Introduction
Liver cancer is now considered a common malignancy
and the second leading cause of cancer-associated mortality
worldwide among males (1).
Hepatocellular carcinoma (HCC) is the most common type of liver
cancer, accounting for 90% of all cases (2). Of all the available treatment methods,
hepatectomy is the most effective treatment for HCC; however, only
20% of patients are eligible for surgery (3). For the remaining 80% of patients with
HCC who are at an advanced stage of disease, surgical resection is
not a viable option, and systemic chemotherapy is the principal
treatment option (4). However, due to
multidrug resistance and adverse reactions from traditional
chemotherapy agents, the effective rate of systemic chemotherapy is
only 10–15% (5). With the development
of basic biomedical research for HCC, molecular targeted therapy
has become a novel regimen for the treatment of HCC (6). Sorafenib, at present, is the only FDA
approved drug with the same first-line treatment position to
chemotherapy for the systemic therapy of HCC, and is a multi-target
inhibitor, which may provide more survival benefits for patients
with non-resectable HCC (7). Although
patients with HCC exhibit a positive response to sorafenib,
resistance to sorafenib usually appears in 3.5 months and the
median progression-free survival (mPFS) has become a major obstacle
for the extension of the median overall survival (mOS) (8). Consequently, the discovery of novel
therapeutic drugs for the treatment of HCC has become an urgent
requirement. A novel antiangiogenic agent, Apatinib, is a highly
selective inhibitor of vascular endothelial growth factor
receptor-2 (VEGFR-2) tyrosine kinase and possesses 10 times more
binding ability than sorafenib (9).
It may block downstream signaling through highly selective
competition with intracellular VEGFR-2 ATP binding sites and
prevent VEGF-mediated endothelial cell migration and proliferation
(10). In pre-clinical research,
Apatinib was demonstrated to have an inhibitory effect on
neovascularization and exhibited potential antitumor activity
against a series of tumor cells (11). A phase III clinical trial demonstrated
that Apatinib improved the mPFS and mOS of patients with advanced
gastric cancer (12). Simultaneously,
Phase II clinical trials confirmed the antitumor effect of Apatinib
in numerous cancer types, including non-small cell lung cancer
(NSCLC), breast cancer and HCC (13–15). In
patients with HCC, a small sample clinical trial demonstrated that
Apatinib improved OS (16). It
appears to be a possible treatment strategy for patients with
advanced HCC. However, the potential antitumor mechanism in HCC
remains unclear. A number of studies (17–19) have
demonstrated that monomer drugs, including Tripterine from the
traditional Chinese medicine Tripterygium, exhibited strong
antitumor activity against different types of tumors. Tripterine,
also known as celastrol, is one of the main active components of
Tripterygium and has been confirmed to have antitumor
effects in vitro and in vivo (20,21).
However, whether the traditional Chinese medicine monomer drug
Tripterine may improve the antitumor activities of Apatinib is
unknown. Therefore, the present study was designed to elucidate the
mechanism of the antitumor effect of Apatinib on HCC cells. In
addition, the synergistic antitumor effect of Tripterine with
Apatinib and the molecular mechanism will also be investigated.
Materials and methods
Cell lines and reagents
Human hepatoma Hep3B cells were obtained from the
Liver Disease Experimental Center of Beijing Friendship Hospital
affiliated, Capital Medical University (Beijing, China). HUVECs
were purchased from ScienCell Research Laboratories, Inc. (San
Diego, CA, USA). The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM; Corning Incorporated, Corning, NY, USA)
supplemented with 10% fetal bovine serum (FBS; ExCell Biology,
Shanghai, China) at 37°C in the presence of 5% CO2.
Apatinib was purchased from Jiangsu Hengrui Medicine Co., Ltd.
(Lianyungang, China). Tripterine was purchased from Shanghai
Aladdin Company Bio-Chem Technology Co., Ltd. (Shanghai, China).
MTS was purchased from Promega Corporation (Madison, WI, USA). The
Annexin V-FITC/PI Apoptosis Detection kit was purchased from
Nanjing Kaiji Bio Tech Co., Ltd. (Nanjing, China). Antibodies
against protein kinase B (Akt; A5523), phosphorylated Akt (p-Akt;
AP0274), extracellular signal-regulated kinase (ERK; A11116) and
phosphorylated ERK (p-ERK; AP0472) were purchased from ABclonal
(ABclonal Biotech Co., Ltd., Woburn, MA, USA). Antibodies against
Caspase-3 (9662S) and B-cell lymphoma 2-associated X protein (Bax;
5023S), Antibodies against VEGFR-2 (2479S) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Antibodies against
GADPH (41549) were purchased from Signalway Antibody LLC (College
Park, MD, USA). The dilution factor of the antibodies was
1:1,000.
Human hepatoma Hep3B cells were assigned into four
different groups: Control group, Apatinib group, Tripterine group
and combination group. The treatments in the four groups were no
drug for the control group, 30 µmol/l Apatinib for the Apatinib
group, 2.5 µmol/l Tripterine for the Tripterine group, and 30
µmol/l Apatinib with 2.5 µmol/l Tripertine for the combination
group.
Western blot analysis
Human hepatoma Hep3B cells were seeded onto 6-well
plates. After 24 h, the cells were harvested. Cells were lysed with
radioimmunoprecipitation assay lysis buffer (Nanjing Kaiji Bio Tech
Co., Ltd. Nanjing, China), premixed with phenylmethanesulfonyl
fluoride (dilution, 1:100). The protein concentration of the cell
extracts was quantified using a bicinchoninic acid assay. Equal
amounts of protein (40–60 µg) were separated by SDS-PAGE gels (8%,
10% or 12%) and then electrotransferred onto polyvinylidene
difluoride membranes, followed by blocking with bovine serum
albumin (BSA, Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China) at room temperature for 30 min and incubation with
primary antibodies at 4°C overnight. TBST (0.025%) was used to wash
the membrane three times. The membranes were incubated with the
secondary antibody at room temperature for 1 h. The bands were
detected using an enhanced chemiluminescence kit (Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and signal intensities were
analyzed with a Gel-pro 4.5 Analyzer (Media Cybernetics, Rockville,
MD, USA).
Immunofluorescence staining
Hep3B cells were plated at a density of
3×104 cells per well in 12-well plates with coverslips.
The coverslips were washed three times in PBS and fixed with 4%
formaldehyde solution at room temperature for 20 min. Next, the
coverslips were washed three times in PBS and incubated with a
membrane-permeation solution (1 ml Triton X-100 in 100 ml PBS) at
room temperature for 10 min. Following washing three times in PBS,
the coverslips were placed into 2% BSA at room temperature for 30
min. The coverslips were then incubated with a primary VEGFR-2
antibody (dilution, 1:100) at 4°C overnight. The coverslips were
washed three times with PBS and incubated at room temperature for
60 min with a horseradish peroxidase-conjugated goat anti-rabbit
immunoglobulin G secondary antibody (L3012-1; 1:100; Signalway
Antibody LLC, College Park, MD, USA). The coverslips were washed
three times with PBS and the nuclei were stained with DAPI at room
temperature for 5 min. Finally, stained cells were visualized under
a confocal laser-scanning microscope(magnification, ×40; oil,
NA=1.25; zoom=3) TCS SP5 (Leica Microsystems GmbH, Wetzlar,
Germany).
MTS assay
Hep3B cells in the logarithmic growth phase were
collected and inoculated into 96-well plates. A total of 50 µl DMEM
was then added to 50 µl cell suspension (containing
1×104 cells) per well to reach a final volume of 100 µl.
After the cells were adhered to the plates, they were treated with
different concentrations (0, 10, 30, 50 and 100 µmol/l) of Apatinib
for 24 h (Fig. 1A). The final optimal
concentration of Apatinib was 30 µmol/l. Similarly, Hep3B cells
were treated with different concentrations (0, 1, 2.5, 5 and 10
µmol/l) of Tripterine for 24 h (Fig.
1B). The final optimal concentration of tripterine was 2.5
µmol/l. After determining the optimal concentration of the
aforementioned two drugs, Hep3B cells were treated with Apatinib at
a final concentration of 30 µmol/l and Tripterine at a
concentration of 2.5 µmol/l. Four duplicate wells were set for each
sample. Dimethyl sulfoxide (50 µl) was added to the blank control
group. At the end of the experiment, 20 µl MTS was added to each
well and incubated at 37°C for 4 h. Following aspiration of the
supernatant, DMEM was added to the culture medium of each well. The
OD value at a wavelength of 490 nm was measured using a microplate
reader. The formula is cell proliferation rate (%)=OD 490
nmexperimental group/OD 490 nmcontrol group ×100. The inhibition
rate (%)=(average of OD 490 nmcontrol group-average of OD 490
nmexperimental group)/OD 490 nmcontrol group ×100.
Wound healing assay
Hep3B cells were seeded at a density of
1×105 cells per well in 6-well flat-bottomed microplates
and then cultured to 90% confluency at 37°C in a humidified
atmosphere with 5% CO2. A thin scratch (wound) was made in the
central area using pipette tips (200 μl) and cells were carefully
washed three times with DMEM. Different concentrations of Apatinib
and Tripterine were then added to the culture medium of scratched
Hep3B cells. Wound closure was monitored by light microscopy
(magnification, ×40). Images were acquired at 0, 24 and 48 h
post-scratching. The migration rate (%)=(scratch distance-distance
after growing)/scratch distance.
Transwell assay
Matrigel diluted in cold serum-free DMEM was added
to the bottom of the pre-cooled chamber prior to incubation at
37°C. A total of 3×104 Hep3B cells were added to the
coated upper chamber containing DMEM. DMEM supplemented with 10%
FBS was added to the lower chamber. After 24 h of incubation, the
cells remaining on the upper chamber were wiped gently with cotton
swab. Methanol (20%) and 0.1% crystal violet at room temperature
for 20 min were used to stain the cells that invaded through the
membrane. Light microscopy was used to observe migrated cells in
the lower chamber (magnification, ×100).
Flow cytometry assay
Apoptotic cells were quantified by flow cytometry
using an Annexin V-FITC/PI Apoptosis Detection kit (BD Biosciences,
Franklin Lakes, NJ, USA), according to the manufacturer's protocol.
Briefly, Hep3B cells were treated with 30 µmol/l Apatinib, 2.5
µmol/l Tripterine or a combination of 2.5 µmol/l Tripterine and 30
µmol/l Apatinib for 24 or 48 h. Cells were then collected and
washed twice with cold PBS followed by resuspension with 500 µl
Annexin V binding buffer containing 5 µl fluorescein isothiocyanate
(FITC)-labeled Annexin V. The cell suspension was transferred into
round-bottom tubes and incubated for 15 min in the dark at room
temperature. Finally, 5 µl propidium iodide (PI) was used and the
percentage of apoptotic cells was measured using a flow cytometer
and BD facs divaTM software (version 7.0; BD Biosciences).
Statistical analysis
Data are presented as the mean ± standard deviation.
The differences between the control and experimental groups were
analyzed by one-way analysis of variance, followed by the
Student-Newman-Keul's (SNK) post hoc test. The statistical
differences were evaluated by SPSS 19.0 software (IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
The expression of VEGFR-2 in Hep3B
cells
HUVECs were used as a positive control and VEGFR-2
was demonstrated to be expressed in Hep3B cells as detected by
western blot analysis (Fig. 2A). An
immunofluorescence assay also demonstrated that VEGFR-2 was located
on the Hep3B cells (Fig. 2B).
Therefore, VEGFR-2 was demonstrated to be expressed in Hep3B
cells.
Tripterine enhances the inhibitory
effect of Apatinib on Hep3B cell proliferation
An MTS assay was used to detect the effects of
Apatinib, Tripterine and the combination of drugs on the
proliferation of Hep3B cells. According to our pre-experiment, 30
µmol/l Apatinib combined with 2.5 µmol/l Tripterine may be the
optimal therapeutic concentration for Hep3B cells (Fig. 1). At 24 h post-treatment, Apatinib,
Tripterine and a combination of the two significantly inhibited the
proliferation of Hep3B cells compared with the control group
(P<0.001). The inhibitory effect of the combination group was
more pronounced than that of the Apatinib and Tripterine groups
(Fig. 3A). At 48 h post-treatment,
the inhibitory effects were more evident in all three groups.
However, the inhibitory capacity of the combination group was
significantly stronger than that of the Apatinib group (P<0.05).
However there was no significant difference in the combination
group at 48 h post-treatment compared with the Tripterine group
(P>0.05; Fig. 3B). The results
demonstrated that the inhibition rates were 69.1 and 60.4% of
Apatinib and Tripterine alone at 24 h post-treatment, respectively.
However, following treatment with the combination of two drugs for
24 h, the inhibition rate was as high as 75.8%. At 48 h
post-treatment, the inhibition rates of Apatinib and Tripterine
were 78.8 and 82.2%, respectively, while the inhibition rate of the
combination of the two drugs was 83.5%. Therefore, Tripterine
enhanced the inhibitory effect of Apatinib on Hep3B cell
proliferation.
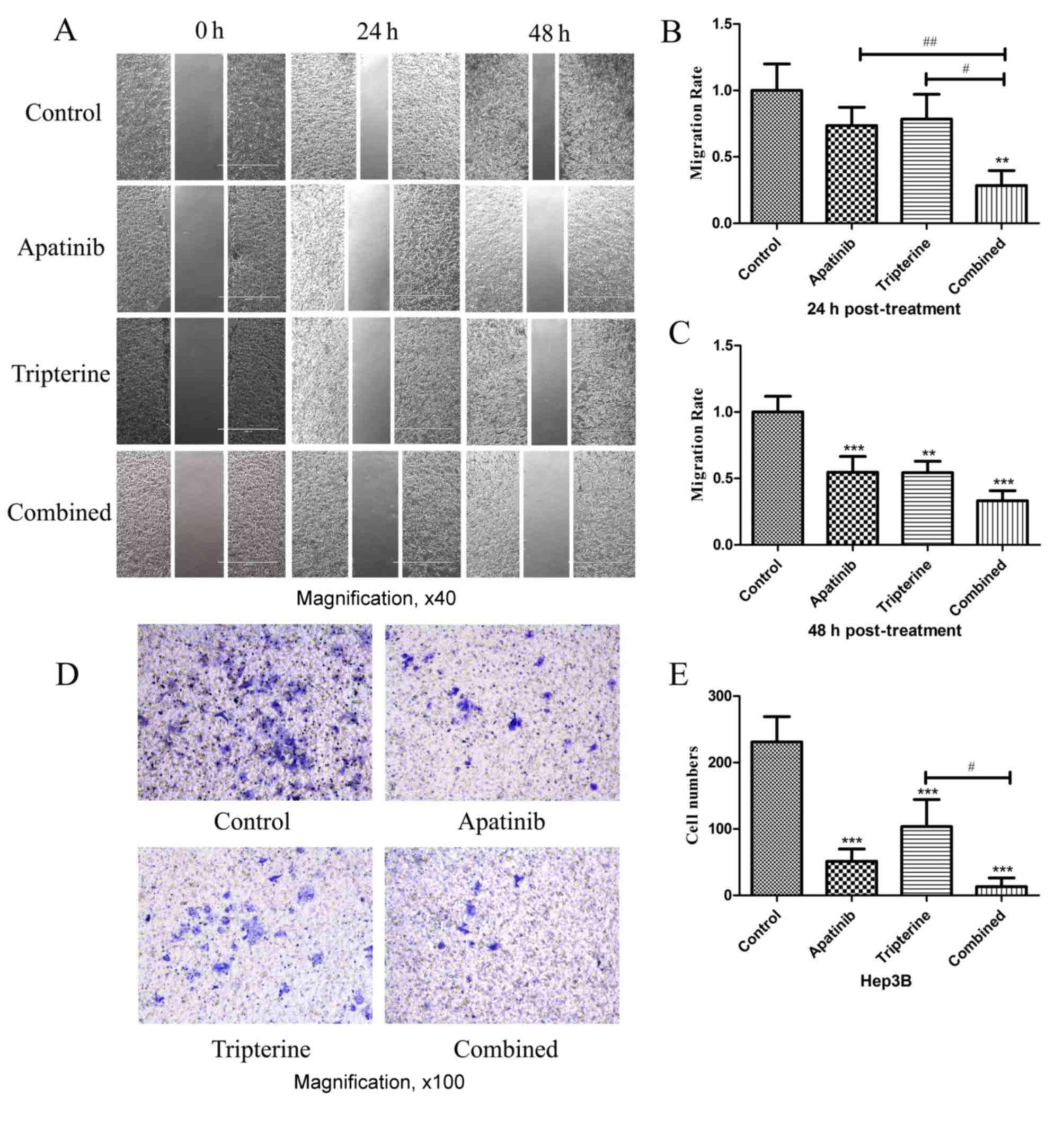
The migration and invasion of Hep3B
cells were inhibited by Apatinib and Tripterine
Identical drug concentrations were selected
according to the MTS assay as previously described. Wound healing
assays were performed to determine the migration effect of Hep3B
cells under different drug treatments (Fig. 4A). A total of 24 h after scratching
compared with the control group, Apatinib and Tripterine inhibited
Hep3B cell migration; however, the difference was not statistically
significant (P>0.05; Fig. 4B). By
contrast, the combination group significantly inhibited Hep3B cell
migration (P<0.01). Compared with Apatinib or Tripterine alone,
the combination group exhibited a significantly more evident
inhibitory migration effect (P<0.01 and P<0.05, respectively;
Fig. 4A and B). At 48 h
post-treatment, the inhibitory effect of Hep3B cell migration was
significantly enhanced in all three groups (all P<0.01).
Furthermore, the combination group and the Apatinib group exhibited
a significantly stronger inhibitory migration effect (P<0.001;
Fig. 4A and C). Transwell assays were
performed to determine the invasive effect of Hep3B cells under
different drug treatments. After 24 h of the Transwell assay,
compared with the control group, all treatments significantly
inhibited Hep3B cell invasion (P<0.001). The inhibitory effect
was the strongest in the combination group and the difference was
statistically significant compared with the Tripterine group
(P<0.05; Fig. 4D and E). Therefore
Tripterine enhanced the inhibitory effect of Apatinib on Hep3B cell
migration and invasion.
Tripterine combined with Apatinib
promoted the apoptosis of Hep3B cells
The present study evaluated the effectiveness of the
treatment in terms of apoptosis by performing Annexin V and PI
staining on Hep3B cells from different treatment groups at 24 and
48 h after treatment. The present study identified a larger
population of apoptotic cells in the Apatinib, Tripterine and
combination groups at 24 and 48 h post-treatment (P<0.001,
respectively), while the pro-apoptotic effect of the combination
treatment was evident compared with that of Apatinib or Tripterine
treatment alone (P<0.05, respectively; Fig. 5A-D). Tripterine promoted the apoptosis
of Hep3B cells via Apatinib.
Apatinib and Tripterine downregulated
the expression of p-Akt and p-ERK and upregulated the expression of
cleaved Caspase-3 and Bax
The present study demonstrated that the combination
of Apatinib and Tripterine promoted the caspase-dependent apoptosis
of Hep3B cells. As demonstrated in Fig.
6A and B, the expression of cleaved caspase-3 and Bax was
elevated in the Apatinib, Tripterine and combination groups, and
the increases in cleaved caspase-3 and Bax were more evident in the
combination group at 24 and 48 h post-treatment (Fig. 6C and D). ERK and Akt are downstream
regulators of VEGFR-2 that serve a vital role in the process of
apoptosis (22). Therefore, the
expression of p-Akt and p-ERK were detected via western blotting.
The results demonstrated that the expression of p-Akt and p-ERK
were significantly decreased in the Apatinib, Tripterine and
combination groups (Fig. 6A and C).
The expression of p-Akt and p-ERK in the combination group was
significantly lower than that in the Apatinib group or the
Tripterine group (Fig. 6B and D).
These findings suggested that the reduced phosphorylation of Akt
and ERK may be the key to inducing caspase-dependent apoptosis in
Hep3B cells. The combination of Apatinib and Tripterine
downregulated the expression of p-Akt and p-ERK and upregulated the
expression of cleaved Caspase-3 and Bax.
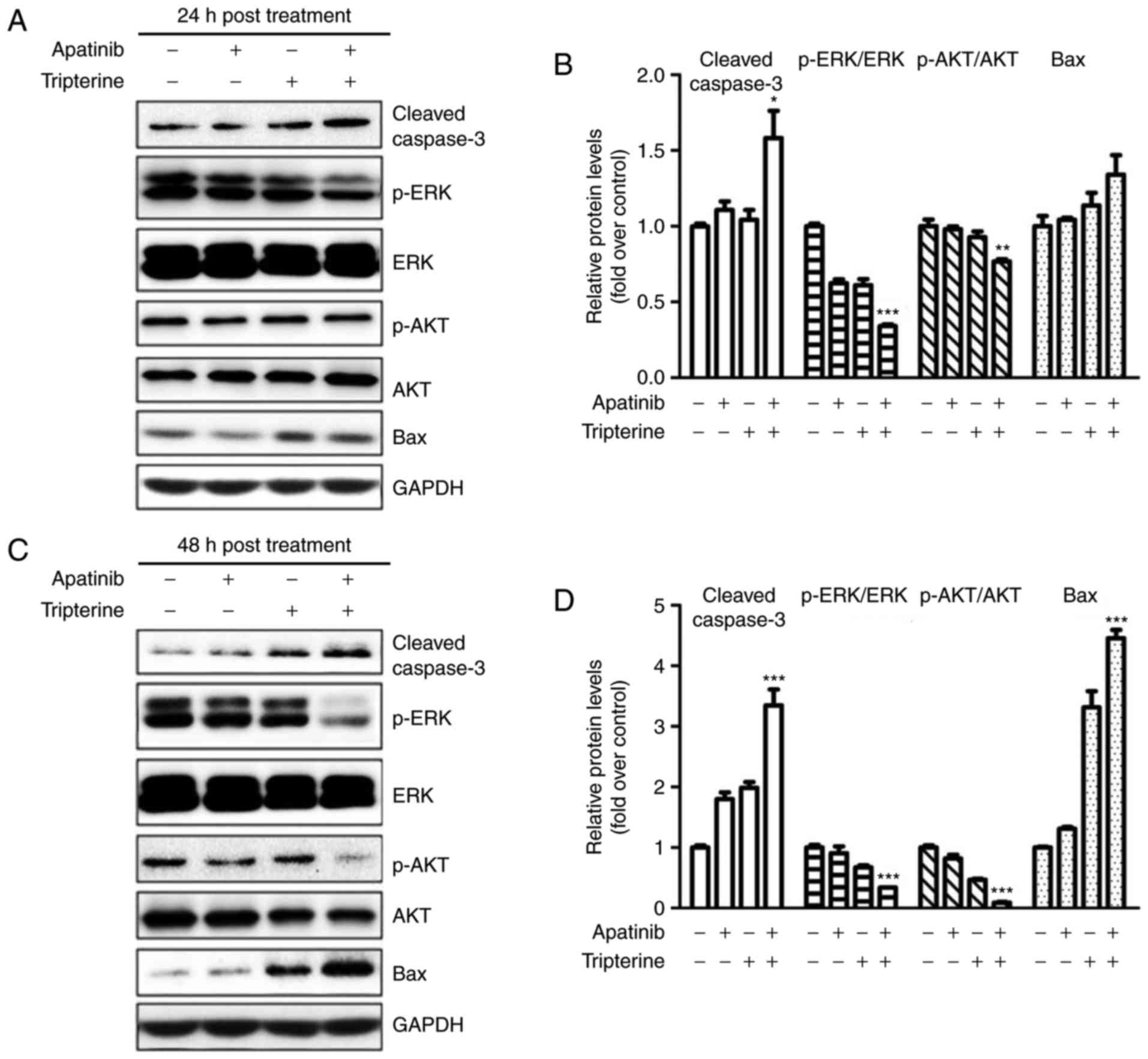 | Figure 6.Protein expression in various
treatment groups (Apatinib 30 µmol/l and Tripterine 2.5 µmol/l)
detected by western blot analysis. (A) Expression of p-Akt, p-ERK,
cleaved caspase-3 and Bax in four different groups at 24 h
post-treatment. (B) Quantification of different protein expression
levels between the four groups at 24 h post-treatment. (C)
Expression of p-Akt, p-ERK, cleaved caspase-3 and Bax in four
different groups at 48 h post-treatment. (D) Quantification of
different protein expression levels between the four groups at 48 h
post-treatment. *P<0.05, **P<0.01 and ***P<0.001. Akt,
protein kinase B; ERK, extracellular signal-regulated kinase;
p-Akt, phosphorylated protein kinase B; p-ERK, phosphorylated
extracellular signal-regulated kinase; Bax, B-cell
lymphoma-associated X protein. |
Discussion
A large volume of evidence has demonstrated that
tumor angiogenesis is an essential event in the process of tumor
growth and metastasis, which lead to the poor prognosis of HCC
(23–25). HCC becomes more aggressive and lethal
once it obtains a sufficient blood supply (26). Therefore, it is of great importance to
block angiogenesis in HCC. Among all angiogenic factors, VEGF is
the key regulator that triggers a series of signaling pathways to
promote endothelial cell proliferation, migration and survival from
pre-existing vasculature (27).
VEGF/VEGFR are highly expressed in the majority of tumor tissues.
Therefore, the VEGF-VEGFR pathway has been a research hotspot in
the field of anti-angiogenesis therapy (28,29).
Apatinib is an orally administered small-molecule
inhibitor that selectively targets VEGFR-2, inhibiting the
activities of platelet-derived growth factor-b receptor, c-kit, and
c-src, which subsequently suppress the formation of new blood
vessels (30). Angiogenesis is an
important mechanism of tumorigenesis (31). The study of Li et al (32) demonstrated that Apatinib significantly
improved the survival outcomes of patients with gastric cancer who
underwent second-line treatment failure in a phase II clinical
trial, which was consistent with the research of Roviello et
al (33). Therefore, the present
study used Apatinib as a novel anti-tumorigenesis agent for the
treatment of HCC.
Tripterine is a traditional medicine monomer
extracted from the Chinese herb Tripterygium wilfordii HOOK
f. Previous studies have demonstrated that Tripterine has
anti-inflammatory, anti-immune and antitumor effects (34–36).
Tripterine has been demonstrated to induce apoptosis in human
triple-negative breast cancer cells by upregulating the expression
of Bax and downregulating the activity of the phosphoinositide
3-kinase enzyme and the phosphorylation of Akt (17). Lee et al (18) also identified an increase in
phosphorylated mitogen-activated protein kinase following a
decrease in all phosphorylated forms of Akt, mechanistic target of
rapamycin and S6K following treatment with Tripterine in gastric
cancer. Therefore, the present study further investigated the
synergistic antitumor effect of Apatinib and Tripterine. The
proliferation, migration and invasion of tumor cells contribute
toward the local infiltration and distant metastasis of
malignancies, which subsequently lead to the lethality of
cancer.
In the present study, the effects of Apatinib
combined with Tripterine on the proliferation, migration and
invasion of Hep3B cells were investigated. Due to the function of
VEGFR-2 as a membrane receptor protein, western blot analysis and
immunofluorescence assays were used to confirm the expression of
VEGFR-2 in Hep3B cells. The results demonstrated that the
proliferation, migration and invasion of HCC cells were
significantly inhibited by Apatinib and Tripterine, and the
inhibitory effect was more evident in the combination group,
suggesting that Apatinib and Tripterine had a synergistic effect in
anti-tumorigenesis. Furthermore, the underlying mechanism of this
process was elucidated. As a key tumor suppression mechanism,
apoptosis is a process of programmed cell death that can be
initiated by the pro-apoptotic factor Bax. Bax is widely expressed
in a variety of cells and is an important apoptotic protein
(37,38). Bax translocation from the cytosol to
the outer mitochondrial membrane alters the permeability of the
mitochondrial membrane and releases several pro-apoptotic factors,
including cytochrome c (39). In
addition to Bax, caspase-3 is a crucial mediator of apoptosis that
catalyzes the specific cleavage of numerous key pro-apoptotic
proteins that leads to DNA fragmentation and the formation of
apoptotic bodies (40). The present
study examined whether Apatinib and Tripterine could induce
apoptosis of HCC and demonstrated that Apatinib and Tripterine
could significantly increase apoptosis. Furthermore, apoptosis was
more evident with a combination of the two drugs. According to the
flow cytometry assay, the number of necrotic cells was large,
culminating in the hypothesis that there were a number of other
mechanisms for cell death in addition to apoptosis that require
further research. The focus was on the effects of different drugs
on the apoptosis of Hep3B cells. In addition, the expression of
cleaved caspase-3 and Bax was significantly increased in the
Apatinib and Tripterine groups and further increased in the
combination group.
The ERK signaling pathway serves a vital role in
numerous cell functions that mediate different
proliferation-related events, including apoptosis, autophagy and
senescence (41). Akt is also a key
apoptosis-associated protein that promotes cell survival via the
inhibition of apoptosis (42,43). The expression of two important
proteins, Akt and ERK, which are downstream of VEGFR-2, was
examined and it was revealed that the expression of p-Akt and p-ERK
was decreased in the Apatinib and Tripterine groups and further
decreased in the combination group at 48 h post-treatment.
A combination of Apatinib and Tripterine
significantly inhibited the proliferation, invasion and migration
of Hep3B cells while promoting caspase-dependent apoptosis.
However, there are certain limitations to the present study. To
begin with, the expression of Bax was examined during an apoptosis
assay. As previously mentioned, Bax translocation from the cytosol
to the outer mitochondrial membrane releases several pro-apoptotic
factors (44); consequently,
detecting cellular Bax localization will be more intuitive and
representative and should be the focus of future study. The
activation of Caspase-3 (cleaved caspase-3) indicates the
progression of apoptosis into an irreversible stage, which can
irrefutably demonstrate that apoptosis occurs (45). In future experiments, a cleaved
caspase-3 and caspase-3 precursor should be detected to further
confirm the results. Furthermore, when the expression of VEGFR-2 in
Hep3B cells was examined, the specific location of VEGFR-2 in Hep3B
cells was not established. However, according to previous reports,
VEGFR-2 should be located on the cell membrane (46). Future studies should use
non-permealibilized cells as a control and a membrane marker to
verify whether VEGFR-2 was expressed on the cellular membrane. In
conclusion, the present study may represent a potential treatment
strategy for patients in the advanced stages of liver cancer.
However, more studies should be conducted in order to evaluate the
safety and the appropriate dose in a clinical setting.
Acknowledgements
The authors would like to thank Dr Zhaoyu Zhong
(Harbin Medical University, Harbin, China) for providing advice and
technical assistance regarding the design of this study.
Funding
The present study was supported by grants from the
Research Foundation of Beijing Friendship Hospital, Capital Medical
University (grant no. yyqdkt 2015-11), the Beijing Natural Science
Foundation (grant no. 7172081) and the Beijing Administration of
Traditional Chinese Medicine (grant no. JJ2016-16).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL and BC designed the study. The data were analyzed
by HL, LZ, FY and YF. The figures were prepared by HL and LZ. HL
wrote the paper. All authors reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Akt
|
protein kinase B
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
p-Akt
|
phosphorylated protein kinase B
|
|
p-ERK
|
phosphorylated extracellular
signal-regulated kinase
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
References
|
1
|
Torre LA, Sauer AM, Chen MS Jr,
Kagawa-Singer M, Jemal A and Siegel RL: Cancer statistics for Asian
Americans, native Hawaiians, and Pacific Islanders, 2016:
Convergence of incidence between males and females. CA Cancer J
Clin. 66:182–202. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molina-Sánchez P and Lujambio A:
Strategies for HCC target discovery. Aging (Albany NY).
9:1088–1089. 2017.PubMed/NCBI
|
|
3
|
Azumi M, Suda T, Terai S, Terai K and
Akazawa K: Prognostic impact of indocyanine green plasma
disappearance rate in hepatocellular carcinoma patients after
radiofrequency ablation: A prognostic nomogram study. Intern Med.
56:1001–1007. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu SJ: A concise review of updated
guidelines regarding the management of hepatocellular carcinoma
around the world: 2010–2016. Clin Mol Hepatol. 22:7–17. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin J, Shen A, Chen H, Liao J, Xu T, Liu
L, Lin J and Peng J: Nitidine chloride inhibits hepatic cancer
growth via modulation of multiple signaling pathways. BMC Cancer.
14:7292017. View Article : Google Scholar
|
|
6
|
Liu Z, Wang T, Zhang Z, Tang S, Feng S,
Yue M, Hu M, Xuan L and Chen Y: Survivin downregulation using siRNA
nanoliposomes inhibits cell proliferation and promotes the
apoptosis of MHCC-97H hepatic cancer cells: An in vitro and
in vivo study. Oncol Lett. 13:2723–2730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kelley RK, Verslype C, Cohn AL, Yang TS,
Su WC, Burris H, Braiteh F, Vogelzang N, Spira A, Foster P, et al:
Cabozantinib in hepatocellular carcinoma: Results of a phase 2
placebo-controlled randomized discontinuation study. Ann Oncol.
28:528–534. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Al-Rajabi R, Patel S, Ketchum NS, Jaime
NA, Lu TW, Pollock BH and Mahalingam D: Comparative dosing and
efficacy of sorafenib in hepatocellular cancer patients with
varying liver dysfunction. J Gastrointest Oncol. 6:259–267.
2017.
|
|
9
|
Peng S, Zhang Y, Peng H, Ke Z, Xu L, Su T,
Tsung A, Tohme S, Huang H, Zhang Q, et al: Intracellular autocrine
VEGF signaling promotes EBDC cell proliferation, which can be
inhibited by Apatinib. Cancer Lett. 373:193–202. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roviello G, Ravelli A, Polom K, Petrioli
R, Marano L, Marrelli D, Roviello F and Generali D: Apatinib: A
novel receptor tyrosine kinase inhibitor for the treatment of
gastric cancer. Cancer Lett. 372:187–191. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li F, Liao Z, Zhao J, Zhao G, Li X, Du X,
Yang Y and Yang J: Efficacy and safety of Apatinib in stage IV
sarcomas: Experience of a major sarcoma center in China.
Oncotarget. 8:64471–64480. 2017.PubMed/NCBI
|
|
12
|
Li K and Li J: Current molecular targeted
therapy in advanced gastric cancer: A comprehensive review of
therapeutic mechanism, clinical trials, and practical application.
Gastroenterol Res Pract. 2016:41056152016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang H: Apatinib for molecular targeted
therapy in tumor. Drug Des Devel Ther. 9:6075–6081. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu X, Cao J, Hu W, Wu C, Pan Y, Cai L,
Tong Z, Wang S, Li J, Wang Z, et al: Multicenter phase II study of
Apatinib in non-triple-negative metastatic breast cancer. BMC
Cancer. 14:8202014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Langer CJ, Mok T and Postmus PE: Targeted
agents in the third-/fourth-line treatment of patients with
advanced (stage III/IV) non-small cell lung cancer (NSCLC). Cancer
Treat Rev. 39:252–260. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kou P, Zhang Y, Shao W, Zhu H, Zhang J,
Wang H, Kong L and Yu J: Significant efficacy and well safety of
apatinib in an advanced liver cancer patient: a case report and
literature review. Oncotarget. 8:20510–20515. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shrivastava S, Jeengar MK, Reddy VS, Reddy
GB and Naidu VG: Anticancer effect of celastrol on human triple
negative breast cancer: Possible involvement of oxidative stress,
mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp Mol
Pathol. 98:313–327. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH
and Chun KH: Celastrol inhibits gastric cancer growth by induction
of apoptosis and autophagy. BMB Rep. 47:697–702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rajendran P, Li F, Shanmugam MK, Kannaiyan
R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP, et al:
Celastrol suppresses growth and Induces apoptosis of human
hepatocellular carcinoma through the modulation of STAT3/JAK2
signaling cascade in vitro and in vivo. Cancer Prev Res. 5:631–643.
2012. View Article : Google Scholar
|
|
20
|
Li H, Li Y, Liu D, Sun H and Liu J:
miR-224 is critical for celastrol-induced inhibition of migration
and invasion of hepatocellular carcinoma cells. Cell Physiol
Biochem. 32:448–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chakravarthy R, Clemens MJ, Pirianov G,
Perdios N, Mudan S, Cartwright JE and Elia A: Role of the eIF4E
binding protein 4E-BP1 in regulation of the sensitivity of human
pancreatic cancer cells to TRAIL and celastrol-induced apoptosis.
Biol Cell. 105:414–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang S and Liu G: Targeting the
Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett.
13:1041–1047. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Popper HH: Progression and metastasis of
lung cancer. Cancer Metastasis Rev. 35:75–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arvelo F, Sojo F and Cotte C: Tumour
progression and metastasis. Ecancermedicalscience. 10:6172016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vilanova G, Colominas I and Gomez H: A
mathematical model of tumour angiogenesis: Growth, regression and
regrowth. J R Soc Interface. 14:201609182017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fontanella C, Ongaro E, Bolzonello S,
Guardascione M, Fasola G and Aprile G: Clinical advances in the
development of novel VEGFR2 inhibitors. Ann Transl Med.
2:1232014.PubMed/NCBI
|
|
27
|
Bertino G, Demma S, Ardiri A, Proiti M,
Gruttadauria S, Toro A, Malaguarnera G, Bertino N, Malaguarnera M,
Malaguarnera M and Di Carlo I: Hepatocellular carcinoma: Novel
molecular targets in carcinogenesis for future therapies. Biomed
Res Int. 2014:2036932014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao Y and Adjei AA: Targeting
angiogenesis in cancer therapy: Moving beyond vascular endothelial
growth factor. Oncologist. 20:660–673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Al-Abd AM, Alamoudi AJ, Abdel-Naim AB,
Neamatallah TA and Ashour OM: Anti-angiogenic agents for the
treatment of solid tumors: Potential pathways, therapy and current
strategies-A review. J Adv Res. 8:591–605. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding J, Chen X, Gao Z, Dai X, Li L, Xie C,
Jiang H, Zhang L and Zhong D: Metabolism and pharmacokinetics of
novel selective vascular endothelial growth factor receptor-2
inhibitor apatinib in humans. Drug Metab Dispos. 41:1195–1210.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schulte N, Ebert MP and Härtel N: Gastric
cancer: New drugs-New strategies. Gastrointest Tumors. 1:180–194.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Qin S, Xu J, Guo W, Xiong J, Bai Y,
Sun G, Yang Y, Wang L, Xu N, et al: Apatinib for
chemotherapy-refractory advanced metastatic gastric cancer: Results
from a randomized, placebo-controlled, parallel-arm, phase II
trial. J Clin Oncol. 31:3219–3225. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roviello G, Ravelli A, Fiaschi AI,
Cappelletti MR, Gobbi A, Senti C, Zanotti L, Polom K, Reynolds AR,
Fox SB and Generali D: Apatinib for the treatment of gastric
cancer. Expert Rev Gastroenterol Hepatol. 10:887–892.
2016.PubMed/NCBI
|
|
34
|
Venkatesha SH and Moudgil KD: Celastrol
and its role in controlling chronic diseases. Adv Exp Med Biol.
928:267–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Z, Zhai Z and Du X: Celastrol
inhibits migration and invasion through blocking the NF-κB pathway
in ovarian cancer cells. Exp Ther Med. 14:819–824. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Figueiredo SAC, Salvador JAR, Cortés R and
Cascante M: Novel celastrol derivatives with improved selectivity
and enhanced antitumour activity: Design, synthesis and biological
evaluation. Eur J Med Chem. 138:422–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brahmbhatt H, Uehling D, Al-Awar R, Leber
B and Andrews D: Small molecules reveal an alternative mechanism of
Bax activation. Boichem J. 473:1073–1083. 2016. View Article : Google Scholar
|
|
38
|
Luna-Vargas MPA and Chipuk JE:
Physiological and pharmacological control of BAK, BAX, and
beyond.trends. Cell Biol. 26:906–917. 2016.
|
|
39
|
Todt F, Cakir Z, Reichenbach F,
Emschermann F, Lauterwasser J, Kaiser A, Ichim G, Tait SW, Frank S,
Langer HF and Edlich F: Differential retrotranslocation of
mitochondrial Bax and Bak. EMBO J. 34:67–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burgess JT, Bolderson E, Adams MN, Baird
AM, Zhang SD, Gately KA, Umezawa K, O'Byrne KJ and Richard DJ:
Activation and cleavage of SASH1 by caspase-3 mediates an apoptotic
response. Cell Death Dis. 7:e24692016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kochetkova EY, Blinova GI, Bystrova OA,
Martynova MG, Pospelov VA and Pospelova TV: Targeted elimination of
senescent Ras-transformed cells by suppression of MEK/ERK pathway.
Aging (Albany NY). 9:2352–2375. 2017.PubMed/NCBI
|
|
42
|
Adlung L, Kar S, Wagner MC, She B,
Chakraborty S, Bao J, Lattermann S, Boerries M, Busch H, Wuchter P,
et al: Protein abundance of AKT and ERK pathway components governs
cell type-specific regulation of proliferation. Mol Syst Biol.
13:9042017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ewald F, Nörz D, Grottke A, Bach J,
Herzberger C, Hofmann BT, Nashan B and Jücker M: Vertical targeting
of AKT and mTOR as well as Dual targeting of AKT and MEK signaling
is synergistic in hepatocellular carcinoma. J Cancer. 6:1195–1205.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Renault TT and Chipuk JE: Death upon a
kiss: Mitochondrial outer membrane composition and organelle
communication govern sensitivity to BAK/BAX-dependent apoptosis.
Chem Biol. 21:114–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McIlwain DR, Berger T and Mak TW: Caspase
functions in cell death and disease. Cold Spring Harb Perspect
Biol. 7:a0086562013.
|
|
46
|
Wu W, Zhang D, Pan D, Zuo G, Ren X and
Chen S: Downregulation of vascular endothelial growth factor
receptor-2 under oxidative stress conditions is mediated by
β-transduction repeat-containing protein via glycogen synthase
kinase-3β signaling. Int J Mol Med. 37:911–920. 2016. View Article : Google Scholar : PubMed/NCBI
|