Introduction
Squamous cell carcinoma (SCC) is a common type of
skin cancer. Although SCC primarily occurs in areas of the skin
that are frequently exposed to sun, it may occur on all areas of
body, including the mucous membranes and genitals (1,2). It is
estimated that ~700,000 incident cases of SCC were diagnosed in the
United States in 2012 (3).
Understanding the mechanism of SCC, and developing novel and
effective therapies are required.
Peroxisome proliferator-activated receptor-γ
(PPAR-γ) activation has been demonstrated to inhibit cell growth in
numerous malignant cell types, suggesting that PPAR-γ agonists may
act as tumor suppressors (4). PPAR-γ
is a transcription factor that participates in metabolism of lipid
and glucose (5). PPAR-γ is expressed
at a high level in adipose tissue, regulating adipocyte
differentiation and glucose utilization (6). PPAR-γ is also expressed in intestinal
epithelial cells and tumor cells in breast, colon, and lung
(7–10). In addition, reduction of the
expression levels of PPAR-γ in PPAR-γ+/− mice is
associated with an increased susceptibility to 7,12-dimethylbenz(a)
anthracene-mediated carcinogenesis in the skin (11). A previous study indicated that topical
treatment of hairless mice with PPAR-γ agonists troglitazone and
ciglitazone enhanced the expression of markers of differentiation
that promoted epidermal barrier recovery (12).
Tumors may be caused by a number of factors,
including genetic mutations that lead to malfunction of the cell
cycle, inhibition of apoptosis and environmental factors that lead
to DNA damage (13). Clinically, the
induction of apoptosis to modulate cell growth has become an
important approach in cancer therapy (14). To examine the function and mechanism
of PPAR-γ agonist in treating malignant skin cancer, the present
study investigated the effect of one of the most potent PPAR-γ
agonists, rosiglitazone, on cell growth and cell apoptosis in
vitro. It was identified that rosiglitazone inhibited cell
growth, potentially through reducing the expression of cell
cycle-associated proteins and without affecting apoptosis.
Materials and methods
Cell culture experiment
Human epidermoid carcinoma A431 cells (American Type
Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's
modified Eagles medium/Ham's (powder, high glucose; cat no.
12800017; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 0.15% sodium bicarbonate, 10% fetal bovine serum
and 100 U/ml penicillin-streptomycin (all from Invitrogen; Thermo
Fisher Scientific, Inc.) at 37°C under 5% CO2. The stock
solution of rosiglitazone (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) was dissolved in dimethyl sulfoxide (DMSO; Cell Signaling
Technology, Inc., Danvers, MA, USA). For all drug assays, an equal
amount of DMSO was added as a control.
MTS assay
Cell viability was evaluated using MTS assay
(Promega Corporation, Madison, WI, USA). Specifically, 5,000 A431
cells were seeded in each well of a 96-well plate and incubated
with different concentrations of rosiglitazone (0, 10, 20, 30, 40
and 100 µM) for 24 h at 37°C. Then, 20 µl MTS/well was added and
incubated at room temperature for 4 h. Cell viability was measured
by absorbance at 490 nm using a microplate reader.
3H-Thymidine incorporation
assay
A431 cells (density, 5×104 cells/well)
were seeded on 6-well plates and cells at 80% confluence were
treated in triplicate with vehicle or rosiglitazone (10, 20, 30 and
40 µM) for 3–24 h at 37°C. A total of 2 h prior to harvesting,
cells were pulsed with 1 µC 1 mCi/ml 3H-thymidine (Merck
KGaA) at 37°C for 30 min. At each time point of harvesting, cells
were washed with cold PBS buffer, and then fixed with cold 10%
(w/v) trichloroacetic acid (three times; firstly for 10 min, then 5
min twice). Next, cell lysis was performed at room temperature by
adding 1 ml lysis buffer (0.3 N NaOH, 1% SDS) for 15 min. Following
cell lysis, the lysates were transferred to scintillation vials,
then mixed with 5 ml scintillation liquid to measure radioactivity,
which was later normalized to protein concentration (15).
Flow cytometric analysis of cell cycle
and apoptosis
A431 cells (density, 5×104 cells/well)
were seeded on 6-well plates and, at 80% confluence, cells were
treated with the rosiglitazone (10, 20, 30 and 40 µM) for 24 h at
37°C. For cell cycle analysis, cells were fixed at 4°C with 70%
ethanol overnight and stained with 500 µl propidium iodide (PI; 40
mg/ml) at room temperature for 30 min, then analyzed by flow
cytometry (BD FACSAria Cell Sorter; BD Biosciences, Franklin Lakes,
NJ, USA). For apoptosis analysis, harvested cells were resuspended
in 400 µl binding buffer (with Ca2+; Thermo Fisher
Scientific, Inc.) and divided into two tubes; one was used as the
blank group and the second one was incubated with 5 µl Annexin V in
the dark for 15 min at room temperature. A total of 10 µl PI was
then added and apoptosis was analyzed by flow cytometry
(excitation, 488 nm; emission, 515 nm). Untreated cells were used
as an additional control in addition to the DMSO control group.
Western blot analysis
A431 cells (density, 5×104 cells/well)
were seeded on 6-well plates and, at 80% confluence, cells were
treated with drugs [40 µM rosiglitazone or 10 µM GW9662 (Tocris
Bioscience, Bristol, UK)] or transfected with PPAR-γ small
interfering (si)RNAs according to a previous study (16). Then, the cells were harvested
following treatment for 24 h. Total cellular proteins were
extracted with radioimmunoprecipitation lysis buffer (Cell
Signaling Technology, Inc.) and protein concentration was measured
with the BCA Assay kit (Shanghai Shenergy Biocolor Bioscience and
Technology Company, Shanghai, China). Protein samples (20–40 µg)
were separated on 8–12% SDS-PAGE gels and then transferred onto
0.22 µm polyvinylidene fluoride membranes. Subsequent to blocking
for 1 h at room temperature with 5% non-fat milk or BSA in
Tris-buffered saline with 0.1% Tween-20, membranes were incubated
with primary antibodies against B-cell lymphoma 2 (Bcl-2; cat no.
15071; 1:1,000), Bcl-2 associated X protein (Bax; cat no. 2274;
1:1,000), cyclin D1 (cat no. 2922; 1:1,000), cyclin-dependent
kinase [(Cdk)2; cat no. 2546; 1:1,000)], Cdk4 (cat no. 12790;
1:1,000), and GAPDH (cat no. 5174; 1:1,000) (all from Cell
Signaling Technology, Inc.) overnight at 4°C. Following washing in
Tris-buffered saline with 0.1% Tween-20 4 times for 5 min each,
membranes were incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (HRP-conjugated rabbit
anti-mouse immunoglobulin G, cat no. ab6728 or HRP-conjugated goat
anti-rabbit immunoglobulin G; cat no. ab6721) (both secondary
antibodies from Abcam, Cambridge, MA, USA; dilution, 1:5,000) for 1
h at room temperature. Western blot membranes were developed using
Immobilon™ Western Chemiluminescent HRP substrate (EMD
Millipore, Billerica, MA, USA), analyzed with Gel Documentation and
Analysis system (G-Box, Syngene Europe, Cambridge, UK). The density
of the protein of interest was normalized to GAPDH with ImageJ
(version 1.46; National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Data were calculated and expressed as the mean ±
standard deviation, unless otherwise specified. For
semi-quantitative western blot analysis, the data were presented as
the mean ± standard error of the mean. To decrease the variance in
analysis, 3 sets of samples were collected from independent
experiments and triplicate western blot experiments were performed
for each set of samples. The mean of each set of samples and the
standard error of the mean of all 3 sets of samples were then
calculated and expressed. For the results of the
3H-Thymidine incorporation assay in Fig. 2, differences among distinct groups
were analyzed by two-way analysis of variance (ANOVA), in order to
examine the effects of treatment time and drug doses. If there was
a significant interaction, post hoc analysis was performed using
Dunnett's test to compare each group. For the data of the MTS
assay, flow cytometry analysis and semi-quantitative western blot
analysis, one-way ANOVA was used to analyze the effects of
different treatment followed by Dunnett's test if there was a
significant interaction. P<0.05 was considered to indicate a
statistically significant difference. All analyses were performed
using SPSS 13.0 statistical software (SPSS, Inc., Chicago, IL,
USA).
Results
Rosiglitazone inhibits cell
proliferation
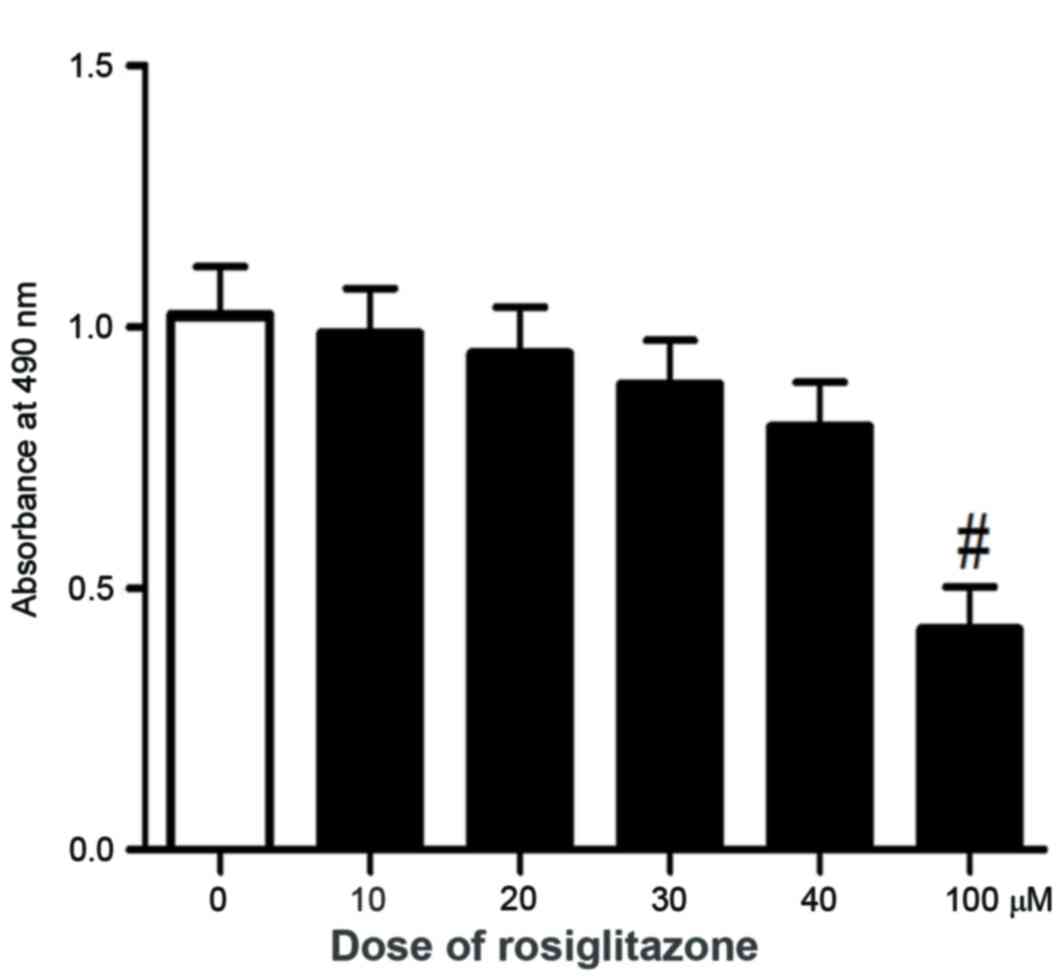
To determine the tolerated treatment concentration
of rosiglitazone, cell viability in response to different doses of
rosiglitazone was primarily examined. As demonstrated in Fig. 1, at a lower concentration of
rosiglitazone (≤40 µM), no significant effect on cell viability was
observed. With higher concentrations (60 and 80 µM) of
rosiglitazone, a decreased but not significant effect on cell
viability (data not shown) was observed. However, there was a
significant inhibition on cell viability with 100 µM rosiglitazone.
It has been demonstrated that a high concentration of rosiglitazone
exhibits side effects on normal skin cells (17). Considering the potential drug safety
issue for treatment, the experiments of the present study were
limited to low concentrations of rosiglitazone, <40 µM, to
maintain the effect of rosiglitazone on cell viability at a low
level. Firstly, to investigate the anticancer potential of
rosiglitazone in human epidermoid carcinoma, the effects of
rosiglitazone on A431 cell proliferation were evaluated. A431 cells
were cultured in Dulbecco's Modified Eagle's Medium for 24 h prior
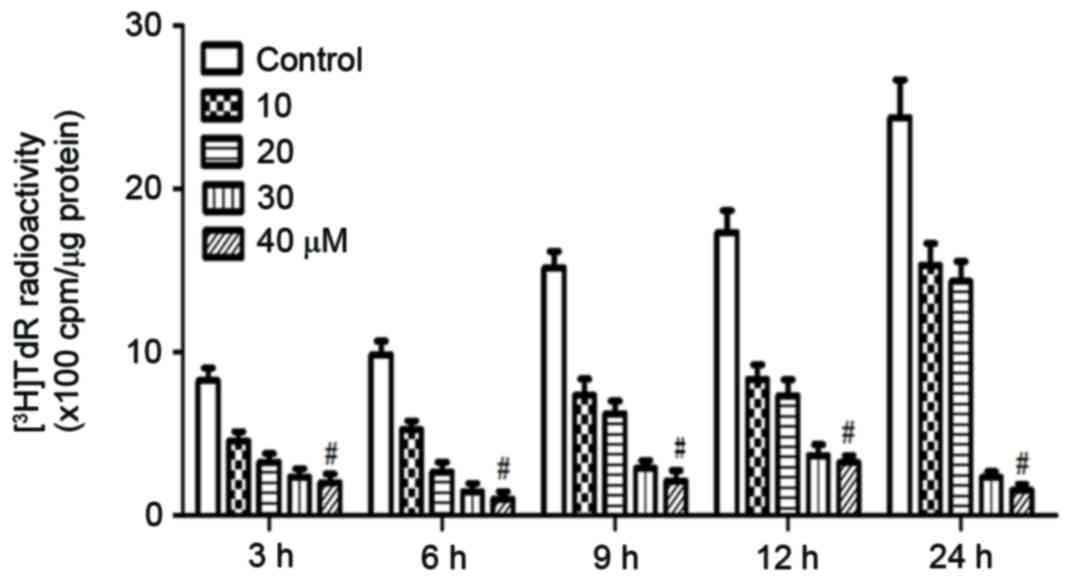
to stimulation with rosiglitazone for 3–24 h. A
3H-thymidine incorporation assay was then performed to
evaluate cell proliferation. As demonstrated in Fig. 2, at the lowest concentration and as
early as 3 h after treatment, rosiglitazone inhibited DNA synthesis
and cell proliferation markedly. This inhibitory effect was dose-
and time-dependent, with higher concentrations of rosiglitazone and
longer treatment times resulting in increased inhibitory
effects.
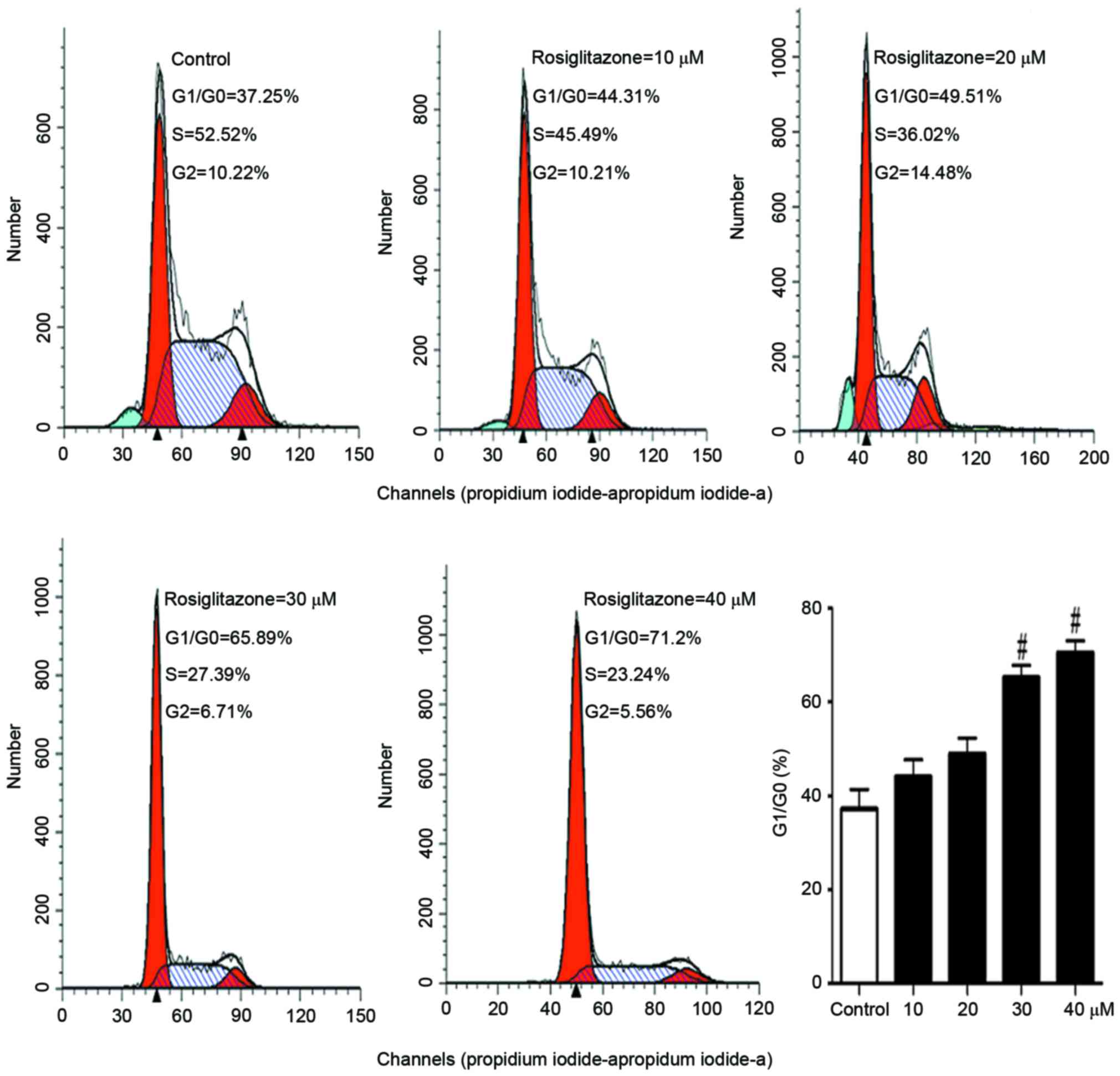
Rosiglitazone inhibits cell cycle
progression
As rosiglitazone suppressed DNA synthesis and cell
proliferation, the present study sought to examine which step of
cell cycle was affected. A431 cells were treated with different
concentrations (10–40 µM) of rosiglitazone for 24 h. Then, the cell
cycle was analyzed with flow cytometry. In Fig. 3, it was identified that with
increasing concentration of rosiglitazone, the ratio of number of
cells at G1 to number of cells at G0 phase increased, and the
number of cells at S phase decreased, indicating that rosiglitazone
inhibited cell cycle progression from G1 to S phase.
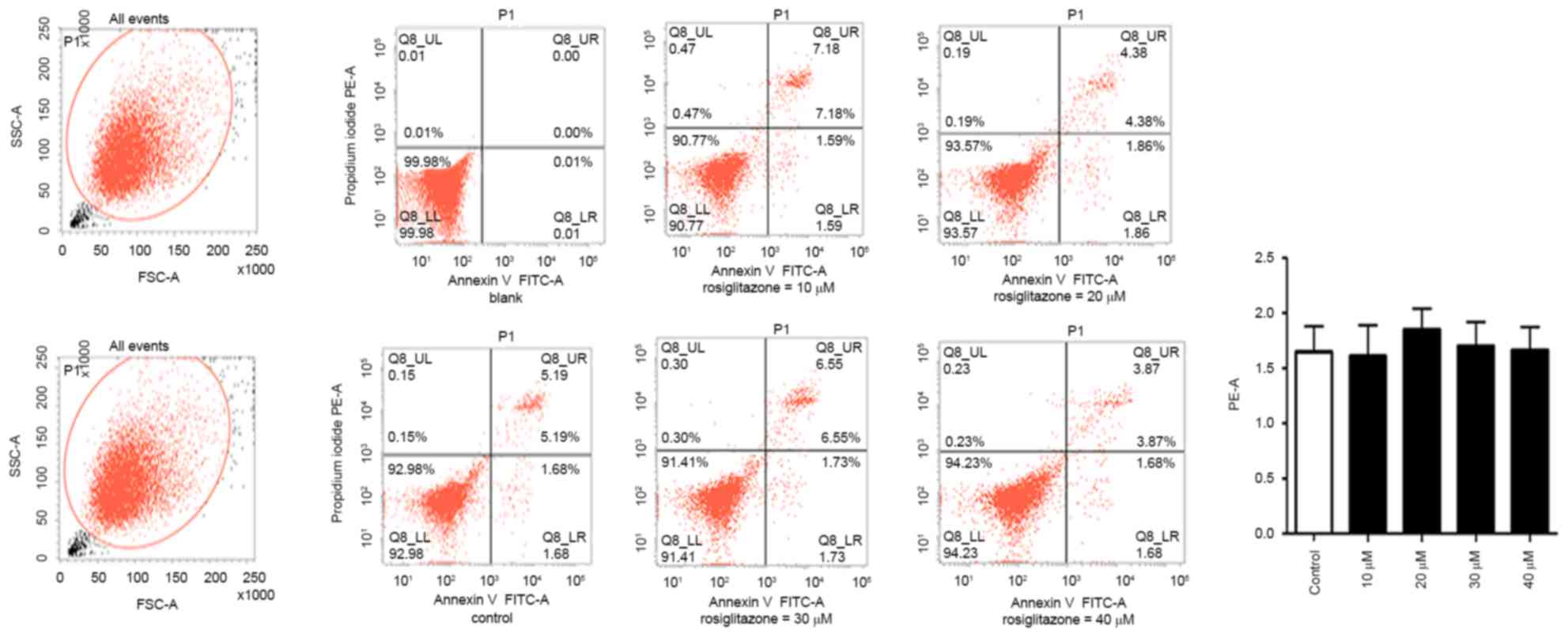
Rosiglitazone exhibits no significant
effect on cell apoptosis
The present study investigated whether there was any
association between inhibition of cell proliferation and apoptosis.
The effect of rosiglitazone on cell apoptosis in A431 cells was
examined by flow cytometry with Annexin V, a marker for apoptosis,
and PI, a DNA stain to evaluate cell viability. As indicated in
Fig. 4, with the increasing
concentration of rosiglitazone, no effect on the distribution of
A431 cells in the upper right and lower right regions was observed,
indicating that there was no increase in apoptosis due to
rosiglitazone treatment.
Rosiglitazone regulates cell
cycle-associated protein expression
To additionally understand how rosiglitazone
regulates cell proliferation, the protein expression levels of cell
cycle-associated proteins, including CyclinD1, Cdk2 and Cdk4 were
examined. Notably, it was identified that the expression levels of
these proteins decreased as the concentration of rosiglitazone
increased (Fig. 5A). However,
rosiglitazone exhibited no effects on the regulation of the ratio
of Bcl-2 to Bax, which is an indicator of cell apoptosis. In
addition, whether rosiglitazone regulated protein expression levels
of cyclin D1, Cdk2 and Cdk4 through PPAR-γ was examined. Firstly, a
selective antagonist (GW9662) was used to inhibit PPAR-γ, and it
was identified that this inhibitor rescued the agonistic effects of
rosiglitazone on the modulation of expression levels of cyclin D1,
Cdk2 and Cdk4 (Fig. 5B). Secondly,
specific knockdown of PPAR-γ also rescued the expression levels of
cyclin D1, Cdk2, Cdk4 in cells treated with rosiglitazone (Fig. 5B). However, these manipulations did
not affect the expression ratio of Bcl-2 to Bax. Taken together,
these results suggest that rosiglitazone may downregulate protein
expression levels of cyclin D1, Cdk2 and Cdk4 to suppress cell
cycle progress through PPAR-γ.
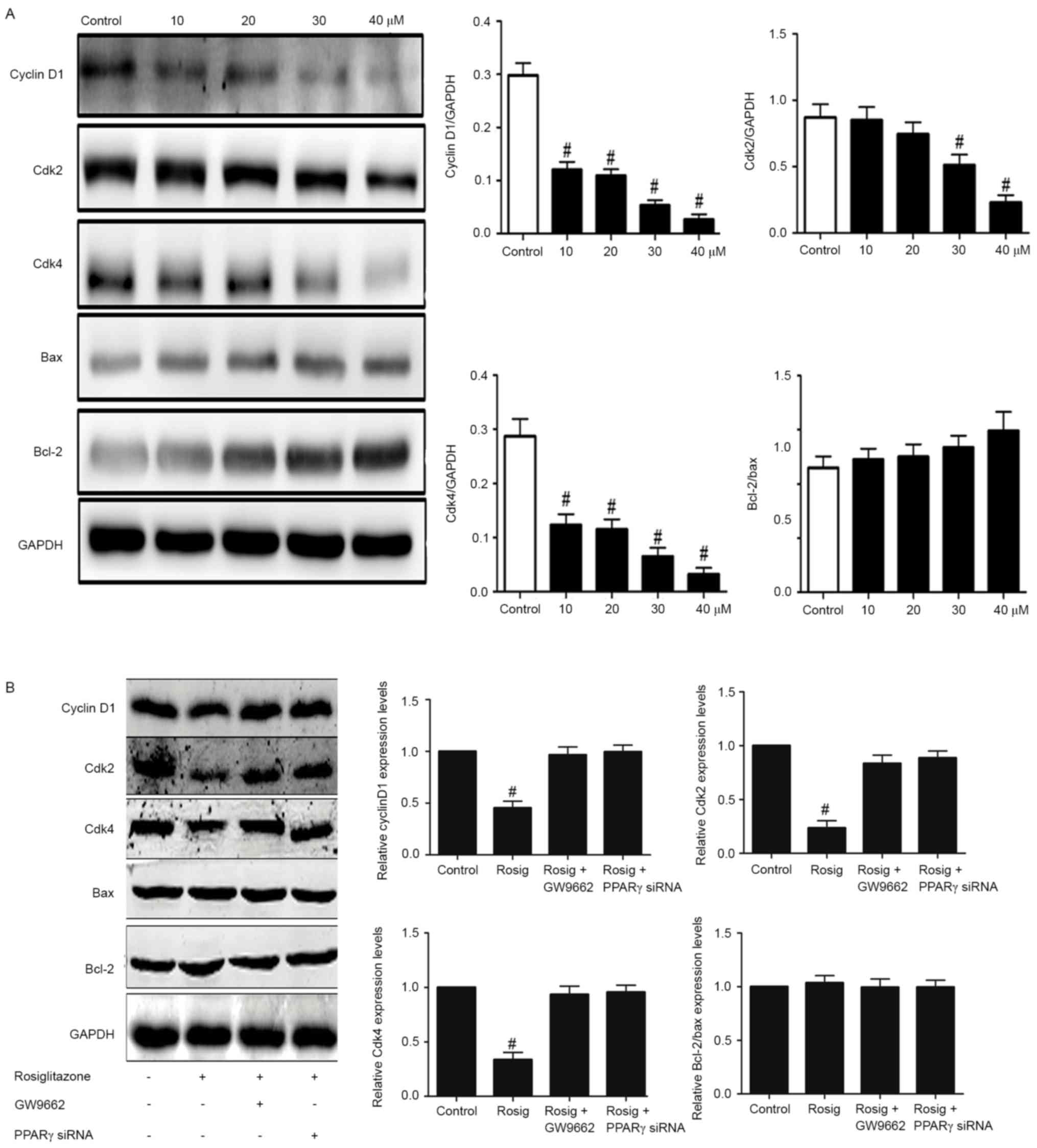 | Figure 5.Effect of rosiglitazone on the protein
level of cell cycle- and apoptosis-associated proteins. (A) A431
cells at 80% confluence were stimulated with rosiglitazone for 24
h, and the effects on Bax, Bcl-2, cyclin D1, cdk2 and cdk4
expression were analyzed by western blotting. Data were quantified
by scanning densitometry, and the ratio of the protein of interest
was normalized to GAPDH. Data are presented as means ± SEM (n=3).
#P<0.05 (10, 20, 30, 40 mM rosiglitazone treatment
compared with control treatment for cyclin D1/GAPDH and Cdk4/GAPDH;
30, 40 mM rosiglitazone treatment compared with control treatment
for Cdk2/GAPDH) as analyzed with one-way ANOVA. (B) A431 cells at
80% confluence were stimulated with rosiglitazone or GW9662 or
transfected with PPAR-γ siRNAs for 24 h and the effects on Bax,
Bcl-2, cyclin D1, cdk2 and cdk4 expression were analyzed by western
blotting. Data were quantified by scanning densitometry and the
ratio of the protein of interest was normalized to GAPDH. Data are
presented as means ± SEM (n=3). #P<0.05
(rosiglitazone vs. control; rosiglitazone vs. rosiglitazone plus
GW9662; rosiglitazone vs. rosiglitazone plus PPAR-γ siRNAs for
cyclin D1/GAPDH, Cdk2/GAPDH and Cdk4/GAPDH) as analyzed with
one-way ANOVA. ANOVA, analysis of variance; SEM, standard error of
the mean; siRNA, small interfering RNA; Cdk, cyclin-dependent
kinase; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated X protein;
Rosig, rosiglitazone; PPAR-γ, peroxisome proliferator-activated
receptor-γ. |
Discussion
PPAR-γ is known to be associated with the
development of malignancies in the breasts, colon and pancreas;
however, its effects on tumor proliferation vary in different types
of cancer (18–24). Previously, multiple studies have
indicated that activating PPAR-γ inhibits cell growth through
arrest of the cell cycle and induction of apoptosis (25–27). In
the present study, the effect of PPAR-γ agonist, rosiglitazone, on
the human epidermoid carcinoma A431 cell line was investigated. The
results demonstrated that rosiglitazone exerted an inhibitory
effect on cell proliferation through inhibiting progression from G1
to S phase in cell cycle. However, rosiglitazone did not induce
apoptosis. Consistent with this, there was a significant reduction
in cyclin D1, Cdk2 and Cdk4 protein levels. Furthermore, this
reduction was rescued with a PPAR-γ antagonist or PPAR-γ agonist
siRNAs. Therefore, these results suggest that the inhibitory
effects of rosiglitazone on cell proliferation are mediated by
downregulation of cyclin D1, Cdk2 and Cdk4 expression through the
activation of PPAR-γ.
Increased DNA synthesis activity was observed in
control cells at 9 h. Therefore, the present study hypothesized
that following rosiglitazone activation, quiescent cells re-entered
the cell cycle. However, DNA synthesis activity was similar between
6 and 3 h in the control cells, suggesting that at least 6 h was
required for these quiescent cells to reach the S phase. At an
early time-point, for example 3 h, rosiglitazone markedly inhibited
DNA synthesis. Therefore, the present study suggested that
rosiglitazone inhibits cell proliferation through inhibiting
dividing cells entering into S phase at an early time point.
Notably, rosiglitazone exhibited no effects on the
Bcl-2/Bax ratio, suggesting that apoptosis is not implicated in the
inhibitory effects of rosiglitazone. Flow cytometry data also
supported this observation, indicating that there was no difference
between the control and treated groups in apoptosis. Previous
studies have demonstrated evidence that PPAR-γ serves important
roles in anti-apoptotic pathways (12–14). In
concordance with this, the present data support that PPAR-γ
agonists inhibit cell proliferation independent of the induction of
apoptosis. However, the effects of PPAR-γ agonists on the human
epidermoid carcinoma cells require additional investigation in
vivo.
To conclude, the present study suggested evidence
that PPAR-γ agonists are implicated in the regulation of cell
growth in human epidermoid carcinoma cells. Rosiglitazone inhibited
cell proliferation of human epidermoid carcinoma. This inhibitory
effect involved the regulation of the expression of cell
cycle-associated proteins, but was independent of apoptosis. These
results provide novel insights into rosiglitazone and the
development of anticancer therapies for epidermoid carcinoma.
However, additional studies are required to improve the
understanding of the molecular and cellular mechanisms of this
potential anticancer drug. For example, it may be useful to further
investigate the mechanisms of inhibition of cell proliferation by
PPAR-γ agonists, and identify essential factors involved in the
modulation of cyclin D1, Ckd2 and Cdk4 expression that contribute
to the regulation of cell cycle arrest. Previously, a study
demonstrated that rosiglitazone activated G protein coupled
receptor 40, additionally regulating the PPAR-γ pathway (28). The present study may suggest a
potential underlying mechanism of how rosiglitazone activates
PPAR-γ to regulate the cell cycle. Furthermore, it is necessary to
investigate the effects of PPAR-γ agonists in animal models, to
confirm the data gathered in vitro. The present study aimed
to contribute to developing effective anticancer therapies by
understanding the cellular and molecular mechanisms of potential
drugs.
Acknowledgements
The present study was partly supported by the
National Natural Science Foundation of China (grant nos. 81100101
and 81270212).
Glossary
Abbreviations
Abbreviations:
|
PPAR-γ
|
peroxisome proliferator activated
receptor-γ
|
|
SCC
|
squamous cell carcinoma
|
|
SD
|
standard deviation
|
|
PI
|
propidium iodide
|
References
|
1
|
Sauter ER, Herlyn M, Liu SC, Litwin S and
Ridge JA: Prolonged response to antisense cyclin D1 in a human
squamous cancer xenograft model. Clin Cancer Res. 6:654–660.
2000.PubMed/NCBI
|
|
2
|
Trakatelli M, Ulrich C, del Marmol V,
Euvrard S, Stockfleth E and Abeni D: Epidemiology of nonmelanoma
skin cancer (NMSC) in Europe: Accurate and comparable data are
needed for effective public health monitoring and interventions. Br
J Dermatol. 156 Suppl 3:S1–S7. 2007. View Article : Google Scholar
|
|
3
|
Karia PS, Han J and Schmults CD: Cutaneous
squamous cell carcinoma: Estimated incidence of disease, nodal
metastasis, and deaths from disease in the United States, 2012. J
Am Acad Dermatol. 68:957–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dong JT: Anticancer activities of PPARγ in
breast cancer are context-dependent. Am J Pathol. 182:1972–1975.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Issemann I and Green S: Activation of a
member of the steroid hormone receptor superfamily by peroxisome
proliferators. Nature. 347:645–650. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kersten S, Desvergne B and Wahli W: Roles
of PPARs in health and disease. Nature. 405:421–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elstner E, Müller C, Koshizuka K,
Williamson EA, Park D, Asou H, Shintaku P, Said JW, Heber D and
Koeffler HP: Ligands for peroxisome proliferator-activated
receptorgamma and retinoic acid receptor inhibit growth and induce
apoptosis of human breast cancer cells in vitro and in BNX mice.
Proc Natl Acad Sci USA. 95:8806–8811. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sarraf P, Mueller E, Jones D, King FJ,
DeAngelo DJ, Partridge JB, Holden SA, Chen LB, Singer S, Fletcher C
and Spiegelman BM: Differentiation and reversal of malignant
changes in colon cancer through PPARgamma. Nat Med. 4:1046–1052.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Motomura W, Okumura T, Takahashi N, Obara
T and Kohgo Y: Activation of peroxisome proliferator-activated
receptor gamma by troglitazone inhibits cell growth through the
increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer
Res. 60:5558–5564. 2000.PubMed/NCBI
|
|
10
|
Inoue K, Kawahito Y, Tsubouchi Y, Kohno M,
Yoshimura R, Yoshikawa T and Sano H: Expression of peroxisome
proliferator-activated receptor gamma in renal cell carcinoma and
growth inhibition by its agonists. Biochem Biophys Res Commun.
287:727–732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nicol CJ, Yoon M, Ward JM, Yamashita M,
Fukamachi K, Peters JM and Gonzalez FJ: PPARgamma influences
susceptibility to DMBA-induced mammary, ovarian and skin
carcinogenesis. Carcinogenesis. 25:1747–1755. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao-Qiang M, Fowler AJ, Schmuth M, Lau P,
Chang S, Brown BE, Moser AH, Michalik L, Desvergne B, Wahli W, et
al: Peroxisome-proliferator-activated receptor (PPAR)-gamma
activation stimulates keratinocyte differentiation. J Invest
Dermatol. 123:305–312. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kundoor V, Zhang X, Bommareddy A, Khalifa
S, Fahmy H and Dwivedi C: Chemopreventive effects of sarcotriol on
ultraviolet B-induced skin tumor development in SKH-1 hairless
mice. Mar Drugs. 5:197–207. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sarfaraz S, Adhami VM, Syed DN, Afaq F and
Mukhtar H: Cannabinoids for cancer treatment: Progress and promise.
Cancer Res. 68:339–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He G, Thuillier P and Fischer SM:
Troglitazone inhibits cyclin D1 expression and cell cycling
independently of PPARgamma in normal mouse skin keratinocytes. J
Invest Dermatol. 123:1110–1119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen Z, He P, Ding X, Huang Y, Gu H and Ni
X: PPARγ stimulates expression of L-type amino acid and taurine
transporters in human placentas: The evidence of PPARγ regulating
fetal growth. Sci Rep. 5:126502015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu M, Melichian DS, Chang E,
Warner-Blankenship M, Ghosh AK and Varga J: Rosiglitazone abrogates
bleomycin-induced scleroderma and blocks profibrotic responses
through peroxisome proliferator-activated receptor-gamma. Am J
Pathol. 174:519–533. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mueller E, Sarraf P, Tontonoz P, Evans RM,
Martin KJ, Zhang M, Fletcher C, Singer S and Spiegelman BM:
Terminal differentiation of human breast cancer through PPAR gamma.
Mol Cell. 1:465–470. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang WG, Redfern A, Bryce RP and Mansel
RE: Peroxisome proliferator activated receptor-gamma (PPAR-gamma)
mediates the action of gamma linolenic acid in breast cancer cells.
Prostaglandins Leukot Essent Fatty Acids. 62:119–127. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Youssef J and Badr M: Peroxisome
proliferator-activated receptors and cancer: Challenges and
opportunities. Br J Pharmacol. 164:68–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitamura S, Miyazaki Y, Shinomura Y, Kondo
S, Kanayama S and Matsuzawa Y: Peroxisome proliferator-activated
receptor gamma induces growth arrest and differentiation markers of
human colon cancer cells. Jpn J Cancer Res. 90:75–80. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsujie M, Nakamori S, Okami J, Hayashi N,
Hiraoka N, Nagano H, Dono K, Umeshita K, Sakon M and Monden M:
Thiazolidinediones inhibit growth of gastrointestinal, biliary, and
pancreatic adenocarcinoma cells through activation of the
peroxisome proliferator-activated receptor gamma/retinoid X
receptor alpha pathway. Exp Cell Res. 289:143–151. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Itami A, Watanabe G, Shimada Y, Hashimoto
Y, Kawamura J, Kato M, Hosotani R and Imamura M: Ligands for
peroxisome proliferator-activated receptor gamma inhibit growth of
pancreatic cancers both in vitro and in vivo. Int J Cancer.
94:370–376. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kawa S, Nikaido T, Unno H, Usuda N,
Nakayama K and Kiyosawa K: Growth inhibition and differentiation of
pancreatic cancer cell lines by PPAR gamma ligand troglitazone.
Pancreas. 24:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Altiok S, Xu M and Spiegelman BM:
PPARgamma induces cell cycle withdrawal: Inhibition of E2F/DP
DNA-binding activity via down-regulation of PP2A. Genes Dev.
11:1987–1998. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ramachandran L, Manu KA, Shanmugam MK, Li
F, Siveen KS, Vali S, Kapoor S, Abbasi T, Surana R, Smoot DT, et
al: Isorhamnetin inhibits proliferation and invasion and induces
apoptosis through the modulation of peroxisome
proliferator-activated receptor γ activation pathway in gastric
cancer. J Biol Chem. 287:38028–38040. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee NJ, Oh JH, Ban JO, Shim JH, Lee HP,
Jung JK, Ahn BW, Yoon DY, Han SB, Ham YW and Hong JT:
4-O-methylhonokiol, a PPARγ agonist, inhibits prostate tumour
growth: p21-mediated suppression of NF-κB activity. Br J Pharmacol.
168:1133–1145. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang S, Awad KS, Elinoff JM, Dougherty EJ,
Ferreyra GA, Wang JY, Cai R, Sun J, Ptasinska A and Danner RL: G
protein-coupled receptor 40 (GPR40) and peroxisome
proliferator-activated receptor γ (PPARγ): AN integrated
two-receptor signaling pathway. J Biol Chem. 290:19544–19557. 2015.
View Article : Google Scholar : PubMed/NCBI
|