Introduction
DNA double-strand breaks (DSBs) represent the most
detrimental form of DNA damage (1,2). Cells
rely on two major pathways to repair DSBs: Homologous recombination
(HR), which is the low-error mechanism for DNA repair; and
non-homologous end joining, which may introduce changes to the DNA
sequence at the repair site (1,2). HR is a
fundamental cellular process that is conserved in all organisms. It
maintains genome integrity by repairing endogenous and exogenous
DSBs (2).
The RAD51 paralogs form two identified complexes:
BCDX2 (RAD51B-RAD51C-RAD51D-XRCC2) and CX3 (RAD51C-XRCC3) (3). These two complexes act at two different
stages of homologous recombinational DNA repair (3). The BCDX2 complex is responsible for
RAD51 recruitment or stabilization at damage sites (3). The BCDX2 complex appears to act by
facilitating the assembly or stability of the RAD51 nucleoprotein
filament (3). The CX3 complex acts
downstream of RAD51 recruitment to damage sites (3). RAD51 paralog C (RAD51C) serves an
important role in the DNA damage response. It acts as a transducer
of the damage signal to ensure that the HR pathway of repair is
engaged. RAD51C localizes to the sites of DNA damage min after
damage occurs, indicating that RAD51C has a role in the early stage
of HR. RAD51C is also required for the phosphorylation of
checkpoint kinase 2 by ataxia telangiectasia mutated protein, which
is required for checkpoint activation (4). This indicates that RAD51C is required
for efficient checkpoint signaling, which delays cell cycle
progression in response to DNA damage. RAD51C foci persist long
after RAD51 can no longer be detected. Therefore, it is possible
that RAD51C is involved in the early and late stages of the HR
reaction (4). Liu et al
(5) demonstrated that RAD51C is
involved in regulating the resolution of Holliday Junctions (HJ)
during the later stages of HR. Furthermore, another study reported
that RAD51C-deficient hamster cells and mouse embryonic fibroblasts
have a reduced level of HJ resolution activity (6).
The HR pathway is a critical repair mechanism for
various lethal forms of DNA damage. Mutations in HR-associated
genes have been observed to cause the accumulation of unrepaired
DSBs, which may lead to carcinogenesis. Previous studies have
demonstrated that mutations in RAD51C increase the risk of breast
and ovarian cancers (7–9). RAD51C mutation is also associated with
Fanconi anaemia-like disorder (10).
Considering the important functions of RAD51C in DNA
repair by HR, the present study aimed to investigate the possible
correlation between RAD51C expression and drug sensitivity in
Eμ-Myc p19Arf−/− lymphoma cells and explore the
association between RAD51C expression and clinicopathological
factors in breast cancer. Arf negative cells were selected as Arg
negatively regulated MDM2, resulting in decreased p53 level which,
when combined with Eµ-driven Myc expression, created an
immortalized cell line; thus, creating a cell line without point
mutations for screening (11).
Knockdown of p53 in these cells leads to resistance to DNA damage
drugs, indicating these was still adequate p53 to trigger cell
arrest (12). A single-cell flow
cytometry-based green fluorescent protein (GFP) competition assay
was used to assess changes in sensitivity to anti-cancer drugs.
Immunohistochemistry (IHC) was used to detect the protein
expression of RAD51C in 213 samples from breast cancer tissue and
99 samples from the adjacent, non-cancerous tissues, and relevant
clinical information was collected from patients for a correlation
analysis.
Materials and methods
Cell culture and drugs
Eμ-Myc p19Arf−/− mouse lymphoma cells
were obtained from the Shanghai Institute for Biological Science
(Shanghai, China) and cultured in B cell medium [45% Dulbecco's
modified Eagle's medium (HyClone; GE Healthcare, Chicago, IL, USA),
45% Iscove's Modified Dulbecco's Medium (HyClone; GE Healthcare),
10% fetal bovine serum (Biochrom; Merck, Germany), L-glutamate and
β-mercaptoethanol] at 37°C in a humidified atmosphere with 5%
CO2. All drugs (Table I)
were obtained from Selleck Chemicals (Shanghai, China). Short
hairpin-RNA (shRNA) vectors were generated as described previously
(13,14).
 | Table I.Names of the 30 drugs used in the
study. |
Table I.
Names of the 30 drugs used in the
study.
| No. | Drug name | RI value |
|---|
| 1 | Camptothecin | 0.46 |
| 2 | Cisplatin | 0.28 |
| 3 | Doxorubicin | 0.70 |
| 4 | Gemcitabine | 1.03 |
| 5 | Methotrexate | 1.04 |
| 6 | Dexamethasone | 0.62 |
| 7 | Tioguanine | 0.69 |
| 8 | Olaparib | 0.29 |
| 9 | Erlotinib | 1.02 |
| 10 | Vismodegib | 0.92 |
| 11 | Actinomycin D | 0.86 |
| 12 | Vincristine | 0.70 |
| 13 | Lovastatin | 0.77 |
| 14 | Vorinostat | 1.06 |
| 15 | Taxol | 1.28 |
| 16 |
5-aza-2′-deoxycytidine | 0.75 |
| 17 | Fluorouracil | 0.84 |
| 18 | Trifluridine | 0.75 |
| 19 | All-trans-retinoic
acid | 0.93 |
| 20 | PP242 | 0.87 |
| 21 | Cytarabine | 0.58 |
| 22 | Fluvastain
sodium | 0.82 |
| 23 | Clofarabine | 1.00 |
| 24 | Pemetrexed
disodium | 1.00 |
| 25 | Axitinib | 0.85 |
| 26 | Albendazole | 1.12 |
| 27 | Bortezomib | 0.73 |
| 28 | Adefovir
dipivoxil | 0.63 |
| 29 | Prasugrel | 1.25 |
| 30 | Vardenafil | 0.88 |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from Eμ-Myc
p19Arf−/− mouse lymphoma cells using the TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. RNA was quantified using
the Nanodrop 1000 (Invitrogen; Thermo Fisher Scientific, Inc.,)
and, in total, 1 µg RNA from each sample was treated with DNAse I
(Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA).
Subsequently, the sample was reverse transcribed using randomized
hexanucleotides and M-MLV reverse transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.). The temperature protocol for reverse
transcription was as follows: 65°C for 5 min, cooled on ice for 5
min, 25°C for 10 min, 50°C for 50 min (cDNA synthesis) and 85°C for
5 min (deactivation). The product from reverse transcription (10
µl) was diluted to 150 µl in ddH2O and, subsequently, 3
µl from the 150 µl sample was added to each qPCR system. qPCR was
performed in triplicate using a CFX Connect Real-Time PCR Detection
System (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with
SYBR-Green Supermix (Bio-Rad Laboratories, Inc.). The sequences of
forward and reverse primers for RAD51C were as follows: Forward
5′-CAACTGCCTGCATTCAGCAC-3′ and reverse
5′-TGCCAGCAGCTCAGTATAATCA-3′. The GAPDH gene primers were as
follows: Forward, 5′-CCTGGAGAAACCTGCCAAGTATG-3′; reverse,
5′-AGAGTGGGAGTTGCTGTTGAAGTC-3′. PCR amplification was performed as
follows: 94°C for 2 min (initial denaturation), followed by 45
cycles of 95°C for 15 sec, 60°C for 15 sec, and 72°C for 20 sec. To
ensure that RNA samples were not contaminated with DNA, negative
controls were obtained by performing PCR on samples that were not
reverse transcribed but were otherwise identically processed.
Retroviral pMSCV–IRES-GFP vector serves as negative control. All
results were normalized by the Pfaffi method (15) using Bio-Rad CFX Manager 3.1.1517.0823
(Bio-Rad Laboratories, Inc.) to the GAPDH internal control and were
expressed as the mean ± standard error of the mean.
Western blot analysis
Equal amounts of protein were extracted from Eμ-Myc
p19Arf−/− mouse lymphoma cells. Cells were centrifuged
at 1,000 × g at 4°C for 2 min, cell pellets were weighed and
resuspended in PBS, mixed with 10 ml 2X SDS sample buffer [2 ml
Tris (1 M, pH 6.8), 4.6 ml glycerol (50%), 1.6 ml SDS (10%), 0.4 ml
bromophenol blue (0.5%) and 0.4 ml β-mercaptoethanol] and boiled
for 15 min at 100°C to generate whole-cell lysates. Were separated
by SDS-PAGE (12% gel) and transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA) via
electroblotting. The membranes were then blocked with 5% skimmed
milk in TBST for 1 h, incubated with specific primary antibodies
against RAD51C (dilution, 1:1,000; catalog no. ab2180; Abcam,
Cambridge, MA, USA) and β-actin (dilution, 1:200; catalog no.
sc-47778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
overnight at 4°C. ProteinFind goat anti-mouse immunoglobulin G
horseradish peroxidase-conjugated secondary antibody (dilution,
1:5,000; catalog no. HS201; TransGen Biotech, Co., Ltd., Beijing,
China) was applied and incubated at 37°C for 1 h. Protein detection
was performed by applying Immunobilon Western Chemiluminescent HRP
Substrate (WBKLS0500; EMD Millipore, Billerica, MA, USA) and images
were captured using Luminescent Image Analyzer (LAS4000; Fujifilm,
Tokyo, Japan). β-actin was used as a loading control for
normalization. Image J software (version 1.49; National Institutes
of Health, Bethesda, MD, USA) was used to quantify western blot
results.
Drug treatment and flow
cytometry-based GFP competition assay
Eμ-Myc p19Arf−/− cells at a density of
1×106 cells/ml were seeded in 48-well plates, as
aforementioned, and treated with various concentrations of distinct
drugs (Table I), according to a
previous study (12). Gemcitabine,
Lovastatin and Taxol were diluted in ethanol, Pemetrexed disodium
was diluted in water and the other drugs were diluted in DMSO. Half
of the drug-containing medium from each experiment was removed and
replenished with fresh medium every 24 h to approximate therapeutic
situations in which drug dose decreases over time. Cells were
analyzed by fluorescence-activated cell sorting (FACS) using
propidium iodide (PI) as a viability marker. The LD80–90
value for each drug was defined as the concentration at which the
lowest viability reading out of three FACS time points (24, 48 and
72 h) was between 10 and 20%. After the drug LD80–90 was
determined, Eμ-Myc p19Arf−/− cells were infected with
retroviruses encoding shRNAs targeting specific genes, according to
a previous study (12). Eμ-Myc
p19Arf -/− cells were infected in the presence of
polybrene (7 µg/ml), by centrifugation at 700 × g for 5 min at room
temperature. The negative control used was the same as previously
described (12). Following infection,
GFP, which was included in the retroviruses as previously described
(12), proportion typically reached
30%, which is desired for the GFP competition assay. Individual
infected cell populations were counted and seeded at
1×106 cells/ml in 48-well plates and treated with drugs,
using the aforementioned protocol. Treated and untreated cells were
analyzed with flow cytometry after 72 h. GFP-expressing percentages
of live (PI-negative) cells were recorded and used to calculate
relative resistance index (RI). To avoid outgrowth of the untreated
control cells, cells were typically seeded at 0.25 million per ml,
and 75% of the medium was replaced at 24 and 48 h (12).
Calculation of RI
To compare the relative level of chemoresistance and
sensitization conferred by gene knockdown, the concept of RI was
used to analyze the GFP competition assay results more accurately.
The biological meaning of RI is that in a mixture of uninfected and
infected (knockdown) cells, the infected cells will be RI-fold as
likely to survive drug treatment when compared with uninfected
cells. By this definition of RI, if 1/m uninfected cells survives a
drug treatment, then RI/n infected cells would survive. If the
total number of uninfected and infected cells is defined as T and
the GFP-containing percentage of the untreated population as G1,
then the number of surviving uninfected cells (un) can be defined
as n-un=T × (1-G1) × 1/m, and the number of surviving infected
cells (in) can be defined as n-in=T × G1 × RI/m. T represents the
total number of uninfected and infected cells, whereas m represents
the number of uninfected cells which resists drug treatment. Thus,
the GFP percentage of the treated surviving population (G2) can be
calculated as G2=(n-in)/[(n-un) + (n-in)]. n-in and n-un represent
the numbers of infected and uninfected cells, respectively. From
these equations, it is derived that RI=(G2-G1 × G2)/(G1-G1 × G2);
this equation was used in the present study to calculate the RI
values (12).
Patients and tissue samples
A total of 213 patients (median age, 53.4 years)
with primary breast carcinomas who had undergone curative surgery
at Changhai Hospital, Ruijin Hospital and Central Hospital of
Huangpu District (Shanghai, China) between 2001 and 2010 were
enrolled for this study. Histological confirmation of primary
breast carcinoma was obtained from the Department of Pathology at
Changhai Hospital. None of the patients had received preoperative
adjuvant chemotherapy. Of the 213 patients, paired non-neoplastic
breast tissues sampled from the resection margins were available
for 99 patients. All of the tissue specimens were obtained for the
present study with informed consent from the patients, and the use
of the human specimens was approved by the Ethics Committee of
Changhai Hospital. The last follow-up date was 31 December 2012,
with a median follow-up time of 72 months (range, 5–115 months).
The clinicopathological features of the tumors and patient survival
times were recorded.
Tissue microarray (TMA) and IHC
A manual arrayer (Beecher Instruments, Sun Prairie,
WI, USA) was used to create paraffin-embedded TMA blocks of normal
breast and breast cancer tissue specimens obtained from the
patients. Each block had at least one 1.5-mm core of non-neoplastic
tissue and two 1.5-mm cores of primary tumor tissue; 4-µm paraffin
sections were then produced using a standard technique (16). A RAD51C mouse monoclonal antibody
(dilution, 1:100; catalog no. ab2180; incubated for 1 h at 37°C;
Abcam) was used to detect RAD51C protein expression in the
sections. An EnVision kit (Dako; Agilent Technologies, Inc., Santa
Clara, CA, USA) was used to visualize antibody binding, and slides
were subsequently counterstained with hematoxylin. Expression of
RAD51C in the TMAs was evaluated by two individuals (Dr. Shao-Guang
Liao and Dr. Lu Liu), who were blinded to the other characteristics
of the patients, using an Olympus CX31 microscope (Olympus
Corporation, Tokyo, Japan). A positive reaction was indicated by a
reddish-brown precipitate in the nucleus of a cell. The scores of
staining depended on the percentage of positive cells and staining
intensity. The percentage of positive cells was divided into five
grades (percentage scores): 0, <10%; 1, 10–25%; 2, 25–50%; 3,
50–75%; and 4, >75%. The intensity of staining was divided into
four grades (intensity scores): 0, no staining; 1, light brown; 2,
medium brown; and 3, dark brown. RAD51C staining positivity was
determined by the following formula: Overall score=percentage score
× intensity score. An overall score of ≤3 was defined as negative,
and >3 as positive. Discrepancies in the scores were resolved by
discussion between the evaluators.
Statistical analysis
All statistical analyses were performed using SPSS
18.0 (SPSS Inc., Chicago, IL, USA) software. The associations
between RAD51C expression and clinicopathological characteristics
of breast cancer were analyzed with Pearson's correlation and
χ2 tests. Survival was defined as the time between
diagnosis and mortality. Survival analysis was performed by the
Kaplan-Meier method and the log-rank test. Cox's regression
analysis was used to estimate hazard ratios and their 95%
confidence intervals. A log-rank test was performed to conduct
survival analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
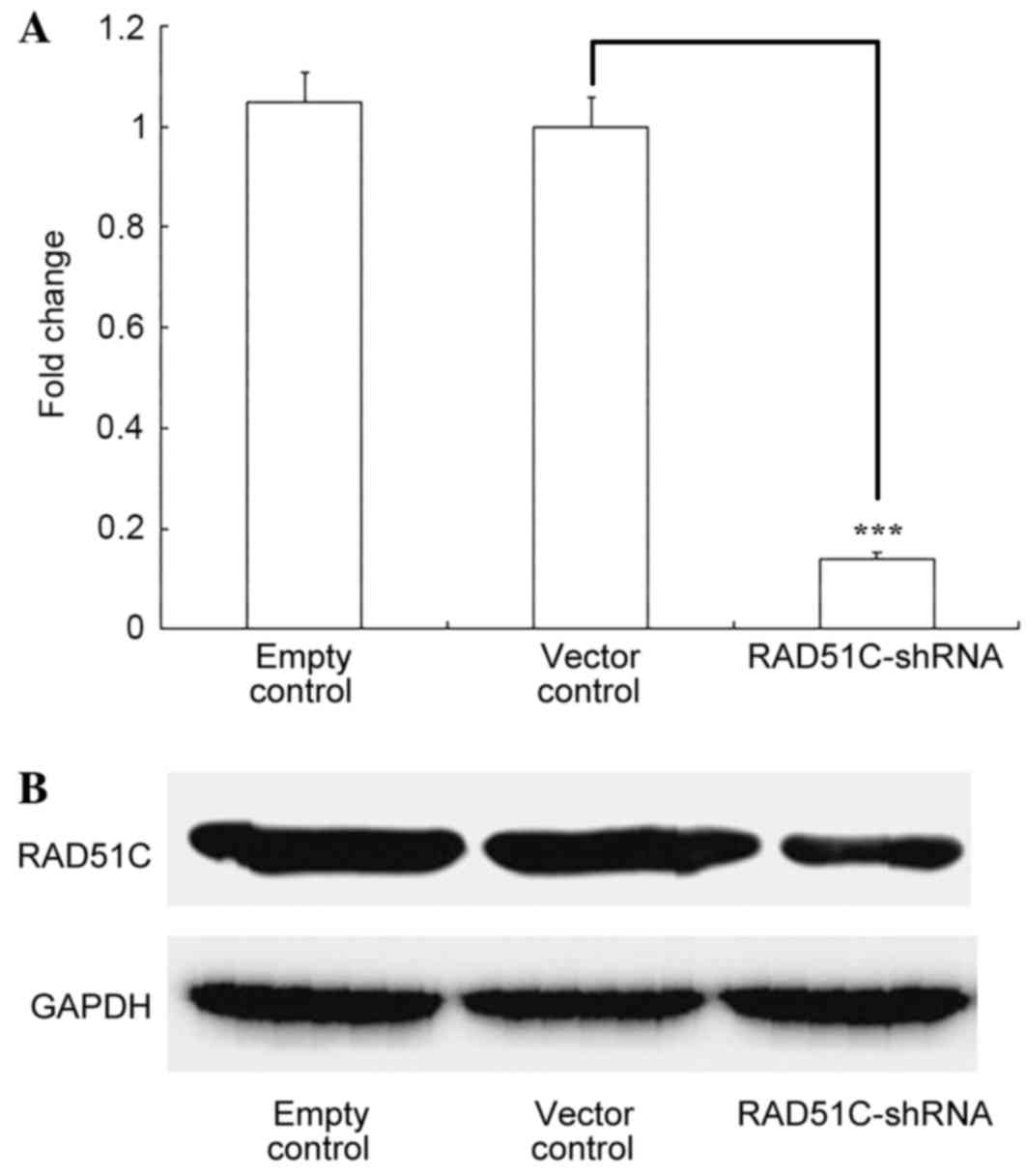
Inhibition of RAD51C expression with
RAD51C-shRNA
Eμ-Myc p19Arf−/− cells were stably
infected with retroviruses coexpressing RAD51C-shRNA and GFP. The
RT-qPCR results showed that RAD51C mRNA was significantly inhibited
(P<0.001) by shRNA targeting RAD51C and not by the negative
control (Fig. 1A). Western blotting
revealed a similar effect of RAD51C shRNA on RAD51C protein levels
in the cells (Fig. 1B).
Effect of knockdown of RAD51C on
sensitivity to anticancer drugs
A total of 30 drugs were selected for this study
(listed in Table I). All drugs were
used at their LD80–90 i.e., the concentration at which
80–90% of uninfected lymphoma cells were killed. A single-cell flow
cytometry-based GFP competition assay was used to determine the
change in sensitivity of the cells to each drug. Lymphoma cells
were infected with retroviruses co-expressing RAD51C-shRNA and GFP,
and subjected to 72 h of drug treatment. GFP-negative cells in the
same population served as an internal control. The effects of the
gene knockdown on chemosensitivity were recorded as the
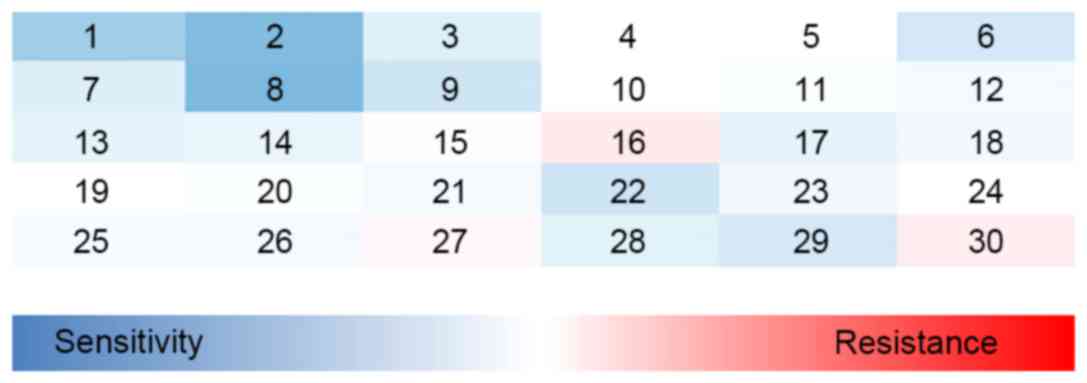
GFP-determined RI values. The results indicated that the
sensitivities of the cells to three drugs, camptothecin (CPT),
cisplatin (DDP) and olaparib, were significantly increased
following RAD51C gene knockdown (Fig.
2). The value of RI for each drug is shown in Tables I and II.
| Table II.RI values for the three drugs that
were more effective against Eμ-Myc p19Arf−/− cells
following RAD51C knockdown by shRNA. |
Table II.
RI values for the three drugs that
were more effective against Eμ-Myc p19Arf−/− cells
following RAD51C knockdown by shRNA.
|
| RI value, mean ±
standard error of mean |
|
|---|
|
|
|
|
|---|
| Drug | RAD51C-shRNA
group | Control group | P-value |
|---|
| Camptothecin | 0.46±0.02 | 0.91±0.08 | <0.001 |
| Cisplatin | 0.28±0.01 | 0.94±0.01 | <0.001 |
| Olaparib | 0.29±0.04 | 0.93±0.12 | <0.001 |
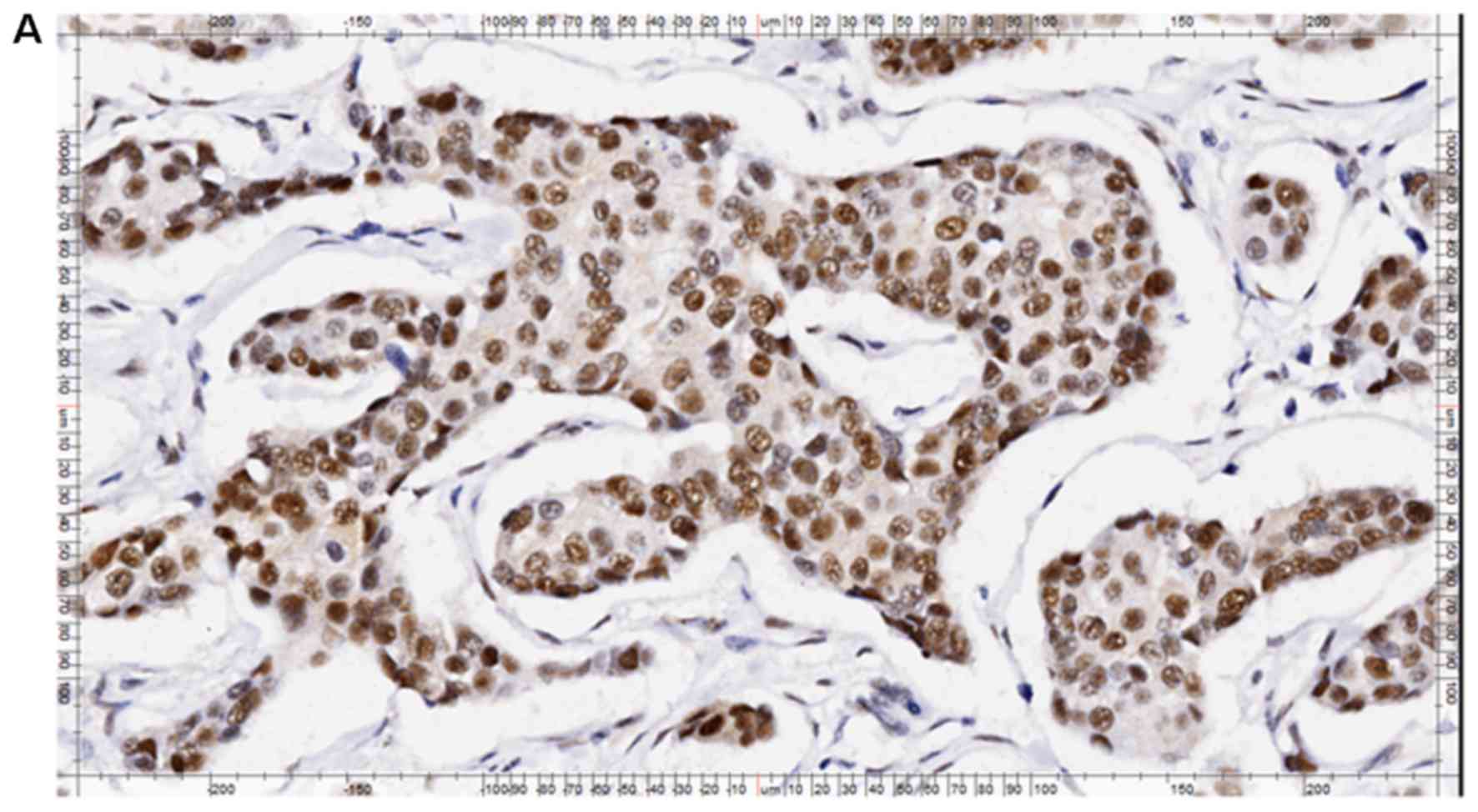
IHC, clinicopathological features, and
survival time analysis
The semi-quantitative results of RAD51C
immunohistochemical staining in breast cancer tissue are presented
in Table III. As is shown in
Fig. 3, RAD51C was predominantly
expressed in the nuclei and cytoplasm of cancer and normal cells.
As RAD51C protein functions primarily in the nucleus (2–3), cells
expressing RAD51C in their nuclei were considered
RAD51C-positive.
| Table III.Expression of RAD51C in breast cancer
tissues and paired non-neoplastic breast tissues. |
Table III.
Expression of RAD51C in breast cancer
tissues and paired non-neoplastic breast tissues.
|
|
| RAD51C expression,
n (%) |
|
|
|---|
|
|
|
|
|
|
|---|
| Breast tissue
type | Total, n | Negative | Positive | χ2 | P-value |
|---|
| Cancer | 213 | 103 (48.4) | 110 (51.6) | 1.72 | 0.22 |
| Non-neoplastic | 99 | 40 (40.4) | 59 (59.6) |
|
|
RAD51C was positively expressed in 51.6% of the
breast cancer tissues and in 59.6% of the paired non-neoplastic
breast tissues. There were no significant differences in RAD51C
expression observed between breast cancer and paired non-neoplastic
breast tissues (Table III).
RAD51C expression was not significantly correlated
with histological type, patient age, tumor size, lymph node
metastasis, TNM stage, histological grade or estrogen receptor
(ER)/progesterone receptor status of the patients with breast
cancer. However RAD51C expression in breast cancer tissues positive
for Erb-B2 receptor tyrosine kinase 2 (HER2) was significantly
higher compared with that in HER2-negative breast cancer tissues
(P=0.009; Table IV).
| Table IV.Associations between RAD51C
expression status and clinicopathological features of breast
cancer. |
Table IV.
Associations between RAD51C
expression status and clinicopathological features of breast
cancer.
|
|
| RAD51C expression,
n |
|
|---|
|
|
|
|
|
|---|
| Variable | Total, n | Negative | Positive | P-value |
|---|
| Total | 213 | 103 | 110 |
|
| Histological
type |
|
|
| 0.443 |
|
Ductal | 181 | 90 | 91 |
|
|
Other | 32 | 13 | 19 |
|
| Age, years (mean ±
SEM) |
| 53.4±12.3 | 53.4±11.6 | 0.994 |
| T stage, n |
|
|
| 0.291 |
| T1 | 65 | 30 | 35 |
|
| T2 | 114 | 60 | 54 |
|
| T3 | 32 | 12 | 20 |
|
| N stage, n |
|
|
| 0.259 |
| − | 86 | 38 | 48 |
|
| + | 118 | 62 | 56 |
|
| TNM stage |
|
|
| 0.489 |
| 1 | 45 | 21 | 24 |
|
| 2 | 105 | 48 | 57 |
|
| 3 | 52 | 29 | 23 |
|
| 4 | 1 | 0 | 1 |
|
| Histological
grade |
|
|
| 0.423 |
|
1–2 | 154 | 74 | 80 |
|
| 3 | 51 | 28 | 23 |
|
| ER status |
|
|
| 0.886 |
| − | 74 | 35 | 39 |
|
| + | 139 | 68 | 71 |
|
| PR status |
|
|
| 0.783 |
| − | 96 | 45 | 51 |
|
| + | 117 | 58 | 59 |
|
| HER2 status |
|
|
| 0.009 |
| − | 142 | 78 | 64 |
|
| + | 71 | 25 | 46 |
|
Cox multivariate regression analysis indicated that
high RAD51C expression (P=0.01) and ER-positive status (P=0.02)
were significant risk factors that may influence the postoperative
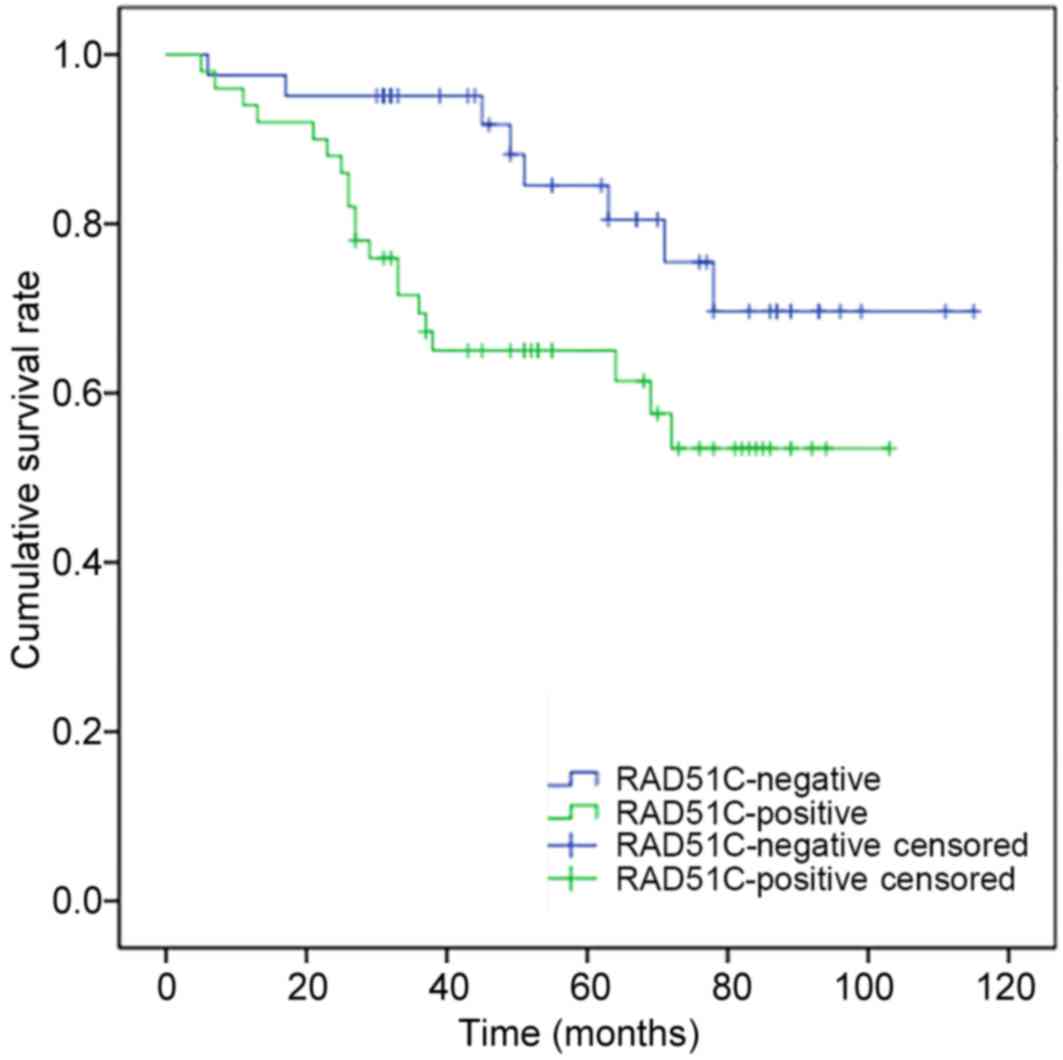
survival time of breast cancer patients (Table V). Survival analysis showed that the
postoperative survival time of patients with expression of RAD51C
was significantly shorter compared with that of patients without
RAD51C expression (P=0.03; Fig.
4).
| Table V.Multivariate analysis of potential
factors affecting the postoperative survival time of patients with
breast cancer. |
Table V.
Multivariate analysis of potential
factors affecting the postoperative survival time of patients with
breast cancer.
| Variable | Hazard ratio | 95% CI | P-value |
|---|
| Age | 1.01 | 0.98–1.04 | 0.73 |
| ER (+ vs. -) | 0.19 | 0.05–0.74 | 0.02 |
| PR (+ vs. -) | 1.70 | 0.46–6.30 | 0.43 |
| HER (+ vs. -) | 1.12 | 0.48–2.60 | 0.79 |
| T stage
(1,2,3) | 1.26 | 0.50–3.16 | 0.63 |
| N stage (+ vs.
-) | 1.13 | 0.39–3.23 | 0.82 |
| Histological type
(ductal vs. other) | 1.00 | 0.34–2.96 | 0.99 |
| Histological grade
(1,2 vs. 3) | 1.49 | 0.52–4.26 | 0.45 |
| RAD51C (+ vs.
-) | 3.34 | 1.37–8.19 | 0.01 |
Discussion
It has been demonstrated that RAD51 paralogs are
involved in two distinct complexes associated with HR: The
RAD51B/RAD51C/RAD51D/x-ray repair cross complementing (XRCC)2
complex (BCDX2) and the RAD51C/XRCC3 complex (CX3) (17,18). As
the only factor found in both the BCDX2 and CX3 complexes, RAD51C
plays an important role in early and late HR. Downregulation of
RAD51C expression may sensitize cells to DNA-damaging agents
(19). In the present study, it was
demonstrated that the sensitivity to three drugs, CPT, DDP and
olaparib, was significantly increased in Eμ-Myc
p19Arf−/− cells following RAD51C gene knockdown.
CPT and DDP are currently first-line
chemotherapeutic drugs for the treatment of various types of
cancer. CPT and DDP kill tumor cells through the induction of DSBs
(20–22). Olaparib is a poly (ADP-ribose)
polymerase (PARP) inhibitor that is in common use against cancer.
Research has shown that PARP inhibition in cells leads to
persistent single-strand breaks in DNA (23). When these breaks encounter a
replication fork, cell cycle arrest occurs and the single-strand
gaps may degenerate into DSBs (24).
Hence, all three drugs can induce DSBs in tumor cells. Normally,
these DSBs would be repaired by RAD51C-dependent HR (25). In the absence of RAD51C, the DSBs
cannot be repaired, leading to cell death. This may explain why the
sensitivity to CPT, DDP and olaparib significantly increased
following RAD51C gene knockdown. This result suggests that
silencing RAD51C may represent a potential therapeutic strategy
against malignant tumors.
Based on the association between RAD51C expression
and drug sensitivity in tumor cells, we hypothesized that cancer
patients who lack RAD51C expression may be more sensitive to
anticancer drugs and have an improved prognosis. To test this
hypothesis, IHC was used to detect the expression of RAD51C protein
in breast cancer tissues. The result indicated that RAD51C
expression in breast cancer tissue was positively correlated with
HER2 expression, which is an effective predictive and prognostic
marker for breast cancer patients. Furthermore, the result showed
that high RAD51C expression is an important risk factor that may
influence the postoperative survival time of breast cancer
patients. The Cox multivariate regression analysis result regarding
ER status was consistent with a previous study (26). Additionally, survival analysis
revealed that the postoperative survival time of patients with
RAD51C expression was significantly shorter compared with that of
patients without RAD51C expression, which confirmed the hypothesis
that the high expression of RAD51C is a marker for an adverse
prognosis in breast cancer.
In conclusion, it was identified that the
sensitivity to three drugs, CPT, DDP and olaparib, was
significantly increased when RAD51C expression was suppressed in
Eμ-Myc p19Arf−/− cells. Additionally, studies on patient
tissues indicated that the expression of RAD51C was positively
correlated with the expression of HER2. RAD51C expression was an
independent prognostic factor for breast cancer patients, and the
overall survival time of the patients without RAD51C expression was
significantly longer. Therefore, RAD51C should be considered as a
marker to predict tumor sensitivity to chemotherapy and a
prognostic factor for breast cancer patients.
Acknowledgements
The present study was supported by The Natural
Science Foundation of Fujian Province (grant no. 2016J05198).
References
|
1
|
Khanna KK and Jackson SP: DNA
double-strand breaks: Signaling, repair and the cancer connection.
Nat Genet. 27:247–254. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chun J, Buechelmaier ES and Powell SN:
Rad51 paralog complexes BCDX2 and CX3 act at different stages in
the BRCA1-BRCA2-dependent homologous recombination pathway. Mol
Cell Biol. 33:387–395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Badie S, Liao C, Thanasoula M, Barber P,
Hill MA and Tarsounas M: RAD51C facilitates checkpoint signaling by
promoting CHK2 phosphorylation. J Cell Biol. 185:587–600. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Masson JY, Shah R, O'Regan P and
West SC: RAD51C is required for Holliday junction processing in
mammalian cells. Science. 303:243–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuznetsov S, Pellegrini M, Shuda K,
Fernandez-Capetillo O, Liu Y, Martin BK, Burkett S, Southon E, Pati
D, Tessarollo L, et al: RAD51C deficiency in mice results in early
prophase I arrest in males and sister chromatid separation at
metaphase II in females. J Cell Biol. 176:581–592. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meindl A, Hellebrand H, Wiek C, Erven V,
Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L,
Schaal H, et al: Germline mutations in breast and ovarian cancer
pedigrees establish RAD51C as a human cancer susceptibilitygene.
Nat Genet. 42:410–414. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pelttari LM, Heikkinen T, Thompson D,
Kallioniemi A, Schleutker J, Holli K, Blomqvist C, Aittomäki K,
Bützow R and Nevanlinna H: RAD51C is a susceptibility gene for
ovarian cancer. Hum Mol Genet. 20:3278–3288. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Loveday C, Turnbull C, Ruark E, Xicola RM,
Ramsay E, Hughes D, Warren-Perry M and Snape K: Breast Cancer
Susceptibility Collaboration (UK), Eccles D, et al: Germline
RAD51C mutations confer susceptibility to ovarian cancer. Nat
Genet. 44:475–476. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vaz F, Hanenberg H, Schuster B, Barker K,
Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, et al:
Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat
Genet. 42:406–409. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Forbes SA, Beare D, Gunasekaran P, Leung
K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et
al: COSMIC: Exploring the world's knowledge of somatic mutations in
human cancer. Nucleic Acids Res. 3:D805–D811. 2015. View Article : Google Scholar
|
|
12
|
Jiang H, Pritchard JR, Williams RT,
Lauffenburger DA and Hemann MT: A mammalian functional-genetic
approach to characterizing cancer therapeutics. Nat Chem Biol.
7:92–100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dickins RA, Hemann MT, Zilfou JT, Simpson
DR, Ibarra I, Hannon GJ and Lowe SW: Probing tumor phenotypes using
stable and regulated synthetic microRNA precursors. Nat Genet.
37:1289–1295. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang H, Reinhardt HC, Bartkova J,
Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB and
Hemann MT: The combined status of ATM and p53 link tumor
development with therapeutic response. Genes Dev. 23:1895–1909.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai MY, Tong ZT, Zheng F, Liao YJ, Wang Y,
Rao HL, Chen YC, Wu QL, Liu YH, Guan XY, et al: EZH2 protein: A
promising immunomarker for the detection of hepatocellular
carcinomas in liver needle biopsies. Gut. 60:967–976. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thacker J: The RAD51 gene family, genetic
instability and cancer. Cancer Lett. 219:125–135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Masson JY, Tarsounas MC, Stasiak AZ,
Stasiak A, Shah R, McIlwraith MJ, Benson FE and West SC:
Identification and purification of two distinct complexes
containing the five RAD51 paralogs. Genes Dev. 15:3296–3307. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suwaki N, Klare K and Tarsounas M: RAD51
paralogs: Roles in DNA damage signalling, recombinational repair
and tumorigenesis. Semin Cell Dev Biol. 22:898–905. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sinha BK: Topoisomerase inhibitors. A
review of their therapeutic potential in cancer. Drugs. 49:11–19.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pommier Y: Topoisomerase I inhibitors:
Camptothecins and beyond. Nat Rev Cancer. 6:789–802. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Altaha R, Liang X, Yu JJ and Reed E:
Excision repair cross complementing-group 1: Gene expression and
platinum resistance. Int J Mol Med. 14:959–970. 2004.PubMed/NCBI
|
|
23
|
Boulton S, Kyle S and Durkacz BW:
Interactive effects of inhibitors of poly (ADP-ribose. polymerase
and DNA-dependent protein kinase on cellular responses to DNA
damage. Carcinogenesis. 20:199–203. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haber JE: DNA recombination: The
replication connection. Trends Biochem Sci. 24:271–275. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Arnaudeau C, Lundin C and Helleday T: DNA
double-strand breaks associated with replication forks are
predominantly repaired by homologous recombination involving an
exchange mechanism in mammalian cells. J Mol Biol. 307:1235–1245.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukutomi T and Akashi-Tanaka S: Prognostic
and predictive factors in the adjuvant treatment of breast cancer.
Breast Cancer. 9:95–99. 2002. View Article : Google Scholar : PubMed/NCBI
|