Introduction
Lung cancer is a malignant type of cancer with a
high incidence rate in China; it is primarily classified into two
groups, small cell lung carcinoma and non-small cell lung carcinoma
(NSCLC). Approximately 80% of patients with lung cancer have NSCLC
(1,2),
furthermore, >50% of these patients present with advanced local
invasion and distant metastasis (3).
Although chemotherapy is the principal treatment modality for the
majority of patients, it is associated with a series of detrimental
side effects, including suppression of the medulla oblongata,
impaired immune function and toxicity in other organs (4–6).
Therefore, improved therapeutic strategies and novel drug targets
for NSCLC are required.
Autophagy is an important catabolic cellular
homeostatic process; its mechanism involves the degradation of
abnormal or dysfunctional cellular components resulting from
digestion in lysosomes, which is associated with survival,
differentiation and development in the normal physiology of cells
(7). A number of studies have
previously reported that autophagy exerts dynamic effects,
including the promotion of apoptosis and the inhibition of
proliferation in tumor cells (8–10). During
the initiation of autophagy, Beclin-1 is able to promote LC3 to
convert it to LC3-II, which is recruited to the major markers
closely associated with autophagy are LC3 and p62. P62 binds to
autophagosomal membrane and has been widely used as a protein
marker to indicate the occurrence of autophagy. Two LC3 via the LC3
interacting region domain and is then degraded during the autophagy
process. Thus, the conversion of LC3 I to LC3 II and clearance of
p62 are considered hallmarks of the autophagic flux (11,12).
However, the underlying molecular mechanisms of autophagy involved
in cancer occurrence and development remain unresolved.
Therefore, drugs targeting autophagy may serve as a
therapeutic strategy for patients with NSCLC. It has been reported
that Cantharidin (CTD), an active chemical compound isolated from
the blister beetle (Coleoptera: Meloidae), serves a notable role in
promoting autophagy and suppressing hepatocellular carcinoma
(13). Consequently, one aim of the
present study was to investigate the association between CTD and
autophagy. A further aim of present study was to characterize the
antitumor effect of CTD, which mediated the inhibition of
metastasis and growth by and its possible underlying mechanism in
NSCLC using A549 cells.
Therefore, the present study aimed to investigate
the effect of CTD on NSCLC cell proliferation and metastasis and
explore the potential molecular mechanism, which may aid in
identifying a novel agent for NSCLC therapy.
Materials and methods
Chemicals and antibodies
CTD was purchased from MedChemExpress (Monmouth
Junction, NJ, USA). Primary antibodies against rabbit active
caspase-3 (#9661), rabbit RAC serine/threonine-protein kinase (AKT;
#9272), rabbit phosphorylated-(p-)AKT: (#9271), rabbit mechanistic
target of rapamycin (mTOR; #2972), rabbit p-mTOR: (#2971), rabbit
phosphorylated p-ribosomal p70S6 protein kinase (p-p70S6K; #9209)
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). Primary antibodies against rabbit B cell lymphoma (Bcl)-2
(12789-1-AP), rabbit Bcl-2-associated X protein (Bax; 50599-2-Ig),
mouse cyclin D1 (60186-1-Ig), rabbit Microtubule-associated protein
1A/1B-light chain 3 (LC3; 14600-1-AP), rabbit Beclin-1
(11306-1-AP), rabbit p62 (18420-1-AP), mouse GAPDH (60004-1-Ig),
anti-rabbit (15134-1-AP) or anti-mouse (30000-0-AP) IgG-horseradish
peroxidase-conjugated antibodies and the enhanced chemiluminescence
(ECL) detection system were purchased from ProteinTech Group, Inc.
(Wuhan, China).
Cell culture
The human lung cancer A549 cell line was purchased
from the Institute of Biochemistry and Cell Biology (Shanghai,
China) and were cultured at 37°C in 5% CO2 in RPMI-1640
medium supplemented with 10% FBS (both GE Healthcare Bio-Sciences,
Pittsburgh, PA, USA), 100 U/ml penicillin G and 100 µg/ml
streptomycin sulfate (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). Experiments were conducted with cells in the logarithmic
growth phase (0.5–1×106 cells/ml). The experimental
groups were treated with 1 µM CTD, which was in accordance with an
effective minimum concentration as investigated by previous studies
(14,15); the negative control group (NC) was
cultured with 1% dimethyl sulfoxide (DMSO) culture media.
Cell proliferation assay
Cell proliferation was evaluated using the Cell
Counting kit-8 (CCK-8) (Beijing Solarbio Science and Technology
Co., Ltd., China) in accordance with the manufacturer's protocol. A
total of 1×103 cells/well were seeded into 96-well
plates for 24 h and then were incubated with CTD (1 µM) and a
control group (1% DMSO in culture RPMI-1640 medium) for 24, 48 or
72 h. Absorbance (optical density) of viable cells was measured at
a wavelength of 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Cell migration and invasion assay
Following treatment of cells with CTD for 48 h, cell
migration and invasion were evaluated using 24-well transwell
chambers (EMD Millipore, Billerica, MA, USA), in the presence
(invasion assay) and absence (migration assay) of Matrigel matrix
(BD Biosciences, Franklin Lakes, NJ, USA), according to the
manufacturer's protocol. A total of 1×105 cells were
resuspended in 100 µl serum-free RPMI-1640 medium and seeded in the
upper chambers. The lower chambers were filled with 500 µl
RPMI-1640 medium with 10% FBS (GE Healthcare Life Sciences,
Shanghai, China). Following incubation at 37°C for 24 h,
non-invading cells were removed using a cotton-tipped swab. The
cells that permeated through the membrane to the bottom chamber
were fixed at room temperature in 4% paraformaldehyde for 10 min
and then stained with 0.5% crystal violet for 15 min at room
temperature. The number of invaded or migrated cells was quantified
by counting five random fields for each membrane under an inverted
microscope (Olympus, Tokyo, Japan) with 10×20 magnification and the
average number of cells per field was calculated.
Cell apoptosis analysis
An Annexin V-Fluorescein Isothiocyanate
(FITC)/propidium iodide (PI) Apoptosis Detection kit (Beijing
Chemclin Biotech Co. Ltd., Beijing, China) was used to detect
apoptosis according to the manufacturer's protocol. Cells were
incubated with 1 µM CTD at 37°C for 24 h and then changed to a
culture with serum-free RPMI-1640 medium for 24 h and cells were
harvested by trypsinization without EDTA (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Cells were resuspended in 1X
binding buffer at 1–5×106 cells/ml. A total of 5 µl
annexin V-FITC was added to 100 µl cell suspension and incubated in
the dark for 5 min at room temperature. Following this, 10 µl PI
and 500 µl PBS were added. The samples were then analyzed using a
FACSCalibur cytometer (BD Biosciences) within 1 h. CellQuest Pro
(version 5.1; BD Biosciences) was used for data analysis.
Western blot analysis
CTD groups and NC groups of A549 cells were
incubated with their respective treatments for 24 h. Total protein
was extracted from cells with radioimmunoprecipitation assay buffer
(Cell Signaling Technology) containing protease and phosphatase
inhibitor cocktails (Sigma-Aldrich; Merck KGaA) and the BCA Protein
assay kit (Beijing ComWin Biotech Co., Ltd, Beijing, China) was
used to determine the protein concentration. Equal quantities of
proteins (20 µg) were separated by 8–12% SDS-PAGE and transferred
to a polyvinylidene difluoride membrane (EMD Millipore) for
immunoblotting analysis. Then, membranes were blocked with 5%
non-fat milk at room temperature for 1.5 h. The membranes were
subsequently incubated with primary antibodies against cyclin D1
(dilution 1:1,000), p-p70S6K (dilution 1:1,000 dilution), Bcl-2
(dilution 1:1,000), Bax (dilution 1:1,000), active caspase-3
(dilution 1:1,000), LC3 (dilution 1:1,000), Beclin-1 (dilution
1:1,000), p62 (dilution 1:1,000) and GAPDH (for reference; dilution
1:5,000) (ProteinTech Group, Inc., Wuhan, Sanying, China) at 4°C
overnight followed by the aforementioned horseradish
peroxidase-conjugated secondary antibodies (dilution 1:5,000) at
room temperature in darkness for 2 h and developed with the
aforementioned ECL method (ProteinTech Group, Inc.) in accordance
with the manufacturers protocol. Densitometry analysis was
performed using Image-Pro Plus 6.0 software (Media Cybernetics Inc.
Rockville, MD, USA).
Statistical analysis
SPSS 22.0 (IBM Corp., Armonk, NY, USA) and GraphPad
Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA) were used to
perform statistical analysis. Data are expressed as the mean ±
standard deviation. The differences between the two groups were
analyzed by Student's t-test. All experiments were performed in
triplicate. P<0.05 was considered to indicate a statistically
significant difference.
Results
CTD attenuates the growth, invasion
and migration of A549 cells
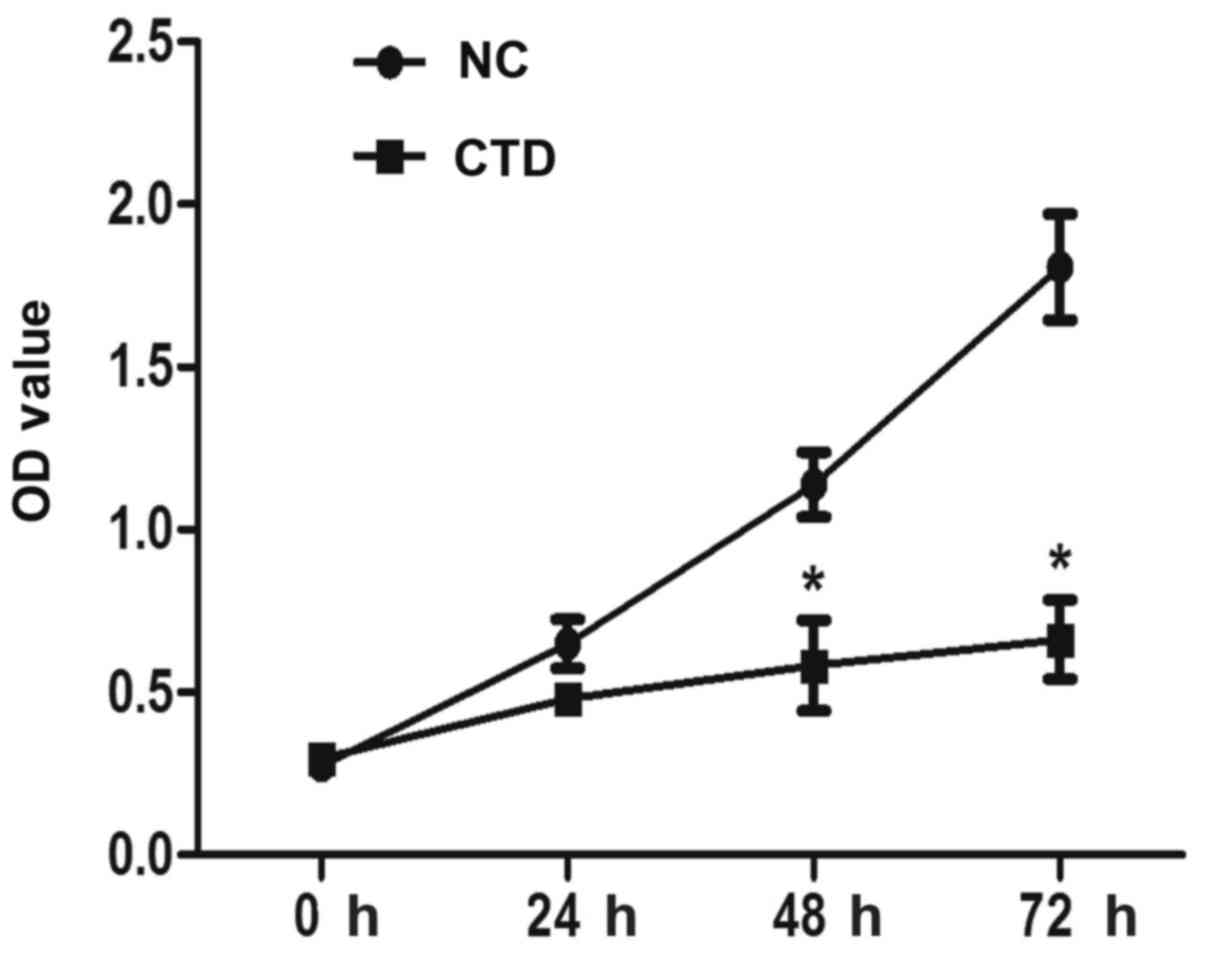
The effect of CTD on proliferation of A549 cells was
determined by CCK-8 assay. Results demonstrated that CTD
significantly inhibited the proliferation of A549 cells at 48 and
72 h, following treatment with 1 µM CTD (Fig. 1, P<0.05).
To confirm the effect of CTD on the migration and
invasion of A549 cells, transwell invasion and migration assays
were conducted. As Fig. 2A depicts,
the number of invading cells was significantly reduced following
treatment with 1 µM CTD. The corresponding cell numbers for A549
cells were 86±6 and 29±2 in the NC and CTD groups, respectively
(Fig. 2B, P<0.05), indicating the
obstruction of invasive ability of A549 cells by CTD. A migration
assay reveled similar results to the invasion assay, as the number
of migrated A549 cells was also significantly suppressed following
CTD treatment (18±5), when compared with the NC groups (32±2)
(Fig. 2C and D; P<0.05).
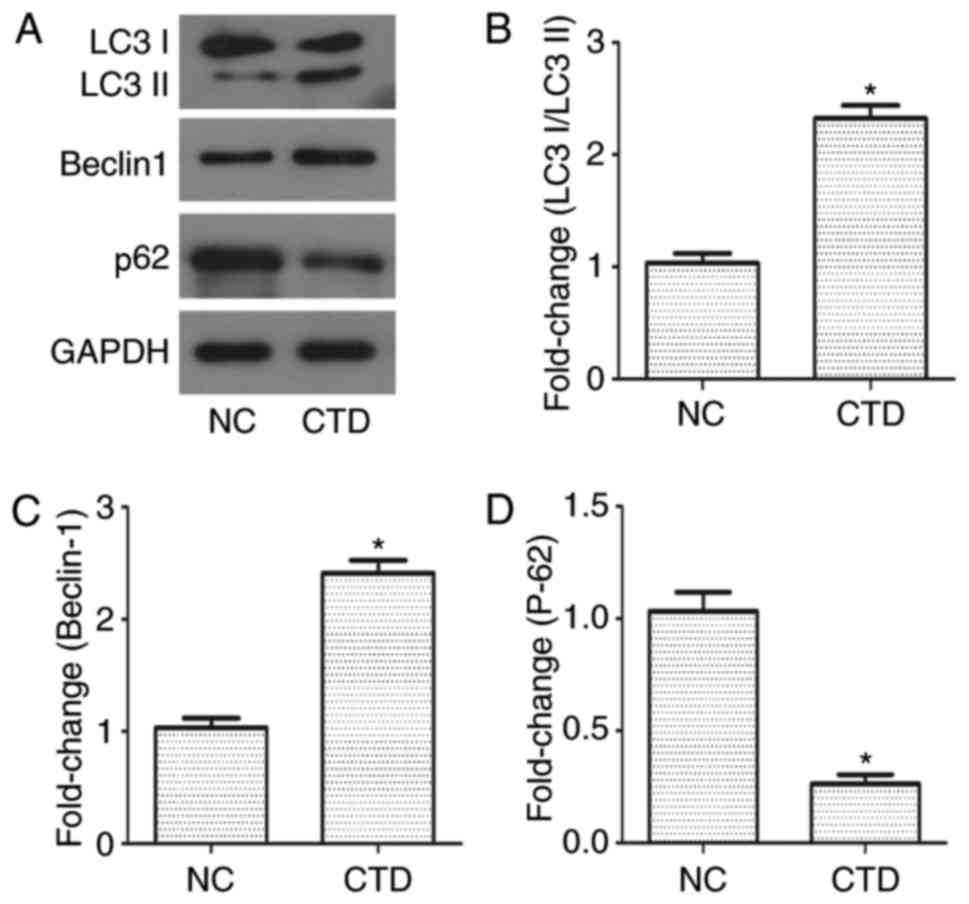
CTD promotes A549 cell autophagy
To investigate whether there was an association
between autophagy and the effects of CTD in A549 cells, western
blotting was performed. The results demonstrated that there was a
significant increase in the expression of LC3 I/LC3 II
(<2.3-fold) and Beclin-1 (<2.5-fold), and a decrease in the
expression of p62 in CTD-treated A549 cells (<0.3-fold), when
compared with the control group (Fig.
3; P<0.05). These results indicated that autophagy may be
involved in the inhibiting role of CTD in A549 cell migration and
growth.
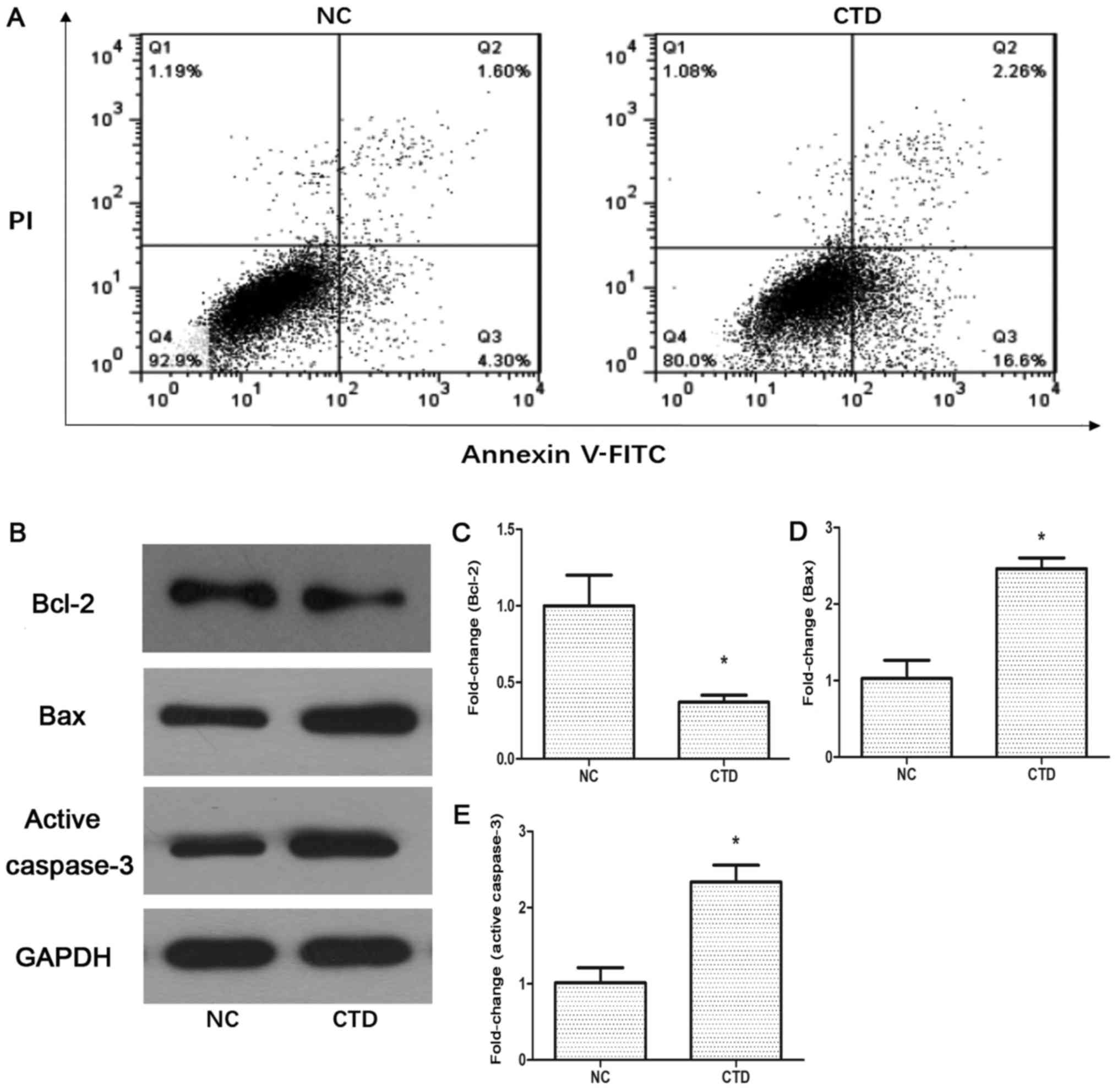
CTD induces A549 cell apoptosis
To demonstrate the role of CTD in apoptosis further,
A549 cells were treated with 1 µM CTD and underwent flow cytometry
analysis using annexin-V/PI. As demonstrated in Fig. 4A, the proportion of A549 cells
undergoing apoptosis significantly increased in the CTD-treated
group (18.8±0.5%) compared with the NC group (5.9±0.3%)
(P<0.05). Western blot analysis was then used to detect any
changes in the levels of apoptosis-associated proteins. The results
demonstrated that the expression of Bcl-2 was significantly
decreased (<0.4-fold) in the CTD-treated groups (Fig. 4B and C; P<0.05), whereas the
expression level of Bax was upregulated (<2.5-fold; Fig. 4B and D; P<0.05). The level of
active caspase-3, a paramount cleavage enzyme associated with the
intrinsic and extrinsic apoptosis pathways, was also measured by
western blotting. The results demonstrated that the expression
level of active caspase-3 was increased (<2.2-fold) following
CTD treatment (Fig. 4B and E;
P<0.05). Taken together these results indicated that CTD may
activate apoptotic pathways in A549 cells.
CTD inhibits activity of
phosphatidylinositol 3-kinase (PI3K) signaling in A549 cells
Previous studies have demonstrated that the
PI3K/Akt/mTOR pathway has a marked effect on tumor growth and
survival (16,17). To confirm whether the PI3K/Akt/mTOR
pathway was associated with the role of CTD in A549 cells, the
present study examined the changes in the levels of total and
p-Akt, and p-mTOR (Fig. 5A-E). The
downstream factors p-p70S6K and cyclin D1 of the PI3K/Akt/mTOR
pathway were also examined (Fig. 5F and
5G). Treatment with CTD led to the markedly decreased
expression of p-Akt (<0.7-fold), p-mTOR (<0.6-fold), p-p70S6K
(<0.3-fold) and cyclin D1 (<0.2-fold) levels in A549 cells
(Fig 5B, D and G). These results
demonstrated that CTD might inhibit the proliferation and
metastasis of A549 cells via the PI3K/AKT/mTOR signaling
pathway.
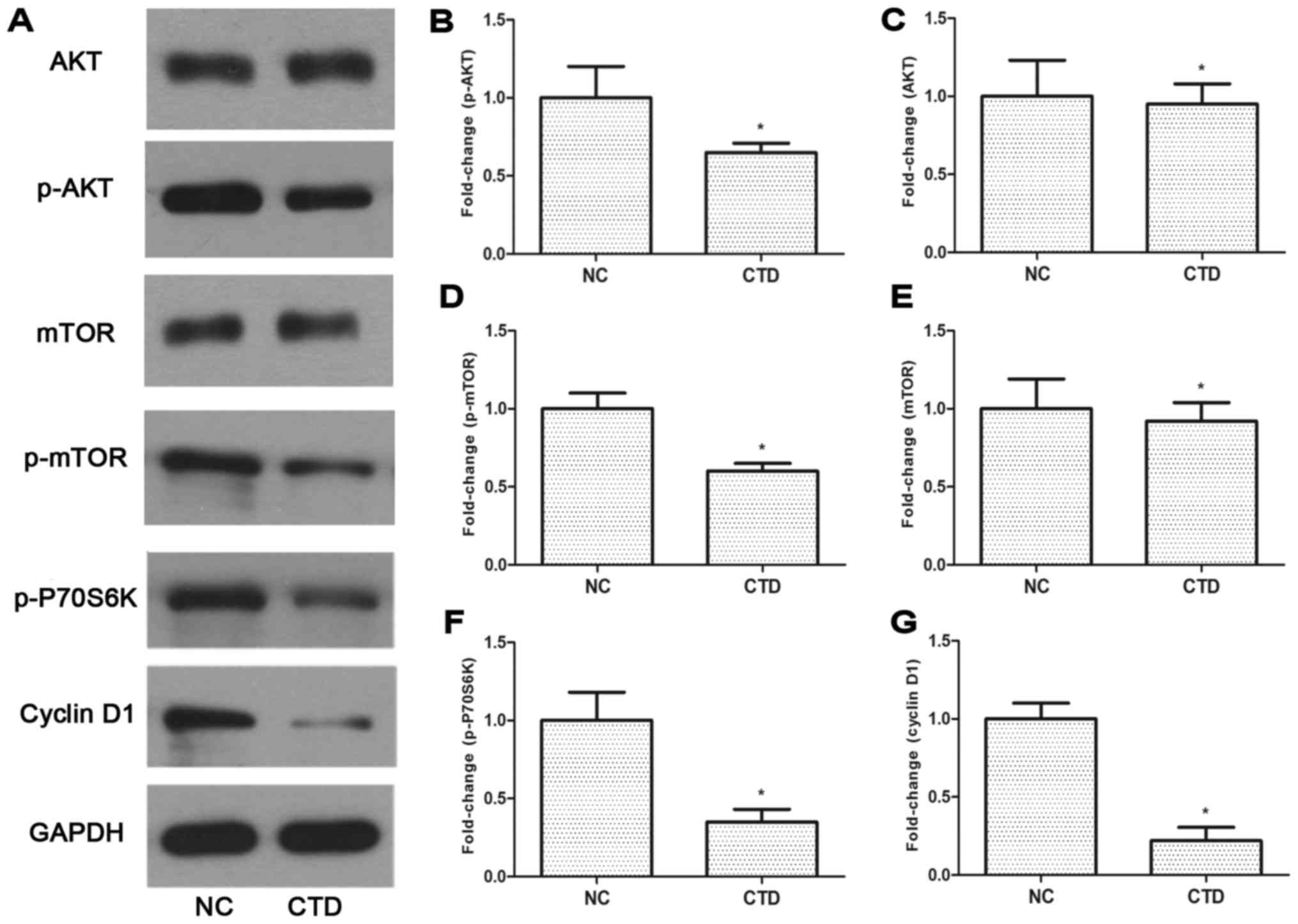 | Figure 5.Effect of cantharidin on the relative
protein levels of PI3K/Akt/mTOR signaling molecules in A549 cells.
(A-G) Western blotting detected proteins involved in the
PI3K/Akt/mTOR signaling pathway, including expression (B) p-Akt,
(C) Akt, (D) mTOR, (E) p-mTOR, (F) p-p70S6K and (G) cyclin D1.
*P<0.05, compared with NC groups. PI3K, phosphoinositide
3-kinase; p-Akt, phosphorylated RAC serine/threonine protein
kinase; mTOR, mechanistic target of rapamycin; p70S6K, p7-S6
kinase; CTD, cantharidin; NC, negative control. |
Discussion
In the present study, CTD was identified to
significantly suppress the proliferation, migration and invasion of
A549 cells. It was also observed that CTD may potentiate A549 cell
autophagy and apoptosis. Furthermore, the present study provided
evidence to indicate that CTD may exert its effects via inhibition
of the PI3K/Akt/mTOR pathway.
Previous studies have reported that CTD efficiently
inhibited proliferation and induced apoptosis in various cancer
cells, including oral squamous cell carcinoma, renal carcinoma and
gastric cancer cells (18–20). The results of the present study also
demonstrated that CTD inhibited A549 cell proliferation and
migration, which is consistent with the results of previous studies
(18–20). The combined results of these studies
provide evidence that CTD is a promising therapeutic candidate for
the treatment of NSCLC.
A number of studies have reported that CTD exerted
an inhibitory effect on cell proliferation by inducing apoptosis in
a variety of tumor cells, including gastric, colorectal and
pancreatic cancer cell lines (20–22). The
results of the present study also demonstrated that CTD had a
similar effect in promoting the apoptosis of NSCLC cells, by
detecting levels of apoptotic markers, including anti-apoptosis
protein Bcl-2 and pro-apoptosis proteins like Bax and active
caspase-3. Based on the background that autophagy is a
survival-promoting process that captures, degrades, and recycles
intracellular constituents in lysosomes and is considered to serve
a distinct role in the suppression of tumorigenesis and promotion
of mortality (23,24), to investigate the underlying
mechanism, autophagy markers were detected, namely, LC3, p62 and
Beclin 1. The results revealed that CTD also exhibited an ability
to induce autophagy. Previous studies have demonstrated that
autophagy is able to accelerate the incidence of apoptosis and
suppress tumor cell multiplication and growth (25,26), which
is line with the present results. The pathways involved in
autophagy progression are complicated. The PI3K/Akt/mTOR signaling
pathway is closely associated with tumor growth and survival, was
implicated to be involved in suppressing autophagy (26). It is established that PI3K is regarded
as a key regulator in various essential cellular processes,
including cell survival, growth, and differentiation. Once PI3K is
activated, its catalytic subunit activates AKT by phosphorylating
AKT and successively activates mTOR by phosphorylating mTOR. The
activation of mTOR leads to phosphorylation of p70S6K1, a mediator
of protein translation and cell growth (27–29). It
has also been demonstrated that Cyclin D1, a PI3K/AKT pathway
downstream factor, is associated with abnormal proliferation,
invasion, and thus prognosis of cancer (30). In the present study, western blot
analysis revealed that the PI3K/Akt/mTOR signaling pathway was
inhibited in CTD-treated A549 cells, which may have enhanced
autophagy in addition to its induction of apoptosis. However, there
remained certain limitations of the study. Only one cell line and
concentration of CTD were examined and no animal models were used.
Future studies may focus on using an expanded variety of cell
lines, or developing animal models.
In conclusion, the results of the present study
indicate that CTD impeded cell growth and migration by promoting
autophagy and apoptosis, which may be regulated by inhibiting the
PI3K/Akt/mTOR signaling pathway in NSCLC. Therefore, this work
provides a novel insight that CTD may serve as a potential
candidate for development of a naturally derived antitumor
agent.
References
|
1
|
Siegel R, DeSantis C, Virgo K, Stein K,
Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al:
Cancer treatment and survivorship statistics. CA Cancer J Clin.
64:220–241. 2012. View Article : Google Scholar
|
|
2
|
Blackhall F and Thatcher N: Chemotherapy
for advanced lung cancer. Eur J Cancer. 40:2345–2348. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grilli R, Oxman AD and Julian JA:
Chemotherapy for advanced non-small-cell lung cancer: How much
benefit is enough? J Clin Oncol. 11:1866–1872. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwamoto T: Clinical application of drug
delivery systems in cancer chemotherapy: Review of the efficacy and
side effects of approved drugs. Biol Pharm Bull. 36:715–718. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Olaussen KA and Postel-Vinay S: Predictors
of chemotherapy efficacy in non-small-cell lung cancer: A
challenging landscape. Ann Oncol. 27:2004–2016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamamoto Y and Iwase H: [Management for
treatment-induced adverse reaction-chemotherapy]. Nihon Rinsho. 70
Suppl 7:S672–S676. 2012.
|
|
7
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Duffy A, Le J, Sausville E and Emadi A:
Autophagy modulation: A target for cancer treatment development.
Cancer Chemother Pharmacol. 75:439–447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li C, Wang Y, Wang C, Yi X, Li M and He X:
Anticancer activities of harmine by inducing a pro-death autophagy
and apoptosis in human gastric cancer cells. Phytomedicine.
28:10–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang B, Lu D, Xuan M and Hu W: Antitumor
effect of sunitinib in human prostate cancer cells functions via
autophagy. Exp Ther Med. 13:1285–1294. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiong X, Wu M, Zhang H, Li J, Lu B, Guo Y,
Zhou T, Guo H, Peng R, Li X, et al: Atg5 siRNA inhibits autophagy
and enhances norcantharidin-induced apoptosis in hepatocellular
carcinoma. Int J Oncol. 47:1321–1328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim YM, Ku MJ, Son YJ, Yun JM, Kim SH and
Lee SY: Anti-metastatic effect of cantharidin in A549 human lung
cancer cells. Arch Pharm Res. 36:479–484. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang WD, Zhao HR, Yan Y, Wang XH, Zong ZH
and Liu Y: Apoptosis induced by cantharidin in human pulmonary
carcinoma cells A549 and its molecular mechanisms. Zhonghua Zhong
Liu Za Zhi. 27:330–334. 2005.(In Chinese). PubMed/NCBI
|
|
16
|
Vanhaesebroeck B, Leevers SJ, Panayotou G
and Waterfield MD: Phosphoinositide 3-kinases: A conserved family
of signal transducers. Trends Biochem Sci. 22:267–272. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pellegatta F, Chierchia SL and Zocchi MR:
Functional association of platelet endothelial cell adhesion
molecule-1 and phosphoinositide 3-kinase in human neutrophils. J
Biol Chem. 273:27768–27771. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren Y, Zhang SW, Xie ZH, Xu XM, Chen LL,
Lou ZG, Weng GB and Yao XP: Cantharidin induces G2/M arrest and
triggers apoptosis in renal cell carcinoma. Mol Med Rep.
14:5614–5618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su CC, Lee KI, Chen MK, Kuo CY, Tang CH
and Liu SH: Cantharidin induced oral squamous cell carcinoma cell
apoptosis via the JNK-regulated mitochondria and endoplasmic
reticulum stress-related signaling pathways. PLoS One.
11:e01680952016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang C, Chen Z, Zhou X, Xu W, Wang G,
Tang X, Luo L, Tu J, Zhu Y, Hu W, et al: Cantharidin induces G2/M
phase arrest and apoptosis in human gastric cancer SGC-7901 and
BGC-823 cells. Oncol Lett. 8:2721–2726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu B, Gao HC, Xu JW, Cao H, Fang XD, Gao
HM and Qiao SX: Apoptosis of colorectal cancer UTC116 cells induced
by Cantharidinate. Asian Pac J Cancer Prev. 13:3705–3708. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y,
Xu Z and Han X: Cantharidin, a potent and selective PP2A inhibitor,
induces an oxidative stress-independent growth inhibition of
pancreatic cancer cells through G2/M cell-cycle arrest and
apoptosis. Cancer Sci. 101:1226–1233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anding AL and Baehrecke EH: Autophagy in
cell life and cell death. Curr Top Dev Biol. 114:67–91. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Denton D, Xu T and Kumar S: Autophagy as a
pro-death pathway. Immunol Cell Biol. 93:35–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang YX, Xu SQ, Chen XH, Liu RS and Liang
ZQ: Autophagy involvement in olanzapine-mediated cytotoxic effects
in human glioma cells. Asian Pac J Cancer Prev. 15:8107–8113. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mihalache CC, Yousefi S, Conus S, Villiger
PM, Schneider EM and Simon HU: Inflammation-associated
autophagy-related programmed necrotic death of human neutrophils
characterized by organelle fusion events. J Immunol. 186:6532–6542.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hassan B, Akcakanat A, Holder AM and
Meric-Bernstam F: Targeting the PI3-kinase/Akt/mTOR signaling
pathway. Surg Oncol Clin N Am. 22:641–664. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sheppard K, Kinross KM, Solomon B, Pearson
RB and Phillips WA: Targeting PI3 kinase/AKT/mTOR signaling in
cancer. Crit Rev Oncog. 17:69–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsuoka T and Yashiro M: The role of
PI3K/Akt/mTOR signaling in gastric carcinoma. Cancers (Basel).
6:1441–1463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alao JP: The regulation of cyclin D1
degradation: Roles in cancer development and the potential for
therapeutic invention. Mol Cancer. 6:242007. View Article : Google Scholar : PubMed/NCBI
|