Prostate cancer (PCa) is the most frequently
diagnosed cancer in men from the Western world (1). The incidence of PCa has exceeded lung
cancer, and is the second leading cause of mortality in malignant
tumors in males (1). The number of
novel diagnosed cases in 2016 was 180,890 in the USA alone
(2). In China, the incidence of PCa
is increasing year by year with the change in living habits and the
aging population (3). In 2015, there
were 60,300 novel cases of PCa and 26,600 patients with PCa
succumbed (3). Androgen deprivation
therapy (ADT) has been the primary treatment option for male
patients with advanced symptomatic PCa for almost seven decades and
generally results in decreased prostate specific antigen (PSA)
levels. By contrast, following a median of 18–24 months of
endocrine therapy, almost all patients progressed to a poor
prognosis stage-castration resistant prostate cancer (CRPC), which
was previously called hormone-refractory prostate cancer. However,
the stage of CRPC is not hormone-refractory based on the
understanding that the androgen axis continues to be activated and
promotes the growth of CRPC (4).
Almost all patients succumbed due to no existing treatment to
control or cure the mortality-causing CRPC; therefore, the
mechanisms underlying the initiation and development of CRPC
remains challenging to decipher, which requires focus from
international academic research, and will help to improve the
survival and the quality of life of patients with PCa. In the
present review, the current studies regarding the molecular and
cellular mechanisms leading to CRPC are summarized for the
development of future molecularly targeted therapies for PCa.
CRPC refers to the continuous progression of PCa
following ADT. In recent years, the definition of castration
resistance has changed substantially. With the emergence of novel
CRPC therapy, it is particularly important to determine the exact
definition of castration resistance. In March 2015, Gallen gathered
41 experts in the field from 17 countries and regions around the
world, and held the first session of the advanced PCa Consensus
Conference (5). Almost all experts in
the session agreed that the diagnosis of CRPC should meet the
following 2 conditions: i) The serum testosterone level of the
castrated is <1.7 nmol/l; and ii) indicating biochemical
progression. Biochemical progression is characterized that the PSA
expression levels have increased twice in a row from an interval of
1 week or >3 consecutive measurements with the lowest value
increased >50% and >2 g/l, and ≥2 increases in novel lesions
based on bone scanning or soft tissue lesions with the
corresponding evaluation criteria of the solid tumor. Currently,
only symptom progression is not sufficient to diagnose CRPC. The
understanding of PCa biology and underlying disease resistance
mechanisms has grown over the last few decades, and has been
translated into improved and clinically meaningful treatment
strategies for males with advanced PCa (6). Understanding the mechanisms underlying
the progression of PCa from hormone sensitive to castration
resistant is the key to develop future therapy. In the present
review, a number of the major underlying mechanisms leading to
castration resistance are discussed.
The growth and survival of PCa tumors depend on
androgens, the male sex steroid hormones, of which testosterone and
dihydrotestosterone (DHT) are the principal members that activate
and bind to the AR (7). High
expression of AR occurs in the majority of patients with CRPC, and
the expression of AR-regulated genes recovered in tumors after
patients received ADT implies that AR transcription activity is
activated again (7).
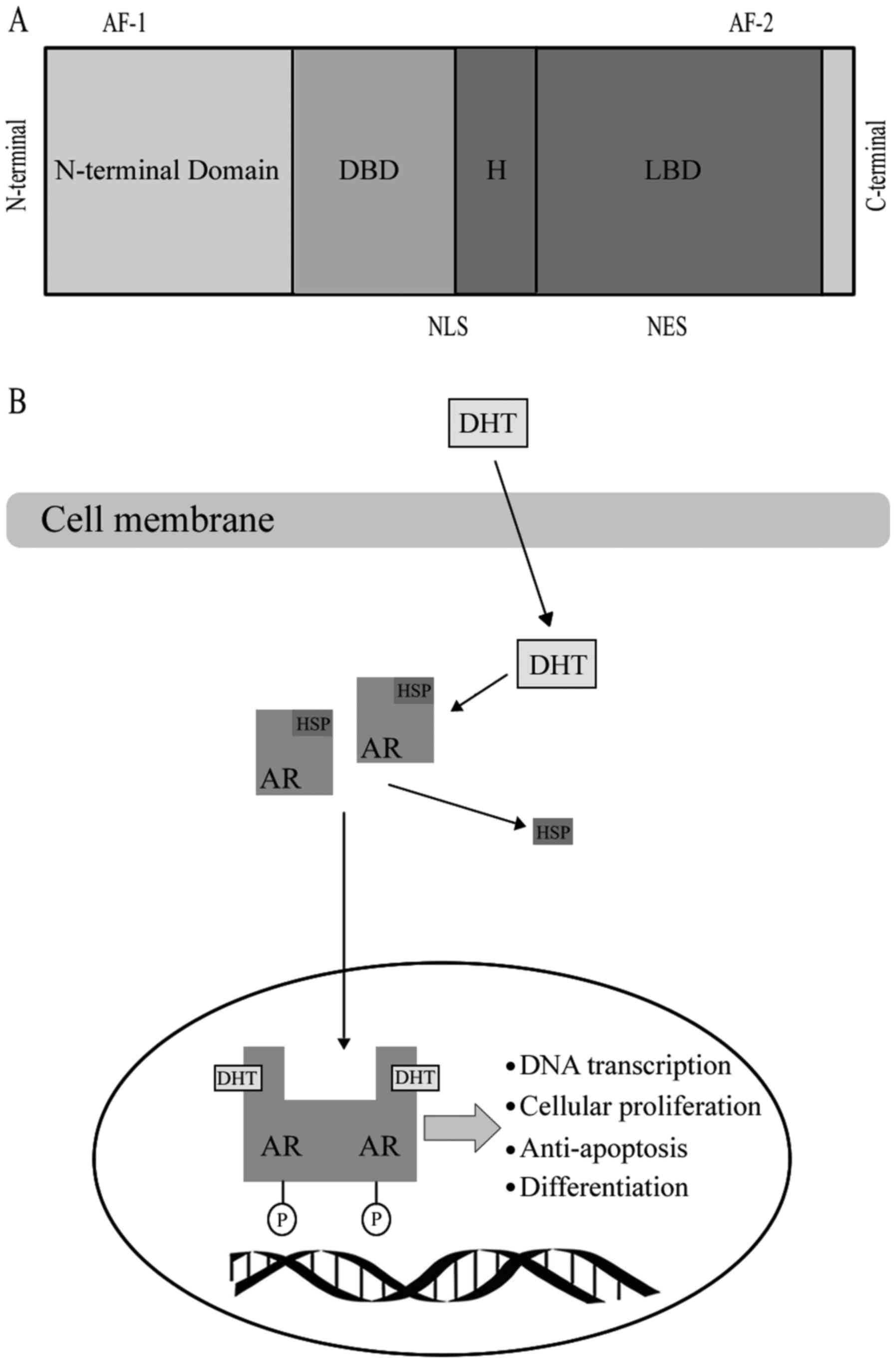
AR, as a member of the steroid hormone super family,
has four functional motifs, including the ligand-binding domain
(LBD), DNA-binding domain (DBD), hinge region and the
amino-terminal domain (N-terminal domain, NTD) that contains the
phosphorylation sites essential for transcriptional activity
(8) (Fig.
1). When AR binds with a steroid ligand, including DHT, it can
prevent ubiquitination and proteasome degradation by interacting
with heat-shock proteins (HSP) (9).
The AR is phosphorylated prior to dimerization, which leads to a
conformational change, which displaces the HSP upon ligand binding
(10). Dimerized ligand-bounding
complex will then be transported into the nucleus (10). Ligand binding is vital to dimerization
and translocation of AR to the nucleus. Following AR being
trafficked into the nucleus, it may mediate transcription and
various growth signaling pathways, including cellular proliferation
or anti-apoptosis and androgen-regulated genes, including PSA,
through binding to androgen response elements in the promoter or
enhancer regions of DNA (11). A
number of studies revealed that the AR may be transported into the
nucleus through the microtubule complex (12). Evacuation of the ligand in
androgen-responsive prostate cells results in an exportation of the
AR from the nucleus (9).
In the process of hormone-responsive PCa to CRPC,
the AR and its cross-signaling pathways exist a number of changes
with various forms, which provides an important clue for the
exploration of the mechanisms underlying CRPC.
AR gene amplification and overexpression of the AR
protein have been frequently observed in clinical studies. The AR
gene amplification leading to an overexpression of the AR protein
is the most common genetic change among patients with CRPC
(13). Of the patients with CRPC,
>80% of them had significant augmentation of AR mRNA and protein
levels (14). By contrast, AR gene
amplification was rarely observed in untreated PCa (14). Research determined that the AR gene
had a high frequency of amplification in circulating tumor cells
(CTCs) of patients with CRPC (15).
Fluorescence in situ hybridization studies utilizing tissue
microarrays discovered that AR gene amplification is present in
only 2% of the primary PCa tumor samples and none of the samples of
benign prostatic hyperplasia, compared with 23.4% of CRPC tumors
(16). Using reverse
transcription-polymerase chain reaction (RT-PCR) analysis, it was
confirmed that the AR gene amplification is indeed reflected at the
mRNA level, where the expression of AR mRNA in CRPC tumors with AR
amplification was increased two fold, compared with those CRPC
tumors without AR amplification (17). On the other hand, increased AR was an
unique gene expression alteration that was sustained in different
CRPC xenograft tumor samples (18).
Elevated AR protein levels are also linked to CRPC.
Using multiple isogenic tumor xenograft models, it was demonstrated
that the expression of the AR protein increased in recurrent tumor
samples, compared with paired androgen-sensitive samples (18,19).
Notably, in a CWR22 xenograft model that imitates the transition
from androgen-sensitive to recurrent growth, the expression of the
AR protein was gradually reduced during the 120-day castration
period, and then regrowth occurred in recurrent tumors (19). In addition, not only was the gene
amplification and elevated mRNA expression directly augmenting the
AR protein levels, increased protein half-life can also contribute
to the elevated levels of AR protein in CRPC (20). Through gene amplification and enhanced
transcriptional induced overexpression of AR causes AR
hypersensitive to low level of androgen (21). Furthermore, it can be concluded that
under the lowest concentration of androgens, tumor cells
proliferate continuously (22), which
indicated that overexpression of AR may contribute to the
development of CRPC formation.
AR mutations in the early stage of PCa are rare, but
it is commonly occurs in CRPC, particularly advanced PCa following
systematic hormone therapy. There are >100 point mutations that
appear in AR, and the majority of them present in the NTD or LBD
region (4,23). In recent years, there are a number of
studies regarding AR mutations including T878A, H875Y/T, W742C,
L702H and F877L (24–26). The mutations in the hinge and LBD
regions of AR result in increased AR transactivation activity and
decreased ligand specificity. A large number of AR mutations can be
activated by adrenal androgens and other steroid hormones;
additionally, a number of mutations can turn AR antagonists
(flutamide and bicalutamide) into potential agonists. Compared with
endocrine therapy, AR antagonists in the treatment of metastatic
PCa will cause the rate of AR mutations in the LBD region to be
increased.
The T878A mutation is one of the most common forms
of AR mutations. It was found that the AR T878A mutation occurs in
patients with CRPC following prolonged ADT; however, not
hormone-sensitive patients (27–29).
Furthermore, it has been reported that this mutant was expressed in
the tumors of patients with CRPC whose PSA levels were
significantly decreased by flutamide withdrawal (29). This discovery has led to the
hypothesis that expression of the AR T878A mutation is responsible
to the beneficial effect of withdrawing anti-androgen therapy in
patients with CRPC. This mutation also leads to an expansion of
binding specificity of the ligand of AR, improving the sensitivity
of steroid hormones such as progesterone and estrogen (30). A number of the mutations can turn the
anti-androgen substances into agonists. For example, the F877L
mutation activated by enzalutamide or ARN-509 has been demonstrated
to transform AR antagonists into AR agonists in a PCa cell line
following long-term treatment with enzalutamide and ARN-509
(31). Recent studies indicated that
the mutations T878A, H875T/Y, W742C and L702H exist in 15–20% of
CRPC tumor samples, which emphasized that the LBD region is a
mutational hotspot (32). Utilizing
circulating free DNA from patients with CRPC, which has been
determined to contain genomic DNA with the AR mutations,
demonstrated that using sequencing to detect those point mutations
aforementioned could possibly act as biomarkers for patients at
risk of developing CRPC (33).
Additionally, combination therapy may overcome part of the
secondary resistance with AR mutations, an ongoing clinical trial
(34), using combination therapy of
abiraterone and enzalutamide in patients with CRPC may demonstrate
this hypothesis. If those studies using blood-based detection to
distinguish AR mutations in patients with CRPC are clinically
validated, then clinicians may have accessible biomarkers for
individual treatment selection, which may improve the clinical
outcome for patients.
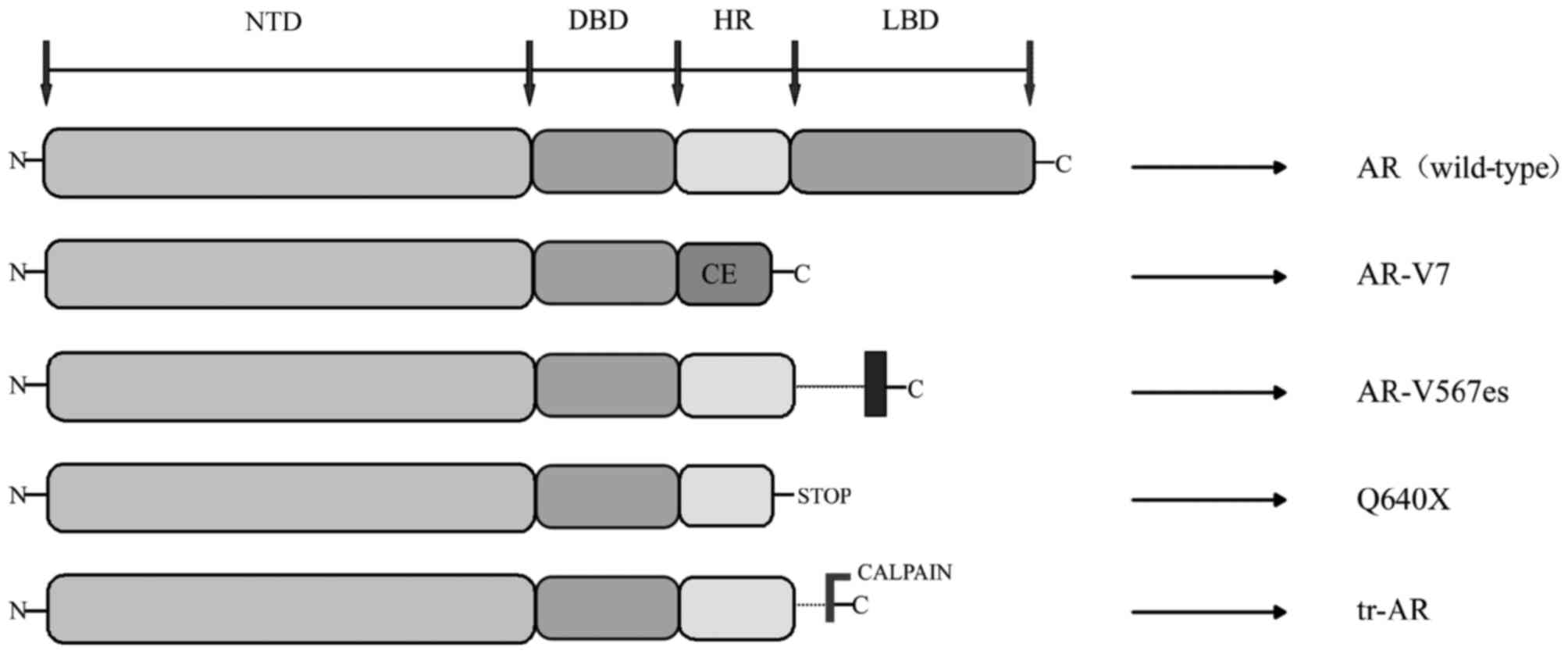
AR-Vs have been demonstrated to be associated with
resistance to ADT of PCa as well as the resistance to abiraterone
and enzalutamide (35). AR-Vs are
truncated forms of AR, which lack the LBD region, where just the
NTD and DBD regions were transcriptionally activated, independent
of the presence or absence of ligand-binding or antagonist effect
(36). Hu et al (37) explored the AR intronic sequences in
the human expressed sequence database, and determined that CE 1–4
and identified AR variants (AR-V 1–7) lacked the LBD region. This
trait of ligand-independence makes truncated AR variants
potentially important to disease progression and patient treatment
response, as they can activate AR target genes independent of the
androgen (36). The current studies
discovered splice variants include AR3, AR4, AR5 and AR-V1V7
(38). The AR transcripts AR-V3 and
AR-V4 have the same sequences to AR1/2/2b and AR1/2/3/2b;
therefore, novel variants were identified as AR-V1, AR-V2, AR-V5,
AR-V6 and AR-V7 (39). A number of
the AR-Vs (including AR-V7) primarily localize in the nucleus,
while others (including AR-V1, AR-V4 and AR-V6) localize mainly in
the cytoplasm. Notably, cytoplasm-predominant AR-Vs frequently
co-expressed with the nucleus-predominant AR-Vs as well as the
full-length AR (AR-FL) (40). Not
only are AR-Vs predominantly products of alternative splicing, but
also can they be products of nonsense mutations (ARQ640X) or
proteolytic cleavage (41) (Fig. 2).
Detected at the mRNA and protein levels, AR-V7 is
the most frequently studied and abundant AR variants in clinical
samples from patients with CRPC (42). A retrospective study have indicated
that the presence of AR-V7 was associated with more advanced
malignance and reduced survival in patients with metastatic CRPC
(mCRPC) (43). Previously, the study
of Antonarakis et al (44)
have carried out a prospective study, which utilized CTC samples of
patients receiving abiraterone or enzalutamide therapy to evaluate
the prognostic role of AR-V7 detected at the mRNA levels by RT-PCR.
They discovered that compared with those patients without
detectable circulating AR-V7, the appearance of AR-V7 in CTCs was
associated with lower PSA response rate, reduced
progression-free-survival (PFS) and reduced overall survival (OS)
(44). Notably, the level of AR-V7
was higher in males who had been treated with enzalutamide and
abiraterone, whilst the level of AR-V7 was lowest in males who had
not received either agent therapy. In addition, when analyzing
serial CTCs over time, the study demonstrated that all males with a
baseline of AR-V7 remained AR-V7 positive during the process of
abiraterone and enzalutamide therapy, whereas 14% of males with
negative AR-V7 level at baseline changed to AR-V7 positive during
treatment, and these patients had an intermediate clinical outcome
(44).
It was confirmed that AR-V1, AR-V4 and AR-V6 can
dimerize with AR-V7 and AR-FL; therefore, AR-V7 and androgen-bound
AR-FL induced nuclear localization of AR-V1, AR-V4 and AR-V6.
Additionally, these variants weakened the efficiency of
enzalutamide to inhibit androgen-induced AR-FL nuclear
localization. It is notable that the impact of nuclear localization
of AR-V4 and AR-V6 on AR transactivation differs from that of AR-V1
(45). Nuclear localization results
in an enhanced ability of AR-V4 and AR-V6 to transactivate
canonical AR targets and AR variants specific targets, and promote
castration resistant cell growth (45); however, when AR-V1, which lacks
inherent transcriptional activity, activates AR-FL in an
androgen-independent way, it significantly inhibits AR-V7
transactivation (45). These data
illustrated the important role of complicated interactions among
different AR-Vs and AR-FL in castration resistant disease, and
dissecting these complex interactions may help to develop effective
strategies to target AR variants signaling (45). In recent research, AR-NTD targeting
drugs have benefits over drugs targeting the AR-LBD due to the NTD
being essential for the transcriptional activities of AR-FL and
AR-Vs; therefore, AR-NTD antagonists, including EPI-002, patients
who are resistant to abiraterone or anti-androgens with
constitutive activated expression of AR-Vs could achieve
therapeutic responses from EPI-002 (46,47). As a
result, with gradual clarification of the AR-Vs formation, it has a
possibility to become a novel targeted therapy for the treatment of
patients with CRPC in the future.
AR co-regulators are protein factors, which were
associated with AR transcription activation, and serve an auxiliary
role in activation or inhibition of AR-mediated transcription
(48). AR co-regulators have the
function of activating transcription activity of AR under an
extremely low concentration of androgens (48). It has also been demonstrated that
co-regulator factors serve an important role in the development of
CRPC formation. Normally, AR recruits a series of co-regulator
complexes, which can either enhance or repress transcriptional
activity. Many of these co-regulators are enzymes that serve to
modulate other proteins through phosphorylation, methylation,
acetylation or ubiquitylation, in a complex form (49,50). They
have also been identified as molecular chaperones, recruiters of
transcriptional machinery and RNA splicing regulators (49,50).
According to their different effects on AR transcription, AR
co-regulators can be divided into co-activators [including P160/SRC
and p300/CREB binding protein (CBP)], co-repressors (including NcoR
1 and NcoR 2), pioneer factors (including forkhead box A1, GATA2
and cardiotrophin 1) and cooperators (including ETS-1, adaptor
associated protein complex 4 and NKX3-1) (51–55). There
are >150 different molecules that have already been identified
as co-activators or co-repressors of AR (56). When AR interacts with a co-activator,
it can lead to a significant abnormal activation of AR, which
results in increased AR transcriptional activity (50); therefore, changing the ability of AR
transcriptional activation is essential for AR to achieve the
maximum transcriptional effect. For example, FKBP51, as an AR
target gene and a co-activator of AR, was determined to be
upregulated in relapsed LAPC-4 tumors that were grown in castrated
mice (57). The formation of a super
chaperone complex was improved by FKBP51 via recruiting p23, to
adenosine 5′-triphosphate (ATP)-bound Hsp90, which in turn keeps AR
in a conformation with high affinity for ligand-binding, thus
promoting androgen-induced transcriptional activity and growth
(58). Other important pathways,
including p300/CBP, which promotes androgen-independent interleukin
(IL)-6 mediated AR activation in the presence of the signal
transducer and activator of transcription 3 (STAT3), lysine
demethylase 1A (LSD1A), jumonji domain-containing 2c (JMJD2c) and
lysine demethylase that demethylates the histone H3 proteins and
then promotes AR induced transcription (59). On the other hand, when AR is combined
with co-repressor, the opposite effect occurs in reducing the AR
transcriptional activity, although they have been observed at
decreased levels in CRPC (60).
Additionally, the majority of cooperators serve the role of
increasing the AR transcription, whilst pioneer factors can
interact with condensed chromatin and directly regulate the
accessibility of chromatin (61). It
is important that the interaction between pioneer factors and
chromatin is prior to the androgen induction, which means that
these factors can direct AR to chromatin aggregation at low or no
levels of androgens (54,55).
Post-translational modifications of AR mainly
include acetylation, methylation, ubiquitination, sumoylation and
phosphorylation (62). The molecular
effects of these transformations induce decreased apoptosis and
elevated transcriptional activity to the androgen-responsive genes
(62). Research indicated that
hypomethylating drugs can be used as a treatment in delaying the
appearance of CRPC (63). Inhibiting
DNA methylation can reverse the mechanism underlying castration
resistance, the potential underlying mechanism may be associated
with the downregulation of DNA methyltransferase 1-dependent STAT3
activity. Many serine/threonine and tyrosine kinases are involved
in AR phosphorylation, including Aurora-A, PIM1 kinase,
cyclin-dependent kinase 1 (CDK1), Src and Ack1 (64–67).
Aurora kinase (68) is the most
extensively studied clinically. In a phase II trial (69), 60 randomly selected patients with
mCRPC were randomized into two different treatment schedules based
on using danusertib or not, an inhibitor of Aurora kinase. All of
the patients had been previously treated with docetaxel. Only two
patients had a PSA response, and the best radiographic response was
an indicated stable disease in 27% of patients. The median PFS was
12 weeks in both groups Due to of the disappointing results, no
further research on this compound is being pursued. The study
indicated that aurora kinase was upregulated in anaplastic or
neuroendocrine PCa (708) More recent clinical trials (NCT01799278
and NCT01848067) are using these agents to target the small cell
and anaplastic PCa. Research for these targets is actively
continuing, although no late-phase clinical trials are currently
planned or ongoing. These phosphorylation modifications, which
enable AR to respond to low levels of androgen, and being the
targets of small molecule inhibitors could be a promising treatment
for patients with CRPC.
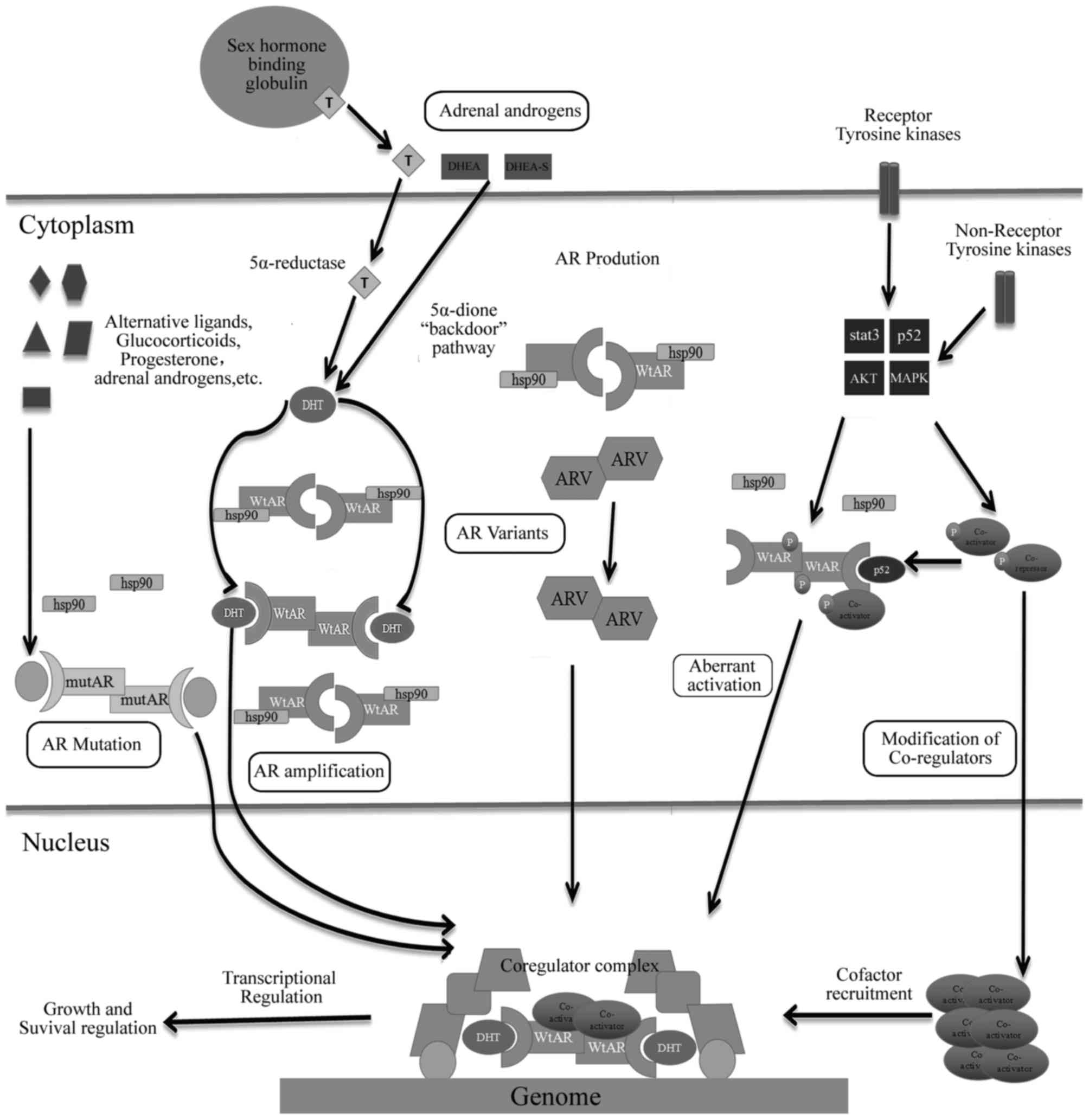
Normally, testosterone mainly originates from the
testes, with 5–10% coming from adrenal glands. In general, medical
or surgical castration can reduce circulating levels of serum
testosterone by >90% to maintain its circulating concentration
below normal levels of castration (1.25 pmol g−1)
(70); however, androgen
concentration in prostate tissues remains sufficient for activation
of AR (70). The androgen
concentration in prostate tissues is significantly lower than that
of serum androgen in patients following ADT (71). PCa may act as synthetic androgen
within tissues through a number of unknown mechanisms; therefore,
PCa tissues can synthesize androgen using prostate tissues and not
rely on androgen cycle following ADT, and this condition may result
in failure of the procedure (71).
Recently, utilizing autopsy and tissue biopsy studies to compare
androgen levels in hormone-sensitive and hormone-resistant tumors
from patients has revealed high levels of continuous androgen
production in tumor tissues of patients with CRPC (72). Studies have considered that sustained
production of androgen within prostate tissues possibly results
from the conversion of weak-bioactivity androgen-like
androstendione (AD) and dehydroepiandrosterone (DHEA) (73,74), which
are produced by adrenal glands or de novo steroidogenesis of
androgen from the cholesterol within tumor tissues (74). Although circulations of AD and DHEA
significantly decrease in patients with CRPC following treatment
with abiraterone (CYP17 inhibition), a sustained pool of DHEA-S,
which is a sulfated form of DHEA, and the predominant adrenal
androgen in circulation can serve as precursors for transformation
into testosterone and DHT in prostate tissues (75). The role of intratumoral androgen in
promoting CRPC cells growth was proven by large phase III clinical
trials, which aimed to test the testosterone concentration of
tumors in patients receiving novel anti-androgen therapies of
enzalutamide and abiraterone (76,77). All
patients with mCRPC were tested and indicated improved OS, compared
with those who received placebo treatment.
Steroid receptors, including AR, estrogen
receptor-α, estrogen receptor-β, PGR, GR and mineralocorticoid
receptor, are derived from the same material and feature high
homology, particularly in the DBD binding region (79). GR and PR are more relevant to AR
(80). Almost all PCa were discovered
to express AR, whereas GR is only present in 30% of PCa cases;
however, GR expression increases in patients following ADT
(81). Recent studies have indicated
that AR and GR possess the same chromatin binding sites and
regulate the expression of a large number of AR-specific genes
(82). Resistance of enzalutamide to
novel AR antagonist may be due to increased expression of GR. ADT
can increase GR expression and demonstrated the potential for GR
signaling (68,83). GR can also act as a substitute for AR
signaling. Montgomery et al have summarized a series of
clinical trials to assess the contributions of glucocorticoids and
GR in patients with CRPC (84,85). In
these trials, analysis of patients in clinical cohorts who received
glucocorticoids indicated poor prognostic features, compared with
patients who did not receive such treatment; therefore, stimulation
of GR signaling may pose negative effects to patients who have
received androgen-targeted therapies (84–86). In
such conditions, the presence of increased glucocorticoids in serum
and absence of androgen possibly enables the selection of
‘promiscuous’ AR variants with mutations in the LBD, allowing their
activation by glucocorticoids (87);
therefore, GR or other nuclear steroid receptors can bypass AR
pathways and promote the development of CRPC.
On the other hand, PGR is also a member of the
steroid hormone nuclear receptor family, and it is structurally
associated with AR. As with GR, PR may have the ability to
transcriptionally regulate a subset of AR target genes in PCa and
thereby bypass AR (88). A large
retrospective analysis carried out recently demonstrated the
association of high PR staining in primary PCa with its clinical
recurrence (89). A recent research
on a cohort of >500 patients also revealed the association of
high PR expression in cancer cells with reduced clinical
failure-free survival (89). This
evidence indicated that PR antagonists may possess therapeutic
effects, indicating that PR signaling can be an important
carcinogenic target in the treatment of PCa.
The PI3K-Akt-mTOR pathway has been known as a
potential driver of CRPC. PI3K is activated by G protein-coupled
receptors or tyrosine kinase receptors. Activation of PI3K leads to
phosphorylation of AKT and activation of mTOR and subsequent
downstream effects, including cellular proliferation, survival and
angiogenesis (90,91). Previous studies have demonstrated that
alterations of components in the PI3K-Akt-mTOR pathway occur in
100% of metastatic tumors and 42% of primary prostate tumors
(4,91,92);
therefore, targeting the PI3K-Akt-mTOR pathway is considered a
promising therapeutic approach for patients with CRPC. Loss of the
phosphatase and tensin homolog (PTEN) gene has been frequently
proven to result in constituting activation of the PI3K-Akt-mTOR
pathway in the majority of advanced PCa cases (93). A recent study used either targeted
drugs or gene knockout in mouse models, whereas cell lines have
indicated alterations in PI3K and PTEN activities, demonstrating
changes in AR expression and AR transcriptional activity (94). Kato et al (92) reported the combined use of the AR-V
inhibitor EPI-002 and PI3K/mTOR inhibitor BEZ235 to evaluate their
therapeutic efficacy on LNCaP95 cells in CRPC models in
vitro and in vivo. Results demonstrated that compared
with single drug cohorts, drug combination can significantly
inhibit LNCaP95 cell growth without increasing toxicity. These data
indicated that co-targeting the PI3K-Akt-mTOR pathway and AR-NTD to
block AR-FL and AR-Vs can be a novel approach for achieving
improved antitumor efficacy for patients with CRPC who have
indicated resistance to abiraterone or other anti-androgen
treatments by a mechanism involving expression of AR-Vs.
The Src kinase family (SFK) comprises of proteins
with protein tyrosine kinase. Src kinase activation has been
indicated to be relevant to androgen-independent cell growth,
inhibition of anti-apoptotic pathways, cell migration and adhesion
and tumor invasion. In PCa, Src signaling upregulates vascular
endothelial growth factor (VEGF) and IL-8 to promote angiogenesis
(95) and activates the nuclear
factor-KB pathway of osteoclasts and various tumor necrosis factor
receptors involved in resistance to apoptosis signal transmission
to participate in bone metastasis of PCa (95). Dasatinib, an inhibitor of Src kinases,
has been confirmed to slow down the growth of PCa tumors and to be
as effective as CRPC treatment based on a phase III clinical trial
(96). Saraeatinib (AZD-0530) is a
dual inhibitor of Src and Abl, and the research of Yang et
al (97) demonstrated that this
drug can inhibit proliferation and invasion in bone metastasis
models of PC3 cell lines and alleviate bone destruction; a phase II
clinical trial with Saraeatinib is currently ongoing.
Activation of the growth factor signaling pathway
can also enhance AR signaling pathway and promote CRPC. Growth
factor receptors, including insulin-like growth factor I receptor,
IL-6 receptor and epidermal growth factor receptor, can mediate the
growth of critical downstream pathways, including mitogen-activated
protein kinase (MAPK), PI3K/AKT and signal transducer and activator
of transcription signaling (98).
Activation of PI3K-Akt and Ras-Raf-MAPK pathway and expression of
receptor tyrosine kinases, including Her-2/neu, have already been
observed to enhance AR stability and activity in CRPC samples
(99). A previous study demonstrated
that Her-2/neu promotes the growth of xenograft cells and androgen
independence through Ack1-kinase, which phosphorylates AR at
tyrosine-267 and activates AR activity (100). Targeting growth factor signaling
pathways has provided a novel therapeutic strategy. In a phase II
clinical trial (101), application
of cabozantinib (XL-184), an inhibitor of tyrosine kinase of c-Met
and VEGF, resulted in PFS of 23.9 weeks in patients with CRPC,
whereas the placebo group yielded a PFS of 5.9 weeks. Following
treatment with cabozantinib for 12 weeks, bone scan of 68% of
patients indicated improvement; whereas, results for 12% of
patients demonstrated that bone metastatic lesions completely
subsided. XL-184 can also relieve pain of patients with CRPC to
improve their quality of life (101).
miRNAs are small, non-encoding, and conserved
genetic single-stranded RNAs. miRNAs can target gene mRNA 3′
untranslated region through complete or incomplete complementary
binding, resulting in degradation of target gene mRNA or inhibition
of translation; which functions as a gene expression regulation
factor in cell metabolism, proliferation, differentiation and cell
death and cell survival (102,103).
Extensive research indicated that expression
profiles of miRNAs in PCa become increasingly important due to of
their application in diagnosis, staging, predicting prognosis and
response to the treatment (104). An
earlier study confirmed upregulation of miRNA-96, −182, −183 and
−375 and downregulation of miRNA-16, −31, −125b, −145, −149, −181b,
−184, −205, −221 and −222 in PCa tumor tissues (105). In a recent study, using miRNA
microarray to detect miRNA expression between primary PCa tumors
and CRPC tumors has indicated divergent results, where 75 miRNAs
were differentially expressed in primary PCa, 88 in CRPC tumors,
and 22 changed expressions of miRNAs overlapping between primary
and CRPC samples (106). Altogether,
these data implicate that changes in miRNA expression can
contribute to resistance to ADT.
The most frequently studied miRNAs associated with
PCa progression include miRNA-16, miRNA-34a/34b, miRNA-143,
miRNA-101 and miRNA-200c. Cell experiments indicated that exogenous
expression of miRNA-16 can inhibit the recurrent growth of 22Rv1
cells, androgen-independent DU145 cells and PC3 cells, but not
androgen-dependent LNCaP cells (107). miRNA-34a has been demonstrated to
specifically downregulate AR expression (108), indicating that loss of miRNA-34a can
increase AR expression, as observed in PCa cell lines, xenograft
models and human CRPC tumors (17).
Observation of patient with PCa samples has demonstrated that high
miRNA-34b expression is correlated with longer OS, whereas the
opposite is correlated with higher Gleason score (15–17,109). One
assumption indicated that induced miRNA-34b expression can increase
the aggressiveness of PCa. Expression levels of miRNA-200c were
significantly reduced in primary PCa tumors of patients who
relapsed following prostatectomy, compared with those without
relapse (110). miRNA-101
expressions notably decreased in metastatic tumors, compared with
those of primary samples (111). All
these data implicated that miRNAs cannot only potentially promote
tumor progression and sustain resistance to ADT but also act as
biomarkers for patient outcomes. Analysis of radical prostatectomy
specimens indicated that miRNA-301a, miRNA-449b and miRNA-182 can
act as biomarkers to predict biochemical recurrence following
prostatectomy (112–114). Recently, combining Gleason scores
and lymph node status with expression levels of miRNA-4516 and
miRNA-601 has demonstrated predictive roles of these miRNAs in
biochemical recurrence following post-prostatectomy salvage
radiation therapy (115), supporting
the utilization of miRNAs in clinically used predictive models.
In conclusion, miRNAs serve important roles in
initial PCa pathogenesis, progression and development of CRPC.
miRNAs can also function as attractive biomarkers due to they are
relatively stable in biological fluids, easy to measure and are
resistant to storage handling (104); therefore, abnormal levels of
expressed miRNA in tumor tissues, serum or plasma and urine can be
a promising biomarker for diagnosis, prognostic prediction, or
treatment efficacy of patients with PCa.
Defects and mutations in DDR genes have been
reported in CRPC and in localized aggressive cancers (116). For example, mutations and loss of
BRCA2 were observed in 12% of PCa cases (117). Germline mutations in BRCA increase
the probability of lymph node involvement and distant metastases,
which confer PCa with more aggressive phenotypes (118). CRPC frequently features homologous
DNA recombination function deletion, expression of TMPRSS2-ERG gene
fusion and the DDR protein poly (ADP-ribose) polymerase (PARP)-1,
which is recruited to sites of AR targets and serves an important
role in AR transcription, which is essential for the activity of
TMPRSS2-EGR in PCa (119). In a
phase II trial (120), patients who
had recurred following >2 rounds of CRPC therapies were treated
with olaparib, a PARP inhibitor and a base excision repair protein.
Results demonstrated that patients who exhibited genomic defects in
DNA repair genes (including BRCA1, BRCA2 and ATM) manifested a
6.3-month increase in OS, compared with those without these
defects. These results indicated that blockade of DNA damage
response may promote CRPC, whereas genomic instability improves
responsiveness of patients with CRPC to treatment for target DNA
repair (120).
AR indicated potential links with DDR. By using a
combination of anti-androgen therapy and radiotherapy to treat
aggressive PCa, large clinical trials demonstrated significant
augmentation in efficacy, indicating the potential role for AR
inhibition in weakening DDR (121,122).
AR enhances the expression and activity of key DDR factors,
including DNA-dependent protein kinase (DNAPK); in turn, DNAPK
sustains AR transcription (122).
ADT induces reduction in transcription of key DDR genes, resulting
in higher levels of DNA damage following radiotherapy, specifically
non-homologous end-joining (121).
Such results indicated that ADT affects genomic instability prior
to castration or promotes development of CRPC through repressing
DDR. MYB protein was observed to replace AR function in regulating
DDR by modulating an overlapping set of genes. Silencing of MYB
gene or a number of its targets (TOPB1, ATR and checkpoint kinase
1) synergizes with PARP inhibitor olaparib, and such condition can
significantly suppress PCa growth in in vitro and in
vivo experiments and increase cytotoxicity of PCa (123); therefore, combination of MYB pathway
inhibitor and PARP activity suppressor can be a viable clinical
strategy.
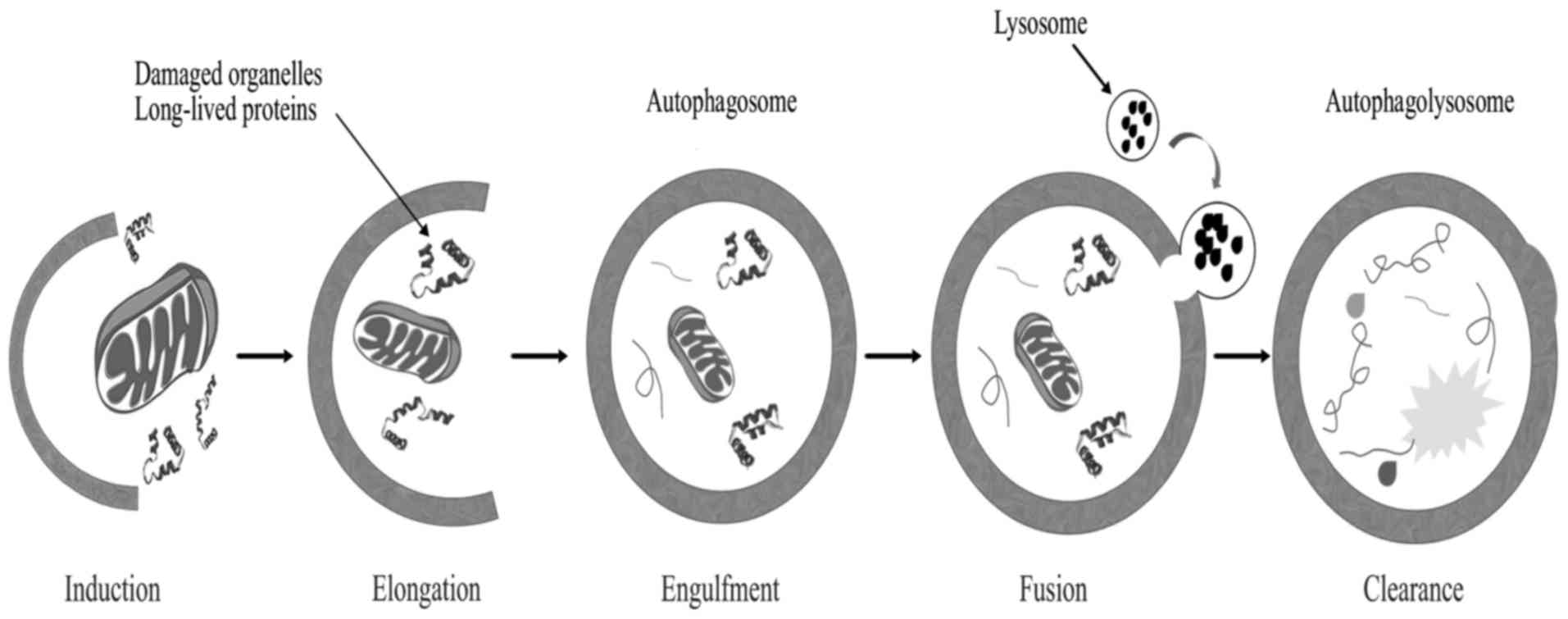
Autophagy is a genetically programmed cellular
stress regulation mechanism occurring in all eukaryotic cells. This
process is one of the major protein degradation processes that
prevail through the lysosomal pathway (124). Autophagy mediates the clearance of
damaged organelles and long-lived proteins, leading to the
formation of double-membrane structures called autophagosomes
(Fig. 4). Cells can recycle amino
acids and other macromolecular materials for biosynthesis via
autophagy. Normal cells clear chemical carcinogens and impaired
organelles caused by radiation or oxidative stress mainly through
the action of mitochondria, thereby protecting cell DNA against
damage from reactive oxygen species, ensuring genomic stability and
reducing incidence of cell malignant transformation (125); therefore, autophagy is crucial in
maintaining hereditary stability.
Recent studies indicated that autophagy may be
associated with resistance to chemotherapy in malignant tumors. For
example, the studies of Jiang et al (126,127)
have reported that overexpression of mitochondrion-associated
leucine-rich pentatricopeptide repeat motif-containing protein, an
inhibitor of autophagy, and low levels of autophagy activator
microtubule-associated protein 1S (MAP1S), a member of MAP1 family,
can be both used as independent biomarkers for patients with
late-stage PCa and poor prognosis. Patients receiving multiple
chemotherapy, particularly for oxygen-deprived tumor cells,
demonstrated significant increase in the degree of nutrient
limitation. Ischemia, hypoxia and lack of nutrition can activate
autophagy. Autophagy modifies metabolism by degradation and
recycling of intracellular substances. Through this mechanism,
tumor cells can induce adaptive responses to stress and survive. In
the late stage of tumor progression, autophagy becomes an important
survival mechanism for tumor cells that cannot directly obtain
nutrition from circulation in the central body of solid tumors
(128,129). Facial tumors and those determined
that have poor living environments can eliminate large molecules or
mitochondria damaged by ionizing radiation and cell toxicity
through enhancing autophagic activity and blocking transmission of
the mitochondrial apoptosis signal cascade (129). Autophagy can also prevent
pro-apoptotic factors, including cytochrome and apoptosis-inducing
factor diffusion by separating mitochondria. These mechanisms may
form a resistance of tumor cells to radiotherapy and chemotherapy
and prevent their apoptosis. Patients with CRPC frequently use
chemotherapy based on docetaxel and mitoxantrone as palliative
therapy. However, with repeated administration, resistance to
chemotherapy becomes prevalent in patients, but overall effect is
poor (130,131). Existing studies imply that autophagy
serves an important role in development of CRPC (130–133).
Using enzalutamide to block AR as a long-term
treatment can activate AMP-dependent protein kinase [AMP-activated
protein kinase (AMPK)], whereas mTOR is inhibited by mTOR mudulin
(132,133). Studies have indicated that AMPK can
directly regulate mTOR activities to match energy imbalance, thus
increasing the ratio of AMP/ATP (132,133).
Hypoxia is one of the indirect causes by which anti-androgen
therapy can improve autophagic activities (130), whereas blockage of angiogenesis by
androgen withdrawal and nutrient synthesis under hypoxia further
stimulates autophagy. The information aforementioned can confirm
the effects of AR on autophagy surges but fail to fully elucidate
autophagy mechanisms. For example, ADT can still be used to develop
PC3 cells that lack AR for upregulating autophagy (132). Possible reasons include changes in
proto-oncogenes, including PTEN, altered tumor suppressor genes,
including cell tumor antigen P53 and abnormal metabolism of AR
downstream (134). Bennett et
al (131) indicated that the use
of synergistic autophagy inhibitor 3-methyladenine with
bicalutamide results in 1.5-fold higher cell mortality rate than
using bicalutamide alone. Colquhoun et al (135) also demonstrated that using
bicalutamide with metformin in treating LNCaP cells will
significantly decrease the colony formation rate. This phenomenon
also occurs in PC3 cell lines although the latter requires higher
doses. Notte et al (136)
discovered that two key pathways of taxol inducing autophagy; the
first is paclitaxel-inhibited mTOR, whereas the second participates
in activation of c-Jun N-terminal kinase, Bcl-2 and Bcl-XL
phosphorylation and activation of autophagy. Notably, although
these pathways perform similar functions, treatment time with
paclitaxel and environmental pressures required for activation of
autophagy remarkably differ. LNCaP-AI is an androgen-dependent PCa
cell line. When cultured in an androgen-deficient environment,
tolerance of the LNCaP-AI cell line to docetaxel is 2.5 times
greater than that of common LNCaP cell lines (131). Recovery of androgen of the LNCaP-AI
cell line in culture medium also indicated sensitivity to docetaxel
(131). When re-adding
3-methyladenine to inhibit autophagy, cytotoxicity of docetaxel
increases; thus, autophagy is a factor influencing docetaxel
resistance (131).
Kinases serve important roles in the metabolism and
proliferation of PCa cells and thus affect the progression of the
disease. A previous study established PCa models in xenogeneic
mice, with one group receiving saracatinib treatment and another
receiving saracatinib combined with chloroquine treatment.
Following 14 days of observation, the rate of tumor growth
inhibition reached 64%, whereas that of single-saracatinib group
totaled 26% (137). Current
limitation of inhibitors of SFK may be due to simultaneous
emergence of therapeutic effects and therapeutic resistance caused
by overlapping molecular pathways (137). An autophagy inhibitor may overcome
this limitation.
At present, many clinical trials focus on regulation
of autophagy in PCa. However, regulation of autophagy still lacks
specificity, and influences on non-tumor tissues are also
uncertain. In the future, the aim is to gain further insights into
dynamic changes in autophagy during PCa development and treatment.
Through specific autophagy regulation, inducing cell apoptosis and
malignization may be a viable strategy for treatment of PCa.
Stem cells are a type of cell that indicate multiple
differentiation potential, self-renewal, absence of differentiation
or low differentiation and exist in multicellular organisms
(138). In recent years, constant
research demonstrated that prostate stem cells are closely
associated with the occurrence and development of PCa. Prostate
epithelial stem cells exist in the basal layer, which generates
intermediate amplification cells, resulting in highly
differentiated luminal epithelial cells of the prostate (139). Mesenchymal stem cells exist in the
prostatic stroma, and they are induced by the microenvironment to
generate various prostate stromal cells, including fibroblasts and
muscle cells. Prostate epithelial stem cells and mesenchymal stem
cells do not depend on the presence of androgen but can respond to
this hormone (139,140). Using Hoechst33342 staining, Rupesh
first isolated prostate stem cells in 2003. Richardson et al
(141) confirmed that ~1% of human
prostate basal cells express cell surface markers, CD133, cells
with phenotype of α2β1 and hi/CD133+ possess high proliferative
potential in vitro and can reconstruct prostatic acini in
immunodeficient male nude mice. A previous study also isolated a
type of PCa cell similar to prostatic basal cells from
androgen-dependent PCa; these cells can also survive without
androgen (140). The study of
Collins et al (142)
successfully isolated PCa stem cells with the phenotype of
CD44+/α2β1 hi/CD133+ from PCa tissues utilizing a transplantation
assay based on isolated cells xenografted into immunodeficient
mice; they first proved the existence of PCa stem cells, and this
result may explain why patients with PCa will indicate potential
castration resistance and metastasis following ADT.
PCa stem cells are generally believed to be caused
by mutations of prostate stem cells, and development of PCa is
possibly associated with mutations in stem cells of the prostate
(143). Stem cells from PCa or the
normal prostate possess unlimited self-renewal ability and the
capacity for multipotential differentiation, they improve
self-protection through a variety of ways, including anti
apoptosis. Stem cells activate membrane transport proteins and
strengthen DNA repair capacity and they differentiate into cancer
tissues similar to primary prostate carcinoma following
transplantation in vitro (138). However, PCa stem cells and prostate
stem cells also exhibit differences. Normal prostate stem cell
proliferation is frequently associated with tissue damage or other
conditions, whereas malignant proliferation of PCa stem cell is
usually associated with loss of body control (140,144).
By contrast, normal prostate stem cells can differentiate into
mature tissues and perform corresponding functions unlike PCa stem
cells, which can only differentiate and maturate. Compared with
normal prostate stem cells, PCa stem cells are more prone to
accumulate mutations (140,144). Although prostate stem cells have
thus far not been purified in vitro, based on their previous
experience, Wang et al (145)
retrieved stem cell-like cells from PCa tissues in 2014. At
present, PCa stem cells and prostate basal stem cells express a
number of similar markers, including CD44, integrin α2β1 and CD133,
which can be used to identify and isolate stem cell populations.
Clinically, prostate stem cell surface markers, including prostate
stem cell antigen, can be used to assist in the diagnosis of PCa or
metastatic tumors of the prostate (141,142,146).
Recent evidence has demonstrated that prostate cancer stem-like
cells (PCSLCs) serve a critical role in stimulating CRPC evolution
and resistance to abiraterone and enzalutamide (147). For CRPC, the drug control of PCa
stem cells may be a novel targeted therapy direction for future PCa
treatments. A precautionary PCa vaccine can also be developed based
on characteristics of PCa stem cells to prevent the progression of
human PCa.
Currently, PCa, particularly advanced and
metastatic, has been a continuous burden on the healthcare system;
therefore, the clinical research on CRPC has become a hot spot.
Multiple mechanisms underlying the resistance to AR targeted
therapies have been identified, including AR overexpression with or
without amplification, AR mutations, AR-Vs, intratumoral DHT
synthesis, GR overexpression, defects of DDR genes and loss of AR,
with other therapies to be discovered. In addition to the
aforementioned underlying mechanism, including the studies of PSMA,
which contains high expression in androgen resistance prostate
cells and has been demonstrated to promote the proliferation,
invasion and apoptosis of PCa cells (148); additionally, the p100/p52 pathway,
one of the NF-κB pathways, the process of p100 to p52 in PCa via
molecules, including B-cell activating factor, CD40 ligand and
STAT3, leads to significant hyperplasia and induces the growth of
castration-resistant cells (149).
The majority of those advancements in the mechanism understanding
of CRPC have provided numerous potential biological targets for the
CRPC treatment, including AR antagonist abiraterone and
enzalutamide, AR-NTD antagonist EPI-002, PARP inhibitor olaparib,
PI3K/mTOR inhibitor BEZ235, chemotherapy drugs cabazitaxel and
immunotherapy sipuleucel-T, which can improve the OS and
disease-free survival of patients with CRPC (47,92,150–152).
In addition, the majority of these pharmaceutical agents are
undergoing clinical trials and are being approved for the treatment
of CRPC by FDA (6,153); however, all of these agents merely
slow the progression of disease, and all patients will inevitably
deteriorate into an incurable stage. The changes in the AR
signaling pathway, multiple genes and signaling pathways are
involved in the process of PCa from early androgen dependence to
late CRPC (154), and the underlying
mechanism remains at a limited understanding. The interconnection
of these signaling pathways form a molecular signaling network,
inhibiting a target alone or blocking the occurrence and
progression of a signaling pathway is not sufficient to prevent
CRPC. To conclude, the focus of the study is to identify and
control the key genes targeting these pathways, and effectively
prevent PCa from CRPC transformation and evolution, which has an
important role in adjustment of CRPC therapeutic strategies,
discovery of novel drug targets and improvement of the therapeutic
effect.
Not applicable.
This study was supported by the Guangzhou Municipal
Science and Technology Project (Grant no. 1563000448)
Not applicable.
JXH and HYQ drafted the manuscript; LX and JGG were
responsible for the collection of the relevant literature. All
authors have read and approved the final manuscript.
This article does not contain any studies with
human participants performed by any of the authors.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Society AC: Cancer facts & figures
2016. Atlanta: American Cancer Society; 2016
|
|
3
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taylor BS, Schultz N, Hieronymus H,
Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva
B, et al: Integrative genomic profiling of human prostate cancer.
Cancer Cell. 18:11–22. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomas C, Bögemann M, König F, Machtens S,
Schostak M, Steuber T and Heidenreich A: Advanced prostate cancer
consensus conference (APCCC) 2015 in St. Gallen. Critical review of
the recommendations on diagnosis and therapy of metastatic prostate
cancer by a German expert panel. Urologe A. 55:772–782. 2016.(In
German).
|
|
6
|
Ozono S and Furuse H: Progress of the
treatment for CRPC. Nihon Rinsho. 74 Suppl 3:S615–S618. 2016.(In
Japanese).
|
|
7
|
Lian F, Sharma NV, Moran JD and Moreno CS:
The biology of castration-resistant prostate cancer. Curr Probl
Cancer. 39:17–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maughan BL and Antonarakis ES: Androgen
pathway resistance in prostate cancer and therapeutic implications.
Expert Opin Pharmacother. 16:1521–1537. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roy AK, Tyagi RK, Song CS, Lavrovsky Y,
Ahn SC, Oh TS and Chatterjee B: Androgen receptor: Structural
domains and functional dynamics after ligand-receptor interaction.
Ann N Y Acad Sci. 949:44–57. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gelmann EP: Molecular biology of the
androgen receptor. J Clin Oncol. 20:3001–3015. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andersen RJ, Mawji NR, Wang J, Wang G,
Haile S, Myung JK, Watt K, Tam T, Yang YC, Bañuelos CA, et al:
Regression of castrate-recurrent prostate cancer by a
small-molecule inhibitor of the amino-terminus domain of the
androgen receptor. Cancer Cell. 17:535–546. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Darshan MS, Loftus MS, Thadani-Mulero M,
Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa
ST, et al: Taxane-induced blockade to nuclear accumulation of the
androgen receptor predicts clinical responses in metastatic
prostate cancer. Cancer Res. 71:6019–6029. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aggarwal RR, Thomas G, Youngren J, Foye A,
Olson S, Paris P, Beer TM, Ryan CJ, Witte O, Evans CP, et al:
Androgen receptor (AR) amplification in patients (pts) with
metastatic castration resistant prostate cancer (mCRPC) resistant
to abiraterone (Abi) and enzalutamide (Enz): Preliminary results
from the SU2C/PCF/AACR West Coast prostate cancer dream team
(WCDT). J Clin Oncol. 33:50682015.
|
|
14
|
Haapala K, Kuukasjärvi T, Hyytinen E,
Rantala I, Helin HJ and Koivisto PA: Androgen receptor
amplification is associated with increased cell proliferation in
prostate cancer. Hum Pathol. 38:474–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Attard G, Swennenhuis JF, Olmos D, Reid
AH, Vickers E, A'Hern R, Levink R, Coumans F, Moreira J, Riisnaes
R, et al: Characterization of ERG, AR and PTEN gene status in
circulating tumor cells from patients with castration-resistant
prostate cancer. Cancer Res. 69:2912–2918. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bubendorf L, Kononen J, Koivisto P,
Schraml P, Moch H, Gasser TC, Willi N, Mihatsch MJ, Sauter G and
Kallioniemi OP: Survey of gene amplifications during prostate
cancer progression by high-throughout fluorescence in situ
hybridization on tissue microarrays. Cancer Res. 59:803–806.
1999.PubMed/NCBI
|
|
17
|
Linja MJ, Savinainen KJ, Saramäki OR,
Tammela TL, Vessella RL and Visakorpi T: Amplification and
overexpression of androgen receptor gene in hormone-refractory
prostate cancer. Cancer Res. 61:3550–3555. 2001.PubMed/NCBI
|
|
18
|
Chen CD, Welsbie DS, Tran C, Baek SH, Chen
R, Vessella R, Rosenfeld MG and Sawyers CL: Molecular determinants
of resistance to antiandrogen therapy. Nat Med. 10:33–39. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim D, Gregory CW, French FS, Smith GJ and
Mohler JL: Androgen receptor expression and cellular proliferation
during transition from androgen-dependent to recurrent growth after
castration in the CWR22 prostate cancer xenograft. Am J Pathol.
160:219–226. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Santer FR, Erb HH and McNeill RV: Therapy
escape mechanisms in the malignant prostate. Seminars Cancer Biol.
35:133–144. 2015. View Article : Google Scholar
|
|
21
|
Waltering KK, Helenius MA, Sahu B, Manni
V, Linja MJ, Jänne OA and Visakorpi T: Increased expression of
androgen receptor sensitizes prostate cancer cells to low levels of
androgens. Cancer Res. 69:8141–8149. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koivisto P, Kononen J, Palmberg C, Tammela
T, Hyytinen E, Isola J, Trapman J, Cleutjens K, Noordzij A,
Visakorpi T and Kallioniemi OP: Androgen receptor gene
amplification: A possible molecular mechanism for androgen
deprivation therapy failure in prostate cancer. Cancer Res.
57:314–319. 1997.PubMed/NCBI
|
|
23
|
Barbieri CE, Baca SC, Lawrence MS,
Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van
Allen E, Stransky N, et al: Exome sequencing identifies recurrent
SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet.
44:685–689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mayeur GL, Kung WJ, Martinez A, Izumiya C,
Chen DJ and Kung HJ: Ku is a novel transcriptional recycling
coactivator of the androgen receptor in prostate cancer cells. J
Biol Chem. 280:10827–10833. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sarkar S, Brautigan DL, Parsons SJ and
Larner JM: Androgen receptor degradation by the E3 ligase CHIP
modulates mitotic arrest in prostate cancer cells. Oncogene.
33:26–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Veldscholte J, Ris-Stalpers C, Kuiper GG,
Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J,
Brinkmann AO and Mulder E: A mutation in the ligand binding domain
of the androgen receptor of human LNCaP cells affects steroid
binding characteristics and response to anti-androgens. Biochem
Biophys Res Commun. 173:534–540. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gaddipati JP, McLeod DG, Heidenberg HB,
Sesterhenn IA, Finger MJ, Moul JW and Srivastava S: Frequent
detection of codon 877 mutation in the androgen receptor gene in
advanced prostate cancers. Cancer Res. 54:2861–2864.
1994.PubMed/NCBI
|
|
28
|
Suzuki H, Sato N, Watabe Y, Masai M, Seino
S and Shimazaki J: Androgen receptor gene mutations in human
prostate cancer. J Steroid Biochem Mol Biol. 46:759–765. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suzuki H, Akakura K, Komiya A, Aida S,
Akimoto S and Shimazaki J: Codon 877 mutation in the androgen
receptor gene in advanced prostate cancer: Relation to antiandrogen
withdrawal syndrome. Prostate. 29:153–158. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steketee K, Timmerman L, Ziel-van der Made
AC, Doesburg P, Brinkmann AO and Trapman J: Broadened ligand
responsiveness of androgen receptor mutants obtained by random
amino acid substitution of H874 and mutation hot spot T877 in
prostate cancer. Int J Cancer. 100:309–317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Balbas MD, Evans MJ, Hosfield DJ,
Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y and
Sawyers CL: Overcoming mutation-based resistance to antiandrogens
with rational drug design. Elife. 2:e004992013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard G, et al: Integrative clinical genomics of advanced prostate
cancer. Cell. 161:1215–1228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Azad AA, Volik SV, Wyatt AW, Haegert A, Le
Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, et
al: Androgen receptor gene aberrations in circulating cell-free
DNA: Biomarkers of therapeutic resistance in castration-resistant
prostate cancer. Clin Cancer Res. 21:2315–2324. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alliance for Clinical Trials in Oncology,
. Enzalutamide with or without abiraterone and prednisone in
treating patients with castration-resistant metastatic prostate
cancer. ClinicalTrials.gov Identifier: NCT01949337. https://clinicaltrials.gov/ct2/show/NCT01949337September
24–2013
|
|
35
|
Nakazawa M, Antonarakis ES and Luo J:
Androgen receptor splice variants in the era of enzalutamide and
abiraterone. Horm Cancer. 5:265–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haile S and Sadar MD: Androgen receptor
and its splice variants in prostate cancer. Cell Mol Life Sci.
68:3971–3981. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hu R, Dunn TA, Wei S, Isharwal S, Veltri
RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guo Z, Yang X, Sun F, Jiang R, Linn DE,
Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al: A novel
androgen receptor splice variant is up-regulated during prostate
cancer progression and promotes androgen depletion-resistant
growth. Cancer Res. 69:2305–2313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dehm SM, Schmidt LJ, Heemers HV, Vessella
RL and Tindall DJ: Splicing of a novel androgen receptor exon
generates a constitutively active androgen receptor that mediates
prostate cancer therapy resistance. Cancer Res. 68:5469–5477. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fenner A: Prostate cancer: Unravelling AR
splice variant signalling in CPRC. Nat Rev Urol. 9:4102012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ware KE, Garcia-Blanco MA, Armstrong AJ
and Dehm SM: Biologic and clinical significance of androgen
receptor variants in castration resistant prostate cancer. Endocr
Relat Cancer. 21:T87–T103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Penel N: Splicing variant of androgen
receptors (AR-V7): New paradigms. Bull Cancer. 103:711–713.
2016.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z,
Yang X, Zhang H, Zhu Y and Shi G: Constitutively active AR-V7 plays
an essential role in the development and progression of
castration-resistant prostate cancer. Sci Rep. 5:76542015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhan Y, Zhang G, Wang X, Qi Y, Bai S, Li
D, Ma T, Sartor O, Flemington EK, Zhang H, et al: Interplay between
cytoplasmic and nuclear androgen receptor splice variants mediates
castration resistance. Mol Cancer Res. 15:59–68. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang YC, Mawji N, Wang J and Sadar M:
Preclinical evaluation of novel androgen receptor N-terminal domain
inhibitor EPI-002 for the treatment of castration-resistant
prostate cancer. Proceedings of the 105th Annual Meeting of the
American Association for Cancer Research, San Diego, CA, 2014.
Cancer Res. 74(Suppl 19): Abstract 610. 2014;
|
|
47
|
Ito Y, Banuelos CA and Sadar MD:
Combination therapy with EPI-002 and parp inhibitor for
castration-resistant prostate cancer. J Urol. 197:E11082017.
View Article : Google Scholar
|
|
48
|
Hermanson O, Glass CK and Rosenfeld MG:
Nuclear receptor coregulators: Multiple modes of modification.
Trends Endocrinol Metab. 13:55–60. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Culig Z: Androgen receptor coactivators in
regulation of growth and differentiation in prostate cancer. J Cell
Physiol. 231:270–274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wolf IM, Heitzer MD, Grubisha M and
DeFranco DB: Coactivators and nuclear receptor transactivation. J
Cell Biochem. 104:1580–1586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Massie CE, Adryan B, Barbosa-Morais NL,
Lynch AG, Tran MG, Neal DE and Mills IG: New androgen receptor
genomic targets show an interaction with the ETS1 transcription
factor. EMBO Rep. 8:871–878. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bennett NC, Gardiner RA, Hooper JD,
Johnson DW and Gobe GC: Molecular cell biology of androgen receptor
signalling. Int J Biochem Cell Biol. 42:813–827. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Z, Chang CW, Goh WL, Sung WK and
Cheung E: CENTDIST: Discovery of co-associated factors by motif
distribution. Nucleic Acids Res. 39(Web Server issue): W391–W399.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang Q, Li W, Liu XS, Carroll JS, Jänne
OA, Keeton EK, Chinnaiyan AM, Pienta KJ and Brown M: A hierarchical
network of transcription factors governs androgen
receptor-dependent prostate cancer growth. Mol Cell. 27:380–392.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tan PY, Chang CW, Chng KR, Wansa KD, Sung
WK and Cheung E: Integration of regulatory networks by NKX3-1
promotes androgen-dependent prostate cancer survival. Mol Cell
Biol. 32:399–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Heemers HV and Tindall DJ: Androgen
receptor (AR) coregulators: A diversity of functions converging on
and regulating the AR transcriptional complex. Endocr Rev.
28:778–808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ni L, Yang CS, Gioeli D, Frierson H, Toft
DO and Paschal BM: FKBP51 promotes assembly of the Hsp90 chaperone
complex and regulates androgen receptor signaling in prostate
cancer cells. Mol Cell Biol. 30:1243–1253. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chen S, Sullivan WP, Toft DO and Smith DF:
Differential interactions of p23 and the TPR-containing proteins
Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress
Chaperones. 3:118–129. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wissmann M, Yin N, Müller JM, Greschik H,
Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R,
et al: Cooperative demethylation by JMJD2C and LSD1 promotes
androgen receptor-dependent gene expression. Nat Cell Biol.
9:347–353. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Laschak M, Bechtel M, Spindler KD and
Hessenauer A: Inability of NCoR/SMRT to repress androgen receptor
transcriptional activity in prostate cancer cell lines. Int J Mol
Med. 28:645–651. 2011.PubMed/NCBI
|
|
61
|
Zaret KS and Carroll JS: Pioneer
transcription factors: Establishing competence for gene expression.
Genes Dev. 25:2227–2241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Coffey K and Robson CN: Regulation of the
androgen receptor by post-translational modifications. J
Endocrinol. 215:221–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chen MF, Chen WC, Chang YJ, Wu CF and Wu
CT: Role of DNA methyltransferase 1 in hormone-resistant prostate
cancer. J Mol Med (Berl). 88:953–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Liu Y, Karaca M, Zhang Z, Gioeli D, Earp
HS and Whang YE: Dasatinib inhibits site-specific tyrosine
phosphorylation of androgen receptor by Ack1 and Src kinases.
Oncogene. 29:3208–3216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Willder JM, Heng SJ, McCall P, Adams CE,
Tannahill C, Fyffe G, Seywright M, Horgan PG, Leung HY, Underwood
MA and Edwards J: Androgen receptor phosphorylation at serine 515
by Cdk1 predicts biochemical relapse in prostate cancer patients.
Br J Cancer. 108:139–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ha S, Iqbal NJ, Mita P, Ruoff R, Gerald
WL, Lepor H, Taneja SS, Lee P, Melamed J, Garabedian MJ and Logan
SK: Phosphorylation of the androgen receptor by PIM1 in hormone
refractory prostate cancer. Oncogene. 32:3992–4000. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shu SK, Liu Q, Coppola D and Cheng JQ:
Phosphorylation and activation of androgen receptor by Aurora-A. J
Biol Chem. 285:33045–33053. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xia Y, Wang M, Beraldi E, Cong M, Zoubeidi
A, Gleave M and Peng L: A novel triazole nucleoside suppresses
prostate cancer cell growth by inhibiting heat shock factor 1 and
androgen receptor. Anticancer Agents Med Chem. 15:1333–1340. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Beltran H, Rickman DS, Park K, Chae SS,
Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, et
al: Molecular characterization of neuroendocrine prostate cancer
and identification of new drug targets. Cancer Discov. 1:487–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Massie CE, Lynch A, Ramos-Montoya A, Boren
J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, et al:
The androgen receptor fuels prostate cancer by regulating central
metabolism and biosynthesis. EMBO J. 30:2719–2733. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Kumagai J, Hofland J, Erkens-Schulze S,
Dits NFJ, Jenster G, Bangma CH, Homma Y, De Jong FH and Van Weerden
WM: Intratumoral conversion of adrenal androgens is more important
than De Novo intratumoral steroid synthesis in prostate cancer. Eur
Urol Suppl. 10:2642011. View Article : Google Scholar
|
|
72
|
Montgomery RB, Mostaghel EA, Vessella R,
Hess DL, Kalhorn TF, Higano CS, True LD and Nelson PS: Maintenance
of intratumoral androgens in metastatic prostate cancer: A
mechanism for castration-resistant tumor growth. Cancer Res.
68:4447–4454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yin L and Hu Q: CYP17
inhibitors-abiraterone, C17,20-lyase inhibitors and multi-targeting
agents. Nat Rev Urol. 11:32–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Knuuttila M, Yatkin E, Kallio J,
Savolainen S, Laajala TD, Aittokallio T, Oksala R, Häkkinen M,
Keski-Rahkonen P, Auriola S, et al: Castration induces
up-regulation of intratumoral androgen biosynthesis and androgen
receptor expression in an orthotopic VCaP human prostate cancer
xenograft model. Am J Pathol. 184:2163–2173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tamae D, Mostaghel E, Montgomery B, Nelson
PS, Balk SP, Kantoff PW, Taplin ME and Penning TM: The DHEA-sulfate
depot following P450c17 inhibition supports the case for AKR1C3
inhibition in high risk localized and advanced castration resistant
prostate cancer. Chem Biol Interact. 234:332–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ryan CJ, Smith MR, de Bono JS, Molina A,
Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng
S, et al: Abiraterone in metastatic prostate cancer without
previous chemotherapy. N Engl J Med. 368:138–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Beer TM, Armstrong AJ, Rathkopf DE, Loriot
Y, Sternberg CN, Higano CS, Iversen P, Bhattacharya S, Carles J,
Chowdhury S, et al: Enzalutamide in metastatic prostate cancer
before chemotherapy. N Engl J Med. 371:424–433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Chandrasekar T, Yang JC, Gao AC and Evans
CP: Targeting molecular resistance in castration-resistant prostate
cancer. BMC Med. 13:2062015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang BH: Molecular mechanisms of gene
regulation mediated by nuclear receptor superfamily. Sheng Li Ke
Xue Jin Zhan. 34:369–372. 2003.(In Chinese). PubMed/NCBI
|
|
80
|
Laudet V, Hänni C, Coll J, Catzeflis F and
Stéhelin D: Evolution of the nuclear receptor gene superfamily.
EMBO J. 11:1003–1013. 1992.PubMed/NCBI
|
|
81
|
Szmulewitz RZ, Chung E, Al-Ahmadie H,
Daniel S, Kocherginsky M, Razmaria A, Zagaja GP, Brendler CB,
Stadler WM and Conzen SD: Serum/glucocorticoid-regulated kinase 1
expression in primary human prostate cancers. Prostate. 72:157–164.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Sahu B, Laakso M, Pihlajamaa P, Ovaska K,
Sinielnikov I, Hautaniemi S and Jänne OA: FoxA1 specifies unique
androgen and glucocorticoid receptor binding events in prostate
cancer cells. Cancer Res. 73:1570–1580. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Arora VK, Schenkein E, Murali R, Subudhi
SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis
C, et al: Glucocorticoid receptor confers resistance to
antiandrogens by bypassing androgen receptor blockade. Cell.
155:1309–1322. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Montgomery B, Cheng HH, Drechsler J and
Mostaghel EA: Glucocorticoids and prostate cancer treatment: Friend
or foe? Asian J Androl. 16:354–358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Montgomery B, Kheoh T, Molina A, Li J,
Bellmunt J, Tran N, Loriot Y, Efstathiou E, Ryan CJ, Scher HI and
de Bono JS: Impact of baseline corticosteroids on survival and
steroid androgens in metastatic castration-resistant prostate
cancer: Exploratory analysis from COU-AA-301. Eur Urol. 67:866–873.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Song C, Kim Y, Min GE and Ahn H:
Dihydrotestosterone enhances castration-resistant prostate cancer
cell proliferation through STAT5 activation via glucocorticoid
receptor pathway. Prostate. 74:1240–1248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lorente D, Omlin A, Ferraldeschi R, Pezaro
C, Perez R, Mateo J, Altavilla A, Zafeirou Z, Tunariu N, Parker C,
et al: Tumour responses following a steroid switch from prednisone
to dexamethasone in castration-resistant prostate cancer patients
progressing on abiraterone. Br J Cancer. 111:2248–2253. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chen R, Yu Y and Dong X: Progesterone
receptor in the prostate: A potential suppressor for benign
prostatic hyperplasia and prostate cancer. J Steroid Biochem Mol
Biol. 166:91–96. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Grindstad T, Andersen S, Al-Saad S, Donnem
T, Kiselev Y, Nordahl Melbø-Jørgensen C, Skjefstad K, Busund LT,
Bremnes RM and Richardsen E: High progesterone receptor expression
in prostate cancer is associated with clinical failure. PloS One.
10:e01166912015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Statz CM, Patterson SE and Mockus SM: mTOR
inhibitors in castration-resistant prostate cancer: A systematic
review. Target Oncol. 12:47–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Kato M, Banuelos CA, Imamura Y, Leung JK,
Caley DP, Wang J, Mawji NR and Sadar MD: Cotargeting androgen
receptor splice variants and mTOR signaling pathway for the
treatment of castration-resistant prostate cancer. Clin Cancer Res.
22:2744–2754. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
McMenamin ME, Soung P, Perera S, Kaplan I,
Loda M and Sellers WR: Loss of PTEN expression in paraffin-embedded
primary prostate cancer correlates with high Gleason score and
advanced stage. Cancer Res. 59:4291–4296. 1999.PubMed/NCBI
|
|
94
|
Zhang W, Zhu J, Efferson CL, Ware C,
Tammam J, Angagaw M, Laskey J, Bettano KA, Kasibhatla S, Reilly JF,
et al: Inhibition of tumor growth progression by antiandrogens and
mTOR inhibitor in a Pten-deficient mouse model of prostate cancer.
Cancer Res. 69:7466–7472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Park SI, Shah AN, Zhang J and Gallick GE:
Regulation of angiogenesis and vascular permeability by Src family
kinases: Opportunities for therapeutic treatment of solid tumors.
Expert Opin Ther Targets. 11:1207–1217. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Araujo JC, Trudel GC, Saad F, Armstrong
AJ, Yu EY, Bellmunt J, Wilding G, McCaffrey J, Serrano SV, Matveev
VB, et al: Docetaxel and dasatinib or placebo in men with
metastatic castration-resistant prostate cancer (READY): A
randomised, double-blind phase 3 trial. Lancet Oncol. 14:1307–1316.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yang JC, Bai L, Yap S, Gao AC, Kung HJ and
Evans CP: Effect of the specific Src family kinase inhibitor
saracatinib on osteolytic lesions using the PC-3 bone model. Mol
Cancer Ther. 9:1629–1637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bettedi L and Foukas LC: Growth factor,
energy and nutrient sensing signalling pathways in metabolic
ageing. Biogerontology. 18:913–929. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Neto AS, Tobias-Machado M, Wroclawski ML,
Fonseca FL, Pompeo AC and Del Giglio A: Molecular oncogenesis of
prostate adenocarcinoma: Role of the human epidermal growth factor
receptor 2 (HER-2/neu). Tumori. 96:645–649. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wen Y, Hu MC, Makino K, Spohn B,
Bartholomeusz G, Yan DH and Hung MC: HER-2/neu promotes
androgen-independent survival and growth of prostate cancer cells
through the Akt pathway. Cancer Res. 60:6841–6845. 2000.PubMed/NCBI
|
|
101
|
Smith DC, Smith MR, Sweeney C, Elfiky AA,
Logothetis C, Corn PG, Vogelzang NJ, Small EJ, Harzstark AL, Gordon
MS, et al: Cabozantinib in patients with advanced prostate cancer:
Results of a phase II randomized discontinuation trial. J Clin
Oncol. 31:412–419. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Cannistraci A, Di Pace AL, De Maria R and
Bonci D: MicroRNA as new tools for prostate cancer risk assessment
and therapeutic intervention: Results from clinical data set and
patients' samples. Biomed Res Int. 2014:1461702014. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kojima S, Goto Y and Naya Y: The roles of
microRNAs in the progression of castration-resistant prostate
cancer. J Hum Genet. 62:25–31. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Cannistraci A, Di Pace AL, Di Pace AL, De
Maria R and Bonci D: MicroRNA as new tools for prostate cancer risk
assessment and therapeutic intervention: Results from clinical data
set and patients' samples. Biomed Res Int. 2014:1461702014.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Schaefer A, Jung M, Mollenkopf HJ, Wagner
I, Stephan C, Jentzmik F, Miller K, Lein M, Kristiansen G and Jung
K: Diagnostic and prognostic implications of microRNA profiling in
prostate carcinoma. Int J Cancer. 126:1166–1176. 2010.PubMed/NCBI
|
|
106
|
Jalava SE, Urbanucci A, Latonen L,
Waltering KK, Sahu B, Jänne OA, Seppälä J, Lähdesmäki H, Tammela TL
and Visakorpi T: Androgen-regulated miR-32 targets BTG2 and is
overexpressed in castration-resistant prostate cancer. Oncogene.
31:4460–4471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Takeshita F, Patrawala L, Osaki M,
Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG,
Brown D and Ochiya T: Systemic delivery of synthetic microRNA-16
inhibits the growth of metastatic prostate tumors via
downregulation of multiple cell-cycle genes. Mol Ther. 18:181–187.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kashat M, Azzouz L, Sarkar SH, Kong D, Li
Y and Sarkar FH: Inactivation of AR and Notch-1 signaling by
miR-34a attenuates prostate cancer aggressiveness. Am J Transl Res.
4:432–442. 2012.PubMed/NCBI
|
|
109
|
Majid S, Dar AA, Saini S, Shahryari V,
Arora S, Zaman MS, Chang I, Yamamura S, Tanaka Y, Chiyomaru T, et
al: miRNA-34b inhibits prostate cancer through demethylation,
active chromatin modifications, and AKT pathways. Clin Cancer Res.
19:73–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Barron N, Keenan J, Gammell P, Martinez
VG, Freeman A, Masters JR and Clynes M: Biochemical relapse
following radical prostatectomy and miR-200a levels in prostate
cancer. Prostate. 72:1193–1199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Varambally S, Cao Q, Mani RS, Shankar S,
Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al:
Genomic loss of microRNA-101 leads to overexpression of histone
methyltransferase EZH2 in cancer. Science. 322:1695–1699. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Mortensen MM, Høyer S, Orntoft TF,
Sørensen KD, Dyrskjøt L and Borre M: High miR-449b expression in
prostate cancer is associated with biochemical recurrence after
radical prostatectomy. BMC Cancer. 14:8592014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Casanova-Salas I, Rubio-Briones J,
Calatrava A, Mancarella C, Masiá E, Casanova J, Fernández-Serra A,
Rubio L, Ramírez-Backhaus M, Armiñán A, et al: Identification of
miR-187 and miR-182 as biomarkers of early diagnosis and prognosis
in patients with prostate cancer treated with radical
prostatectomy. J Urol. 192:252–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nam RK, Benatar T, Wallis CJ, Amemiya Y,
Yang W, Garbens A, Naeim M, Sherman C, Sugar L and Seth A: MiR-301a
regulates E-cadherin expression and is predictive of prostate
cancer recurrence. Prostate. 76:869–884. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bell EH, Kirste S, Fleming JL, Stegmaier
P, Drendel V, Mo X, Ling S, Fabian D, Manring I, Jilg CA, et al: A
novel miRNA-based predictive model for biochemical failure
following post-prostatectomy salvage radiation therapy. PloS One.
10:e01187452015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Gallagher DJ, Gaudet MM, Pal P, Kirchhoff
T, Balistreri L, Vora K, Bhatia J, Stadler Z, Fine SW, Reuter V, et
al: Germline BRCA mutations denote a clinicopathologic subset of
prostate cancer. Clin Cancer Res. 16:2115–2121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Beltran H, Yelensky R, Frampton GM, Park
K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus
DM, et al: Targeted next-generation sequencing of advanced prostate
cancer identifies potential therapeutic targets and disease
heterogeneity. Eur Urol. 63:920–926. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Castro E, Goh C, Olmos D, Saunders E,
Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami
K, Guy M, et al: Germline BRCA mutations are associated with higher
risk of nodal involvement, distant metastasis, and poor survival
outcomes in prostate cancer. J Clin Oncol. 31:1748–1757. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao
Q, Asangani IA, Patel S, Wang X, Liang H, Yu J, et al: Mechanistic
rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene
fusion-positive prostate cancer. Cancer Cell. 19:664–678. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Mateo J, Carreira S, Sandhu S, Miranda S,
Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A,
Tunariu N, et al: DNA-repair defects and olaparib in metastatic
prostate cancer. N Engl J Med. 373:1697–1708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Polkinghorn WR, Parker JS, Lee MX, Kass
EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, et
al: Androgen receptor signaling regulates DNA repair in prostate
cancers. Cancer Discov. 3:1245–1253. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Goodwin JF, Schiewer MJ, Dean JL,
Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng
FY and Knudsen KE: A hormone-DNA repair circuit governs the
response to genotoxic insult. Cancer Discov. 3:1254–1271. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Li L, Chang W, Yang G, Ren C, Park S,
Karantanos T, Karanika S, Wang J, Yin J, Shah PK, et al: Targeting
poly(ADP-ribose) polymerase and the c-Myb-regulated DNA damage
response pathway in castration-resistant prostate cancer. Sci
Signal. 7:ra472014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Farrow JM, Yang JC and Evans CP: Autophagy
as a modulator and target in prostate cancer. Nat Rev Urol.
11:508–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Jiang X, Zhong W, Huang H, He H, Jiang F,
Chen Y, Yue F, Zou J, Li X, He Y, et al: Autophagy defects
suggested by low levels of autophagy activator MAP1S and high
levels of autophagy inhibitor LRPPRC predict poor prognosis of
prostate cancer patients. Mol Carcinog. 54:1194–1204. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Jiang X, Li X, Huang H, Jiang F, Lin Z, He
H, Chen Y, Yue F, Zou J, He Y, et al: Elevated levels of
mitochondrion-associated autophagy inhibitor LRPPRC are associated
with poor prognosis in patients with prostate cancer. Cancer.
120:1228–1236. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Hönscheid P, Datta K and Muders MH:
Autophagy: Detection, regulation and its role in cancer and therapy
response. Int J Radiat Biol. 90:628–635. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Ziparo E, Petrungaro S, Marini ES, Starace
D, Conti S, Facchiano A, Filippini A and Giampietri C: Autophagy in
prostate cancer and androgen suppression therapy. Int J Mol Sci.
14:12090–12106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bennett HL, Stockley J, Fleming JT, Mandal
R, O'Prey J, Ryan KM, Robson CN and Leung HY: Does
androgen-ablation therapy (AAT) associated autophagy have a
pro-survival effect in LNCaP human prostate cancer cells? BJU Int.
111:672–682. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Li M, Jiang X, Liu D, Na Y, Gao GF and Xi
Z: Autophagy protects LNCaP cells under androgen deprivation
conditions. Autophagy. 4:54–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Xu Y, Chen SY, Ross KN and Balk SP:
Androgens induce prostate cancer cell proliferation through
mammalian target of rapamycin activation and post-transcriptional
increases in cyclin D proteins. Cancer Res. 66:7783–7792. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Stambolic V, MacPherson D, Sas D, Lin Y,
Snow B, Jang Y, Benchimol S and Mak TW: Regulation of PTEN
transcription by p53. Mol Cell. 8:317–325. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Colquhoun AJ, Venier NA, Vandersluis AD,
Besla R, Sugar LM, Kiss A, Fleshner NE, Pollak M, Klotz LH and
Venkateswaran V: Metformin enhances the antiproliferative and
apoptotic effect of bicalutamide in prostate cancer. Prostate
Cancer Prostatic Dis. 15:346–352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Notte A, Ninane N, Arnould T and Michiels
C: Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast
cancer cells: Role of autophagy and JNK activation. Cell Death Dis.
4:e6382013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Li Y, Luo P, Wang J, Dai J, Yang X, Wu H,
Yang B and He Q: Autophagy blockade sensitizes the anticancer
activity of CA-4 via JNK-Bcl-2 pathway. Toxicol Appl Pharmacol.
274:319–327. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Reya T, Morrison SJ, Clarke MF and
Weissman IL: Stem cells, cancer, and cancer stem cells. Nature.
414:105–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Maitland NJ, Frame FM, Polson ES, Lewis JL
and Collins AT: Prostate cancer stem cells: Do they have a basal or
luminal phenotype? Horm Cancer. 2:47–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Rizzo S, Attard G and Hudson DL: Prostate
epithelial stem cells. Cell Prolif. 38:363–374. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Richardson GD, Robson CN, Lang SH, Neal
DE, Maitland NJ and Collins AT: CD133, a novel marker for human
prostatic epithelial stem cells. J Cell Sci. 117:3539–3545. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Collins AT, Berry PA, Hyde C, Stower MJ
and Maitland NJ: Prospective identification of tumorigenic prostate
cancer stem cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Maitland NJ and Collins A: A tumour stem
cell hypothesis for the origins of prostate cancer. BJU Int.
96:1219–1223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Nicolis SK: Cancer stem cells and
‘stemness’ genes in neuro-oncology. Neurobiol Dis. 25:217–229.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang S, Huang S, Zhao X, Zhang Q, Wu M,
Sun F, Han G and Wu D: Enrichment of prostate cancer stem cells
from primary prostate cancer cultures of biopsy samples. Int J Clin
Exp Pathol. 7:184–193. 2013.PubMed/NCBI
|
|
146
|
Tárnok A, Ulrich H and Bocsi J: Phenotypes
of stem cells from diverse origin. Cytometry A. 77:6–10. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ojo D, Lin X, Wong N, Gu Y and Tang D:
Prostate cancer stem-like cells contribute to the development of
castration-resistant prostate cancer. Cancers (Basel). 7:2290–2308.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Guo Z, Huang H, Zeng L, Du T, Xu K, Lin T,
Jiang C, Dong W, Cao Y, Chen J, et al: Lentivirus-mediated RNAi
knockdown of prostate-specific membrane antigen suppresses growth,
reduces migration ability and the invasiveness of prostate cancer
cells. Med Oncol. 28:878–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Nadiminty N, Lou W, Lee SO, Lin X, Trump
DL and Gao AC: Stat3 activation of NF-(kappa)B p100 processing
involves CBP/p300-mediated acetylation. Proc Natl Acad Sci USA.
103:pp. 7264–7269. 2006; View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Scher HI, Fizazi K, Saad F, Taplin ME,
Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, et
al: Increased survival with enzalutamide in prostate cancer after
chemotherapy. N Engl J Med. 367:1187–1197. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cindolo L, Natoli C, De Nunzio C, De Tursi
M, Valeriani M, Giacinti S, Micali S, Rizzo M, Bianchi G, Martorana
E, et al: Abiraterone acetate for treatment of metastatic
castration-resistant prostate cancer in chemotherapy-naive
patients: An Italian analysis of patients' satisfaction. Clin
Genitourin Cancer. 15:520–525. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kantoff PW, Higano CS, Shore ND, Berger
ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims
RB, et al: Sipuleucel-T immunotherapy for castration-resistant
prostate cancer. N Engl J Med. 363:411–422. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Inamoto T and Azuma H: Overview of the
ongoing clinical trials for new treatments for castrate-resistant
prostate cancer (CRPC). Nihon Rinsho. 74 Suppl 3:S653–S659.
2016.(In Japanese).
|
|
154
|
Penning TM: Mechanisms of drug resistance
that target the androgen axis in castration resistant prostate
cancer (CRPC). J Steroid Biochem Mol Biol. 153:105–113. 2015.
View Article : Google Scholar : PubMed/NCBI
|