Introduction
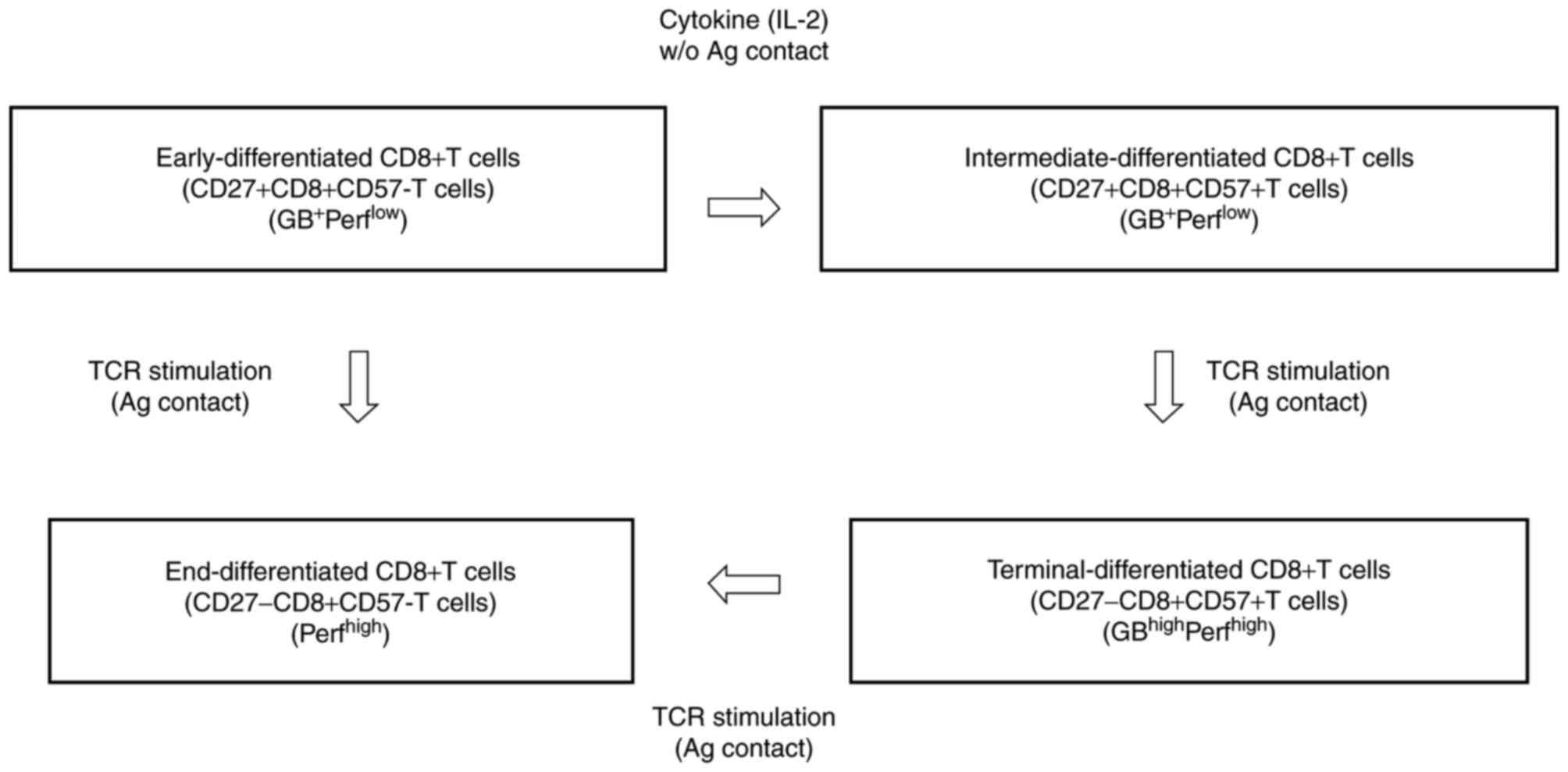
Cluster of differentiation 8 (CD8+)
cytotoxic T lymphocytes (CTLs) typically differentiate linearly,
from early-differentiated
CD27+CD8+CD57− T cells
(early-CD8+ T cells) through intermediate-differentiated
CD27+CD8+CD57+ T cells
(intermediate-CD8+ T cells) to terminally differentiated
CD27−CD8+CD57+ T cells
(terminal-CD8+ T cells) and end-differentiated
CD27−CD8+CD57− T cells
(end-CD8+ T cells) (Fig.
1) (1). Terminal-CD8+
T cells, which are the most mature effector (TMME)
cells, have been recognized as the most potent effector cells. They
express high levels of granzyme B (GB) and perforin, and may induce
effective cytotoxicity against tumor cells (Fig. 1) (2–4).
CD8+ CTL differentiation is essential for maintaining a
pool of mature CTLs in the blood (2–4). However,
differentiation of early-CD8+ T cells toward the
TMME phenotype is inhibited by transforming growth
factor-β, interleukin (IL)-10, and programmed cell death protein-1
in patients with cancer, in whom the accumulation of incompletely
differentiated early- and intermediate-CD8+ T cells
leads to uncontrolled progression of malignant cells (2–4).
A previous study reported that patients with
advanced gastric cancer with a high (>18%) CD57+ T cell
percentage in the peripheral blood had a shorter survival time
compared with those with a low percentage (<18%) (5). In this study, it was demonstrated that a
high CD57+ T cell percentage is an independent poor
prognostic factor in patients with advanced gastric carcinomas
(5). A previous report revealed that
patients with melanoma with a lower percentage (<23%) of
CD8+CD57+ T lymphocytes within the peripheral
blood CD8+ T-cell population prior to receiving adjuvant
interferon (IFN)-α2b treatment survived for longer compared with
patients with a higher percentage (>23%) of
CD8+CD57+ T cells (6); however, the opposite was observed in
patients with advanced renal cell carcinoma (7). In these patients, adjuvant IFN-α2b
therapy significantly increased the survival time of patients with
a higher percentage (>30%) of CD8+CD57+ T
cells, whereas no increase in survival was observed in those with a
lower percentage (<30%) of CD8+CD57+ T
cells (7). Thus, in patients with a
low pre-treatment percentage of CD8+CD57+ T
cells, IFN-α treatment appears to improve the level of
CD8+CD57+ T cells, as IFN-α promotes their
expansion and survival. However, in patients with a higher
pre-treatment CD8+CD57+ T cell percentage,
the level of these cells tends to decrease because IFN-α may
eliminate over-activated T cells (8).
These reports suggest that the cytotoxic subsets of
CD8+CD57+ T cells may predominate in patients
with melanoma, while the expansion of the immunosuppressive subsets
may prevail in patients with renal cell carcinoma. Wu et al
(2) proposed that there are two types
of CD8+CD57+ T cells, based on the expression
of the early effector-memory marker CD27: i) Incompletely
differentiated CD27+CD8+CD57+ T
cells that are GB+perforin−/low (poorly
cytotoxic); and ii) terminally-differentiated
CD27−CD8+CD57+ T cells that are
GBhighperforinhigh (highly cytotoxic), which
may explain the aforementioned seemingly contradictory results.
FOXP3-expressing CD8+ regulatory T cells
(CD8+ Tregs) have been reported to mediate
immunosuppression in prostate, colorectal, hepatocellular and
gastric cancer. This effect is similar to that of
FOXP3+CD4+ T cells, which share a phenotype,
functional features and mechanisms of action with
FOXP3+CD8+ T cells (9–12). By
contrast, during normal CD8+ T cell differentiation,
early-CD8+ T cells
(CD27+CD28+CD57− T cells)
transiently express FOXP3 upon T-cell receptor stimulation in
vitro, but do not necessarily acquire suppressive capabilities
(13,14). Transient expression of FOXP3 during
CD8+ T cell activation may be a mechanism for limiting
excessive immune activation and damage at the site of inflammation
(15,16). Since these reports indicated certain
roles for FOXP3 in CD8+ T cell differentiation, the
present study investigated the expression of FOXP3 in activated
autologous lymphocytes (AALs).
As induction of TMME is the most
important feature of immunotherapy using AALs (IAAL), the present
study aimed to investigate the association between CD57-associated
populations in the CD8+ T cell differentiation pathway
(early-CD8+, intermediate-CD8+,
terminal-CD8+, end-CD8+,
CD57+FOXP3+CD8+ and
CD57−FOXP3+CD8+ T cells) and IAAL
efficacy in the present study.
Patients and methods
Patients
All participants provided written informed consent
prior to enrollment in the present study. Patients with Stage IV
gastric carcinomas diagnosed according to the unified TNM
classification (17) and with a
performance status (Eastern Cooperative Oncology Group Performance
Status) between 0 and 2 were eligible for the present study. The
study protocol was approved by the Institutional Review Board at
each participating center, Tamana Regional Health Medical Center
(Tamana, Japan) and Kumamoto University (Kumamoto, Japan). All
methods and procedures associated with the present study were
conducted in accordance with the Good Clinical Practice guidelines
and accorded ethically with the principles of the Declaration of
Helsinki and local laws.
A total of 35 patients receiving IAAL at the Tamana
Regional Health Medical Center and Hakuzandori Clinic (Tokyo,
Japan) between July 2013 and March 2015 were enrolled in the
current study. The patients with stage IV gastric carcinoma (17 men
and 18 women) ranged between 41 and 84 years in age (mean ±
standard deviation, 59.4±11.0 years).
Sample collection and processing
Peripheral blood samples (50 ml) were collected from
all eligible patients prior to and following IAAL; patient
eligibility was determined by the presence of histologically and
clinically diagnosed carcinoma. Mononuclear cells were separated by
Ficoll-Hypaque density gradient centrifugation as follows: With a
sterile pipette, the Ficoll-Hypaque solution was placed into a
50-ml conical centrifuge tube, using 2 ml of Ficoll-Hypaque/1 ml
blood; anticoagulated blood was mixed with an equal volume of PBS;
the diluted blood was slowly layered over the Ficoll-Hypaque
solution by gently pipetting the diluted blood down the side of the
tube containing the Ficoll-Hypaque; samples were centrifuged for 40
min at 400 × g (22°C) with no brake; mononuclear cells, located at
the interface between the plasma (upper layer) and the
Ficoll-Hypaque (bottom) were carefully removed using a Pasteur
pipette; the aspirated mononuclear cells were transferred to a
15-ml conical tube; mononuclear cells were cultured for 2 weeks
with recombinant IL-2 (700 IU/ml; Shionogi & Co., Ltd., Osaka,
Japan) and immobilized using anti-CD3 monoclonal antibody for 2
weeks at 37°C (MAb) (cat. no. 12277, 5 µg/ml; Janssen-Kyowa;
Johnson & Johnson, New Brunswick, NJ, USA). Sixteen-well cell
cultures were performed for cells obtained from patients assigned
to immunotherapy, preserving ~2×1010 cells as the source
for the second to the fifth infusions (18); however, cultures were discontinued for
patients assigned to other treatments (18). Briefly, mononuclear cells are
separated from approximately 30 ml of peripheral blood and cultured
with immobilized OKT3 and IL-2 for 3–5 days (RPMI-1640+9 Sin and
RPMI-1640 HSR Sin, Lymphotec Inc). This step was designated the
activation step. The activation step was followed by additional
culture in a medium containing only IL-2 for 9–11 days in gas
permeable bags (CP-4 and bagpack medium, Lymphotec Inc., Tokyo,
Japan). This second step is designated the expansion step.
Lymphocytes prior to and following the in vitro culture were
phenotyped with MAbs against CD8 (RPA-T8; cat. no. 560917), CD27
(M-T271; cat. no. 557330), CD57 (NK-1; cat. no. 560844), and FOXP3
(259D/C7; cat. no. 560082) obtained from BD Biosciences (Franklin
Lake, NJ, USA). These antibodies were diluted with IsoFlow (cat.
no. 8599600) obtained from Beckman Coulter, Inc.
Samples were centrifuged at 652 × g at room
temperature for 5 min to remove the supernatant, and then suspended
in sheath solution. Antibodies (20 µl) were added to tubes in
accordance with combinations shown in Table I. A total of 1 ml of each sample was
added to each tube. Staining was performed by keeping on ice for
tubes I to VI and at room temperature for tubes I' and VII, for ~20
min. To tubes I–VI, 2 ml sheath solution was added, and tubes were
centrifuged at 652 × g at room temperature for 5 min. The
supernatant was removed, and the pellet was suspended in sheath
solution. Samples were evaluated using 3-color FACS analysis
(Lymphotec, Inc., Tokyo, Japan) according to the manufacturers
standard operating procedure.
 | Table I.Contents of each tube used in sample
processing. |
Table I.
Contents of each tube used in sample
processing.
| Tube no. | FITC | PE | PE-Cy7 |
|---|
| I | Control
(FITC/PE) |
| Control
(PE-Cy7) |
| II | CD3 | CD4 | CD56 |
| III | CD3 | CD4 | CD57 |
| IV | CD3 | CD8 | CD56 |
| V | CD3 | CD8 | CD57 |
| VI | CD57 | CD27 | CD8 |
| I' | Control
(FITC/PE) |
| Control
(PE-Cy7) |
| VII | CD57 | FoxP3 | CD8 |
The contents of tubes I' and VII were fixed by
centrifuging at 250 × g for 10 min at room temperature with 2 ml of
sheath solution. Buffer A (2 ml) was added and incubated in the
dark at room temperature for 10 min. Tubes were centrifuged again
at 500 × g for 10 min at room temperature, and the supernatant was
removed.
The viability of the final cell products was
determined using the dye exclusion test (trypan blue-exclusion
test), and possible contamination by bacteria, fungi, and
endotoxins was assessed twice (Ager medium method: Cell suspension
(500 µl) was seeded on soybean-casein digest agar medium (cat. no.
P94505R200; Nikken Biomedical Laboratory Inc., Kyoto, Japan,).
In order to maintain safety in the laboratory, a
closed system was maintained for the first 7 days of culture. The
growth of bacteria was investigated twice. First, 2 days before
culture ended, and second 2 days post-culture. Only when no growth
of bacteria was recognized were the cell products provided for
patients.
Antibodies and fluorescence activated
cell sorting (FACS)
The following reagents were used for FACS as
described below: Anti-CD3-fluorescein isothiocyanate (FITC) (HIT3a;
cat. no. 555339), anti-CD57-FITC (NK-1; cat. no. 555619),
anti-CD27-phycoerythrin (PE) (M-T271; cat. no. 557330),
anti-FOXP3-PE (259D/C7; cat. no. 560082), anti-CD56-PE (B159; cat.
no. 561903), anti-CD57-PE (NK-1 cat. no. 560844), anti-CD4-PE-Cy7
(SK3; cat. no. 560909), and anti-CD8-PE-Cy7 (RPA-T8; cat. no.
560917) obtained from BD Biosciences. Cells were analyzed on a BD
FACSCalibur 3A using BD FACS Express software (version 10.6.4; BD
Biosciences). All antibodies with the exception of anti-FOXP3 were
prediluted.
To each tube (1 ml) 2 ml sheath solution was added
and tubes were centrifuged at 250 × g at room temperature for 10
min. The cell pellet was loosened by tapping the tubes, then 2 ml
Buffer A was added and left to incubate in the dark at room
temperature for 10 min. The tubes were centrifuged at 500 × g at
room temperature for 10 min, the supernatant was removed, and the
cell pellet loosened by tapping the tubes.
Buffer C (0.5 ml) was added to each tube, and
incubated in the dark at room temperature for 30 min. Sheath
solution (2 ml) was added and tubes were centrifuged at 500 × g for
10 min to remove the supernatant. An appropriate amount (300 µl) of
sample was added to each tube. Staining was performed by keeping at
room temperature for tubes I' and VII, for about 20 min.
FoxP3(PE) antibody was added and incubated in the
dark at room temperature for 30 min. Sheath solution (2 ml) was
added and samples were centrifuged at 500 × g for 10 min to remove
the supernatant. The samples were once again suspended in sheath
solution and centrifuged as aforementioned, then suspended in 300
µl sheath solution. Samples were evaluated using 3-color FACS
analysis according to the manufacturers standard operating
procedure
Flow cytometric analysis
Two-color flow cytometry
Two- and three-color flow cytometric analyses were
performed using the following MAbs: Leu-7-FITC (cat. no. 555619)
and anti-CD3-PE for CD57+ T cells, anti-CD8-FITC and
anti-CD57-PE for CD8+CD57+ T cells, and
anti-CD3-FITC and anti-CD56-PE for CD56+ T cells (all BD
Biosciences). Flow cytometric analysis was performed using a
FACSCalibur flow cytometer (BD Biosciences). The proportions of
CD56+ T cells and CD57+ T cells were
expressed as a percentage of the total number of mononuclear
cells.
Three-color flow cytometry
A total of 1×106 cell/ml of sample was
dispensed into each tube and incubated with the aforementioned MAbs
for 20 min at 4°C (tubes I–VI) for cell surface immunostaining or
at room temperature for intracellular staining (tubes I' and VII).
A total of 2 ml IsoFlow sheath liquid (Beckman Coulter Inc., Brea,
CA, USA) was added to tubes I–IV, and the samples were centrifuged
at 2,700 × g for 5 min at room temperature. Thereafter, the
supernatant was removed and replaced with sheath liquid. For tubes
I' and VII, 2 ml sheath liquid was added, and the samples were
centrifuged at 250 × g for 10 min at room temperature. The pellets
were resuspended in 2 ml Buffer A (10-fold dilution of Human FOXP3
Buffer A from Human FoxP3 Buffer set with water; BD Biosciences)
after removing the supernatant and maintaining for 10 min at room
temperature. These samples were analyzed using a FACSCalibur 3A
flow cytometer and FACS Express software 10.6.4 (both BD
Biosciences).
Intracellular flow cytometry
For intracellular staining, cells were fixed and
permeabilized using the Human FoxP3 Buffer set, according to
manufacturer's protocol. A total of 2 ml sheath liquid was added to
each tube and centrifuged at 250 × g at room temperature for 10
min. The cell pellet was loosened by tapping the tubes, 2 ml Buffer
A was added and the tubes were incubated in the dark at room
temperature for 10 min. Tubes were centrifuged at 500 × g at room
temperature for 10 min, the supernatant was removed and the cell
pellet was loosened by tapping the tubes. Following the addition of
0.5 ml Buffer C (50-fold dilution of Human FOXP3 Buffer B with
Buffer A) to each tube, the samples were incubated for 30 min at
room temperature. Next, 2 ml sheath liquid was added, the samples
were centrifuged at 500 × g for 10 min, and the supernatant was
removed. PE-conjugated FOXP3 antibody (560082; BD Biosciences) was
added and the tubes were incubated for 30 min at room temperature.
Another 2 ml sheath liquid was added, the samples were centrifuged
at 500 × g for 10 min, the supernatant was removed and the pellets
were resuspended in 300 µl sheath liquid. These samples were
analyzed as aforementioned.
Study endpoints and assessments
The primary endpoint of this study was
progression-free survival (PFS). PFS was measured as the duration
between the date of randomization and the date of first recurrence
or mortality regardless of the cause. The secondary endpoints
included overall survival (OS), and cancer-specific survival and
safety. OS was measured as the duration between the date of
randomization and the date of mortality from any cause; and
cancer-specific survival was measured between the date of
randomization and the date of mortality resulting from cancer.
Tumor assessments were performed using dynamic computed tomography
or magnetic resonance imaging every 3 months for 24 months, and
every 3–6 months thereafter in the two groups. All scans at each
site were reviewed by two independent, blinded radiologists, each
with >5 years of experience. In cases of discordance, a third
independent experienced radiologist reviewed the images to achieve
a consensus. Adverse events (AEs) were classified and graded every
2 months according to the Common Terminology Criteria for Adverse
Events (version 3.0; National Cancer Institute, Bethesda, MD, USA)
(19) from the time that patient
treatment was started until the end of the study or drop-out,
continuing at least 30 days after the last dose of immunotherapy.
Multiple events were counted once for each patient and the most
severe event was summarized. The data collection cut-off date was
November 29, 2015.
Statistical analysis
Data are expressed as the mean ± standard deviation.
Comparison of non-normally distributed variables between groups was
performed using the Mann-Whitney U-test. Comparison of categorical
variables between two groups was performed using the χ2
test. The probability of survival was estimated using the
Kaplan-Meier method, and differences in survival rates were
evaluated by log-rank test. Multivariate analysis of prognostic
factors was conducted using the Cox regression model. All
statistical analyses were conducted using SPSS version 13.0 for
Windows (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Increased percentage of
CD8+CD57+ T cells in AALs
The mean proportion of CD57+ T cells
(51.7±2.6%), in particular the CD8+CD57+ T
cells (32.2±2.7%), was markedly increased following the incubation
of lymphocytes obtained from the peripheral blood with anti-CD3 MAb
and IL-2 when compared with the average proportions prior to
culture (30.3±2.9 and 15±1.6%, respectively; Table II). As these results suggested that
CD57+ T cells, particularly
CD8+CD57+ T cells, mediate an important role
in IAAL, all CD57-associated populations in the CD8+ T
cell differentiation pathway (early-, intermediate-, terminal-, and
end-CD8+ T cells) were investigated. In AALs, the
proportion of early-CD8+ T cells significantly decreased
following culture (20.9±2.9% vs. 41.5±2.7% prior to culture;
P<0.0001), whereas the proportion of terminal-CD8+ T
cells increased significantly following culture (35.7±3.0% vs.
21.8±2.5% prior to culture; P=0.001; Table II).
| Table II.Comparison of CD57 T cells prior to
and following culture. |
Table II.
Comparison of CD57 T cells prior to
and following culture.
| T cell
population | n | Mean ± standard
deviation | P-value |
|---|
| CD57+
T |
|
| <0.0001 |
|
Pre | 34 | 30.3±2.9 |
|
|
Post | 35 | 51.7±2.6 |
|
|
CD8+CD57+ T |
|
| <0.0001 |
|
Pre | 34 | 15.0±1.6 |
|
|
Post | 35 | 32.2±2.7 |
|
|
CD4+CD57+ T |
|
| 0.023 |
|
Pre | 34 | 11.2±1.7 |
|
|
Post | 35 | 18.1±2.4 |
|
|
Early-CD8+ T |
|
| <0.0001 |
|
Pre | 34 | 41.5±2.7 |
|
|
Post | 35 | 20.9±2.9 |
|
|
CD27−CD8+CD57+
T (terminal-CD8 |
|
| 0.001 |
|
Pre | 34 | 21.8±2.5 |
|
|
Post | 35 | 35.7±3.0 |
|
|
FOXP3+CD8+CD57−
T |
|
| 0.202 |
|
Pre | 34 | 16.6±3.0 |
|
|
Post | 35 | 21.8±2.7 |
|
|
FOXP3+CD8+CD57+
T |
|
| 0.005 |
|
Pre | 34 | 31.8±3.8 |
|
|
Post | 35 | 13.8±2.4 |
|
Previous reports revealed that
FOXP3+CD8+ T cells functioning as
CD8+ Tregs were the key prognostic factor in determining
the clinical outcome of patients with cancer (8) and these cells do not express CD57
(16); thus, the expression of CD57
on FOXP3+CD8+ T cells were analyzed. In the
present study, the percentage of
CD57+FOXP3+CD8+ T cells was
significantly decreased in AALs following culture compared with
their pre-culture percentage, whereas there were no significant
differences in the proportion of
CD57−FOXP3+CD8+ T cells (Table II).
Univariate and multivariate analysis
of CD57-associated immune cells
Cox proportional-hazards regression analysis was
conducted to determine the factors influencing the PFS rate of IAAL
recipients (Table III). On the
basis of the univariate analysis of eight variables,
CD8+CD57+ T cells [hazard ratio (HR), 0.948;
95% confidence interval (CI), 0.901–0.998; P=0.040],
terminal-CD8+ T cells (HR, 0.088; 95% CI, 0.010–0.744;
P=0.026), and CD57−FOXP3+CD8+ T
cells (HR, 13.526; 95% CI, 1.678–109.0; P=0.014) were significantly
associated with PFS (Table
III).
| Table III.Univariate and Multivariate analysis
of CD57-associated CD8+ T cells. |
Table III.
Univariate and Multivariate analysis
of CD57-associated CD8+ T cells.
| A, Univariate
analysis |
|---|
|
|---|
| Cells | HR | 95% CI | P-value |
|---|
| CD57+
T |
|
| 0.101 |
|
CD8+CD57+ T | 0.948 | 0.901–0.998 | 0.040 |
|
Early-CD8+ T |
|
| 0.058 |
|
Intermediate-CD8+ T |
|
| 0.209 |
|
Terminal-CD8+ T | 0.088 | 0.010–0.744 | 0.026 |
| End-CD8+
T |
|
| 0.384 |
|
FOXP3+CD8+CD57+
T |
|
| 0.288 |
|
FOXP3+CD8+CD57−
T | 13.53 | 1.678–109.0 | 0.014 |
|
| B, Multivariate
analysis |
|
| Cells | HR | 95% CI | P-value |
|
|
FOXP3+CD8+CD57−
T | 18.71 | 1.922–182.1 | 0.012 |
To determine the independent value and the relative
risk of these prognostic factors, multivariate analysis of the four
potential determinants (CD8+CD57+ T cells,
early-CD8+, terminal-CD8+, and
CD57−FOXP3+CD8+ T cells) was
performed using the Cox regression model. The results confirmed
that CD57−FOXP3+CD8+ T cells were
an independent PFS predictor for patients receiving IAAL (HR,
18.71; 95% CI, 1.922–182.1; P=0.012; Table III).
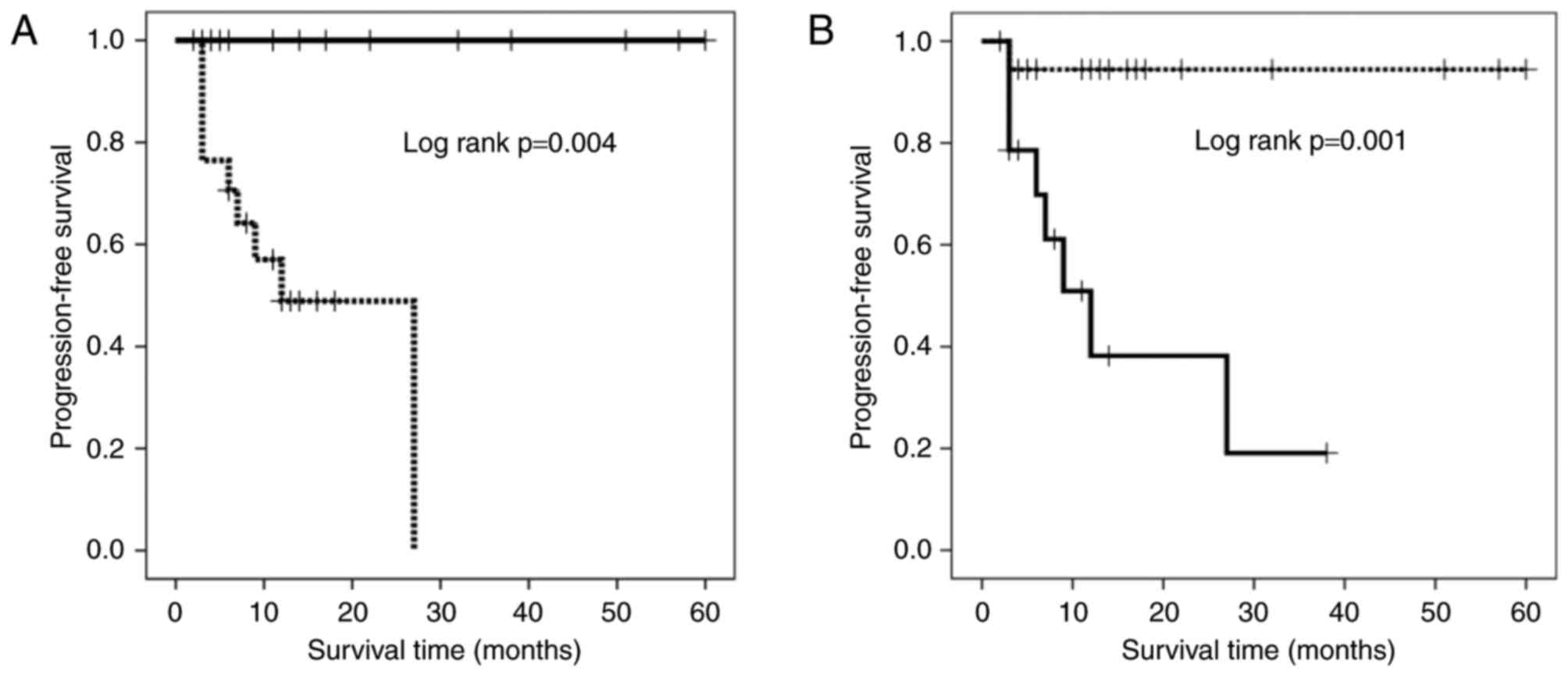
Progression-free survival
The results of univariate analysis demonstrated that
the proportions of terminal-CD8+ and
CD57−FOXP3+CD8+ T cells influenced
the PFS rate. The patients receiving IAAL were divided into groups
based on the proportions of terminal-CD8+ and
CD57−FOXP3+CD8+ T cells with
cutoff values of 33.7, and 21.8%, respectively as determined using
a receiver operating characteristic curve (sensitivity, 68 and
88.9%; specificity, 100 and 75%; area under the curve, 0.788 and
0.819; P=0.009 and 0.005, respectively). A Kaplan-Meier survival
curve revealed that patients with a high percentage of
terminal-CD8+ T cells had a significantly longer PFS
duration compared with those with a low percentage of
terminal-CD8+ T cells (Fig.
2A; P=0.004). By contrast, patients with high percentages of
CD57−FOXP3+CD8+ T cells exhibited
significantly shorter PFS durations compared with those with low
percentage of these cells (Fig. 2B;
P=0.001).
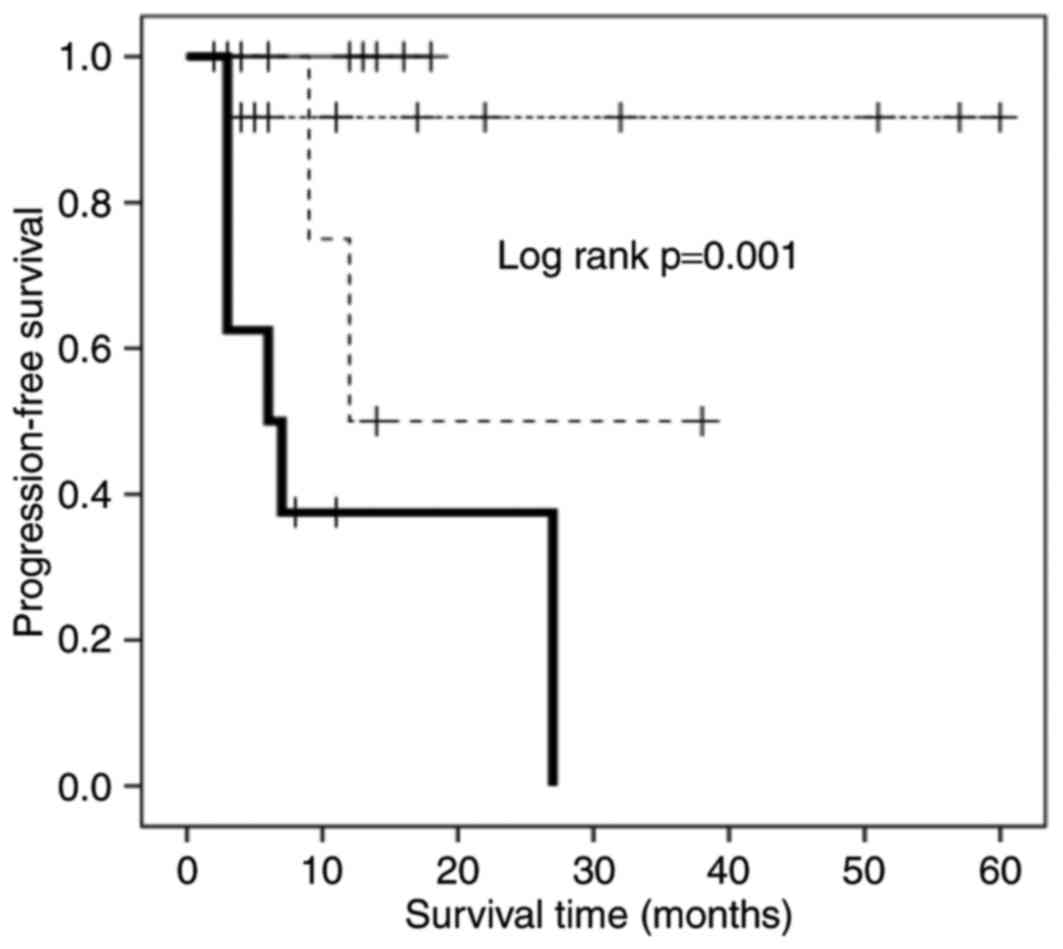
Association between
CD57−FOXP3+CD8+ T cells, CD27
expression, and PFS
The presence of
CD57−FOXP3+CD8+ T cells was an
independent poor prognostic factor as aforementioned, whereas
CD57+FOXP3+CD8+ T cells were not
associated with the PFS of IAAL recipients. These results indicated
that CD57−FOXP3+CD8+ T cells are
the most likely candidates to be CD8+ Tregs. Previous
studies revealed that the CD8+ Treg phenotype of
early-CD8+ T cells expressing FOXP3 is dependent on CD27
expression and the constitutive stimulation of CD70, a ligand of
CD27 (20,21). To investigate the association between
CD57−FOXP3+CD8+ T cells and CD27
expression, the PFS rates of four patient subgroups were analyzed:
FOXP3high/CD27highCD8+CD57−;
FOXP3high/CD27lowCD8+CD57−;
FOXP3low/CD27highCD8+CD57−;
and
FOXP3low/CD27lowCD8+CD57−
T cells (Fig. 3). Among the
subgroups, patients with
FOXP3high/CD27highCD8+CD57−
T cells exhibited the shortest PFS time, and those with
FOXP3low/CD27lowCD8+CD57−
T cells or
FOXP3low/CD27highCD8+CD57−
T cells exhibited the longest (Fig.
3); patients with
FOXP3high/CD27lowCD8+CD57−
T cells exhibited an intermediate PFS time.
High levels of
CD57−FOXP3+CD8+ T cells may impede
CD8+ T cell differentiation
As CD57−FOXP3+CD8+
T cells functioning as CD8+ Tregs were previously
reported to affect CD8+ T cell differentiation (22), the terminal-CD8+ T cell
population of the CD8+ T cell differentiation pathway
was compared between patients with high and low percentages of
CD57−FOXP3+CD8+ T cells. Following
culture, the level of terminal-CD8+ T cells of patients
with high levels of
CD57−FOXP3+CD8+ T cells were
significantly lower compared with those of patients with low
CD57−FOXP3+CD8+ T cells, whereas
there was no significant difference between them prior to culture
(Fig. 4A). This decrease was more
apparent when patients with high and low
FOXP3highCD27highCD8+CD57−
T cells were compared (Fig. 4B).
Discussion
The AALs populations examined in the present study
contained a high percentage of CD8+CD57+ T
cells, which was identified to be significantly associated with
longer PFS times following univariate analysis.
CD8+CD57+ T cells are comprised of two
different populations: Intermediate- and terminal-CD8+ T
cells, the latter of which are associated with longer PFS times.
IAAL reduced the proportion of early- and
intermediate-CD8+ T cells, and increased that of
cytotoxic terminal-CD8+ T cells. These results indicated
that IAAL promoted CD8+ T cell differentiation to
increase the proportion of cytotoxic terminal-CD8+ T
cells (CD27−CD8+CD57+ T cells) as
the TMME. However, data from the present study revealed
that CD57−FOXP3+CD8+ T cells are
an independent poor prognostic factor and ultimately determine the
efficacy of IAAL. In patients with a high percentage of
CD57−FOXP3+CD8+ T cells, the
proportion of cytotoxic terminal-CD8+ T cells was
significantly lower compared with those with a low percentage of
CD57−FOXP3+CD8+ T cells,
indicating that CD57−FOXP3+CD8+ T
cells are likely to function as CD8+ Tregs, and may
disrupt CD8+ T cell differentiation to reduce the
induction of terminal-CD8+ T cells, leading to poor
prognosis (22).
Cosmi et al (9) demonstrated the existence of
FOXP3-expressing CD8+ T cells with immunosuppression
capability (CD8+ Tregs), which was similarly identified
in prostate, colorectal, hepatocellular and gastric cancer
(10–12). However, it has been revealed that
FOXP3 expression is not necessarily associated with regulatory
functions in human CD4+ and CD8+ T cells
(20). In the present study,
CD57+FOXP3+CD8+ T cells were
significantly reduced by IAAL and were not associated with the PFS
of IAAL recipients. Anichini et al (23) reported the existence of
FOXP3+CD8+ T cells expressing an early
effector profile (rather than a regulatory CD8+ T cell
phonotype) that differentiate into terminal-CD8+ T cells
through intermediate-CD8+ T cells. The present study
indicated that FOXP3 expression on
CD57+FOXP3+CD8+ T cells was
transient in the absence of any associated regulatory function
during the natural course of CD8+ T cell differentiation
(24,25). In contrast to
CD57+FOXP3+CD8+ T cells,
CD57−FOXP3+CD8+ T cells were
appropriately classified as CD8+ Tregs in this study for
the following reasons: i)
CD57−FOXP3+CD8+ T cells were an
independent poor prognostic factor in a multivariate analysis; ii)
CD57−FOXP3+CD8+ T cells inhibited
CD8+ T cell differentiation; and iii) like conventional
CD8+ Tregs,
CD57−FOXP3+CD8+ T cells, which
were identified as an independent poor prognostic factor in the
present study, do not express CD57 (26). Taken together, it can be concluded
that CD57−FOXP3+CD8+ T cells were
CD8+ Tregs, and CD57 expression on
FOXP3+CD8+ T cells may be an immunological
marker for discriminating FOXP3+CD8+ T cells
with a regulatory function from those without.
An association between simultaneous expression of
FOXP3 and CD27, and the poorest PFS of the four subgroups was also
demonstrated in the present study, as CD27 expression was reported
to discriminate regulatory from non-regulatory cells (27). CD27, a member of the tumor necrosis
factor receptor superfamily, serves as a co-stimulatory factor, and
CD27 signaling leads either to improved T cell function or to T
cell dysfunction, depending on the level, duration, and timing of
CD27 ligand (CD70) expression (21,28–30).
Consequently, CD70 expression is tightly regulated, and CD70 is
only transiently expressed on activated T and B cells, and on
subsets of professional antigen-presenting dendritic cells and
natural killer cells (21). By
contrast, constitutive CD70 expression has been documented in
various types of cancer, including brain tumors (20), renal cell carcinomas (21) and certain lymphomas (31,32), and
is often associated with FOXP3+ Treg formation (31,32). Thus,
CD27 signaling mediated by constitutive CD70 expression is
essential for the induction of FOXP3+ regulatory
CD8+ T cells. Therefore, we hypothesize that CD27 is
expressed on CD57−FOXP3+CD8+ T
cells functioning as CD8+ Tregs in the current study.
Furthermore, the present study demonstrated that the proportion of
CD57−FOXP3+CD8+ T cells was
unchanged following culture, when compared with the pre-culture
proportion, indicating that once
CD57−FOXP3+CD8+ T cells
(CD8+ Tregs) adopt a regulatory function, their
phenotype cannot be changed despite an improvement in the
microenvironment induced by IAAL. Previous reports demonstrated
that an anti-human CD27 agonist MAb was shown to induce T cell
activation and tumor immunity in human CD27-transgenic mice
(33) through a combination of
co-stimulation, and Treg-depleting activities (34). These results indicated that a
CD27-agonist MAb may deprive
CD57−FOXP3+CD8+ T cells of their
regulatory function as CD8+ Tregs and activate the
co-stimulation signaling for CTLs to induce antitumor immunity.
Thus, combinational immunotherapy using AALs and a CD27-agonist MAb
may lead to more improved outcomes for IAAL.
In conclusion, the results of the present study
demonstrated that terminal-CD8+ T cells are the
TMME of IAAL, and that IAAL may promote CD8+
T cell differentiation to induce cytotoxic terminal-CD8+
T cells. In addition,
CD57−FOXP3+CD8+ T cells were
revealed to be an independent poor prognostic factor for patients,
to be CD8+ Tregs and to inhibit CD8+ T cell
differentiation, which cannot be recovered by IAAL. Thus,
CD57−FOXP3+CD8+ T cells, as
CD8+ Tregs, are essential determinants of IAAL
efficacy.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Strioga M, Pasukoniene V and Characiejus
D: CD8+ CD28- and CD8+ CD57+ T cells and their role in health and
disease. Immunology. 134:17–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu RC, Hwu P and Radvanyi LG: New insights
on the role of CD8(+)CD57(+) T-cells in cancer. Oncoimmunology.
1:954–956. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Brenchley JM, Karandikar NJ, Betts MR,
Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles
SA, Connors M, et al: Expression of CD57 defines replicative
senescence and antigen-induced apoptotic death of CD8+ T cells.
Blood. 101:2711–2720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Papagno L, Spina CA, Marchant A, Salio M,
Rufer N, Little S, Dong T, Chesney G, Waters A, Easterbrook P, et
al: Immune activation and CD8+ T-cell differentiation towards
senescence in HIV-1 infection. PLoS Biol. 2:E202004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Akagi J and Baba H: Prognostic value of
CD57(+) T lymphocytes in the peripheral blood of patients with
advanced gastric cancer. Int J Clin Oncol. 13:528–535. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Characiejus D, Pasukoniene V,
Jonusauskaite R, Azlauskaite N, Aleknavicius E, Mauricas M and
Otter WD: Peripheral blood CD8highCD57+ lymphocyte levels may
predict outcome in melanoma patients treated with adjuvant
interferon-alpha. Anticancer Res. 28:1139–1142. 2008.PubMed/NCBI
|
|
7
|
Characiejus D, Pasukoniene V, Kazlauskaite
N, Valuckas KP, Petraitis T, Mauricas M and Den Otter W: Predictive
value of CD8highCD57+ lymphocyte subset in interferon therapy of
patients with renal cell carcinoma. Anticancer Res. 22:3679–3683.
2002.PubMed/NCBI
|
|
8
|
Dondi E, Roué G, Yuste VJ, Susin SA and
Pellegrini S: A dual role of IFN-alpha in the balance between
proliferation and death of human CD4+ T lymphocytes during primary
response. J Immunol. 173:3740–3747. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cosmi L, Liotta F, Lazzeri E, Francalanci
M, Angeli R, Mazzinghi B, Santarlasci V, Manetti R, Vanini V and
Romagnani P: Human CD8+CD25+ thymocytes share phenotypic and
functional features with CD4+CD25+ regulatory thymocytes. Blood.
102:4107–4114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chaput N, Louafi S, Bardier A, Charlotte
F, Vaillant JC, Ménégaux F, Rosenzwajg M, Lemoine F, Klatzmann D
and Taieb J: Identification of CD8+CD25+Foxp3+ suppressive T cells
in colorectal cancer tissue. Gut. 58:520–529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Billerbeck E, Blum HE and Thimme R:
Parallel expansion of human virus-specific FoxP3-effector memory
and de novo-generated FoxP3+ regulatory CD8+ T cells upon antigen
recognition in vitro. J Immunol. 179:1039–1048. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simone R, Zicca A and Saverino D: The
frequency of regulatory CD3+CD8+CD28-CD25+ T lymphocytes in human
peripheral blood increases with age. J Leukoc Biol. 84:1454–1461.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gavin MA, Torgerson TR, Houston E, DeRoos
P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD and
Rudensky AY: Single-cell analysis of normal and FOXP3-mutant human
T cells: FOXP3 expression without regulatory T cell development.
Proc Natl Acad Sci USA. 103:6659–6664. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Allan SE, Song-Zhao GX, Abraham T,
McMurchy AN and Levings MK: Inducible reprogramming of human T
cells into Treg cells by a conditionally active form of FOXP3. Eur
J Immunol. 38:3282–3289. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pillai V and Karandikar NJ: Human
regulatory T cells: A unique, stable thymic subset or a reversible
peripheral state of differentiation? Immunol Lett. 114:9–15. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo L, Tian J, Marinova E, Zheng B and Han
S: Inhibition of clonal expansion by Foxp3 expression as a
mechanism of controlled T-cell responses and autoimmune disease.
Eur J Immunol. 40:71–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sobin LH and Wittekind CH: TNM
classification of malignant tumours. John Wiley and Sons; New York:
1997
|
|
18
|
Takayama T, Sekine T, Makuuchi M, Yamasaki
S, Kosuge T, Yamamoto J, Shimada K, Sakamoto M, Hirohashi S, Ohashi
Y and Kakizoe T: Adoptive immunotherapy to lower postsurgical
recurrence rates of hepatocellular carcinoma: A randomised trial.
Lancet. 356:802–807. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trotti A, Colevas AD, Setser A, Rusch V,
Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN and
Rubin P: CTCAE v3.0: Development of a comprehensive grading system
for the adverse effects of cancer treatment. Semin Radiat Oncol.
13:176–181. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wischhusen J, Jung G, Radovanovic I, Beier
C, Steinbach JP, Rimner A, Huang H, Schulz JB, Ohgaki H and Aguzzi
A: Identification of CD70-mediated apoptosis of immune effector
cells as a novel immune escape pathway of human glioblastoma.
Cancer Res. 62:2592–2599. 2002.PubMed/NCBI
|
|
21
|
Nolte MA, van Olffen RW, van Gisbergen KP
and van Lier RA: Timing and tuning of CD27-CD70 interactions: The
impact of signal strength in setting the balance between adaptive
responses and immunopathology. Immunol Rev. 229:216–231. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Claus C, Riether C, Schürch C, Matter MS,
Hilmenyuk T and Ochsenbein AF: CD27 signaling increases the
frequency of regulatory T cells and promotes tumor growth. Cancer
Res. 72:3664–3676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anichini A, Molla A, Vegetti C, Bersani I,
Zappasodi R, Arienti F, Ravagnani F, Maurichi A, Patuzzo R and
Santinami M: Tumor-reactive CD8+ early effector T cells identified
at tumor site in primary and metastatic melanoma. Cancer Res.
70:8378–8387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Campbell DJ and Ziegler SF: FOXP3 modifies
the phenotypic and functional properties of regulatory T cells. Nat
Rev Immunol. 7:305–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Billerbeck E, Nakamoto N, Seigel B, Blum
HE, Chang KM and Thimme R: Determinants of in vitro expansion of
different human virus-specific FoxP3+ regulatory CD8+ T cells in
chronic hepatitis C virus infection. J Gen Virol. 90:1692–1701.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duggleby RC, Shaw TN, Jarvis LB, Kaur G
and Gaston JS: CD27 expression discriminates between regulatory and
non-regulatory cells after expansion of human peripheral blood CD4+
CD25+ cells. Immunology. 121:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vlad G, King J, Chang CC, Liu Z, Friedman
RA, Torkamani AA and Suciu-Foca N: Gene profile analysis of CD8
(+) ILT3-Fc induced T suppressor cells. Hum Immunol.
72:107–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matter M, Odermatt B, Yagita H, Nuoffer JM
and Ochsenbein AF: Elimination of chronic viral infection by
blocking CD27 signaling. J Exp Med. 203:2145–2155. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Penaloza-MacMaster P, Rasheed Ur A, Iyer
SS, Yagita H, Blazar BR and Ahmed R: Opposing effects of CD70
costimulation during acute and chronic lymphocytic choriomeningitis
virus infection of mice. J Virol. 85:6168–6174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Borst J, Hendriks J and Xiao Y: CD27 and
CD70 in T cell and B cell activation. Curr Opin Immunol.
17:275–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE
and Ansell SM: CD70+ non-Hodgkin lymphoma B cells induce Foxp3
expression and regulatory function in intratumoral CD4+CD25 T
cells. Blood. 110:2537–2544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jak M, Mous R, Remmerswaal EB, Spijker R,
Jaspers A, Yagüe A, Eldering E, Van Lier RA and Van Oers MH:
Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in
chronic lymphocytic leukemia. Leuk Lymphoma. 50:788–801. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He LZ, Prostak N, Thomas LJ, Vitale L,
Weidlick J, Crocker A, Pilsmaker CD, Round SM, Tutt A and Glennie
MJ: Agonist anti-human CD27 monoclonal antibody induces T cell
activation and tumor immunity in human CD27-transgenic mice. J
Immunol. 191:4174–4183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
He LZ, Wasiuk A, Testa J, Weidlick J,
Sisson C, Crocker A, Widger J, Forsberg E, Gergel L, Thomas L, et
al: The mechanism of anti-tumor immunity induced by varlilumab, a
CD27 agonist mAb, is model dependent. J Immunother Cancer. 3 Suppl
2:P1882015. View Article : Google Scholar
|