients with metastatic
BRAF-mutant thyroid cancer. Thyroid. 25:71–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Murphy DA, Makonnen S, Lassoued W, Feldman
MD, Carter C and Lee WM: Inhibition of tumor endothelial ERK
activation, angiogenesis, and tumor growth by sorafenib
(BAY43-9006). Am J Pathol. 169:1875–1885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lyons JF, Wilhelm S, Hibner B and Bollag
G: Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer.
8:219–225. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hyman DM, Puzanov I, Subbiah V, Faris JE,
Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al:
Vemurafenib in multiple nonmelanoma cancers with BRAF V600
mutations. N Engl J Med. 373:726–736. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Montero-Conde C, Ruiz-Llorente S,
Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA,
Rosen N and Fagin JA: Relief of feedback inhibition of HER3
transcription by RAF and MEK inhibitors attenuates their antitumor
effects in BRAF-mutant thyroid carcinomas. Cancer Discov.
3:520–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ribeiro FR, Meireles AM, Rocha AS and
Teixeira MR: Conventional and molecular cytogenetics of human
non-medullary thyroid carcinoma: Characterization of eight cell
line models and review of the literature on clinical samples. BMC
Cancer. 8:3712008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mercer KE and Pritchard CA: Raf proteins
and cancer: B-Raf is identified as a mutational target. Biochim
Biophys Acta. 1653:25–40. 2003.PubMed/NCBI
|
|
35
|
Baitei EY, Zou M, Al-Mohanna F, Collison
K, Alzahrani AS, Farid NR, Meyer B and Shi Y: Aberrant BRAF
splicing as an alternative mechanism for oncogenic B-Raf activation
in thyroid carcinoma. J Pathol. 217:707–715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Byeon HK, Na HJ, Yang YJ, Kwon HJ, Chang
JW, Ban MJ, Kim WS, Shin DY, Lee EJ, Koh YW, et al: c-Met-mediated
reactivation of PI3K/AKT signaling contributes to insensitivity of
BRAF(V600E) mutant thyroid cancer to BRAF inhibition. Mol Carcinog.
55:1678–1687. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sos ML, Levin RS, Gordan JD, Oses-Prieto
JA, Webber JT, Salt M, Hann B, Burlingame AL, McCormick F,
Bandyopadhyay S and Shokat KM: Oncogene mimicry as a mechanism of
primary resistance to BRAF inhibitors. Cell Rep. 8:1037–1048. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frasca F, Vella V, Nicolosi ML, Messina
RL, Gianì F, Lotta S, Vigneri P, Regalbuto C and Vigneri R: Thyroid
cancer cell resistance to gefitinib depends on the constitutive
oncogenic activation of the ERK pathway. J Clin Endocrinol Metab.
98:2502–2512. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Danysh BP, Rieger EY, Sinha DK, Evers CV,
Cote GJ, Cabanillas ME and Hofmann MC: Long-term vemurafenib
treatment drives inhibitor resistance through a spontaneous KRAS
G12D mutation in a BRAF V600E papillary thyroid carcinoma model.
Oncotarget. 7:30907–30923. 2016. View Article : Google Scholar : PubMed/NCBI
|