Introduction
Malignant glioma is a disease that severely impairs
human health. In our previous study, alkylglycerone phosphate
synthase (AGPS) was found to be an oncogene, and the hypothesis was
made that AGPS may be a target of glioma inhibitors (1,2). The aim
of the present study was to design nitrogenous heterocyclic
compounds via computer-aided design and determine the molecular
biological differences between tumor cells and normal cells, in
order to improve selectivity and enhance the current research on
targetability of the nitrogenous heterocyclic compounds. AGPS was
selected as the target around which to structure a two-dimensional
(2D) and 3D structure-activity model via computer-aided drug design
(CADD) technology to make macromolecular docking. Nitrogenous
heterocyclic compounds were designed and a database was established
in an effort to obtain a comprehensive understanding of the
structure-activity relationship among candidate compounds.
It has been found that the onset and progression
course of a tumor is often accompanied with lipid metabolic
confusion (3). The inactivation of
key enzymes synthesized by AGPS can lower the ether ester level of
tumor cells and reduce the pathogenicity of the cancer, while their
overexpression can raise the ether ester level of the tumor cells
and enhance cell proliferation and movement ability, thereby
promoting growth and invasion of tumor (4). This indicates the potential of AGPS to
become a novel target for antineoplastic agents, and its specific
inhibitor will provide advantages which surpass those provided by
traditional chemotherapy. Our previous study also validated the use
of RNA interference technology to silence the expression of AGPS in
glioma and hepatoma cells in order to inhibit their proliferation
and invasion, and to improve the sensitivity of cells that are
resistant to medicine (5).
Based on the aforementioned study (6), AGPS was selected as target to structure
2D and 3D structure-activity association models via CADD technology
in the present study. A total of 12 nitrogenous heterocyclic
compounds were designed and a database was established, in order to
obtain a comprehensive understanding of the structure-activity
associations among the candidate compounds and then further
optimize the lead compound structure.
Materials and methods
Protein structure and the
database
A 3D structure model [Protein Data Bank (PDB) ID:
2UUV] of human AGPS was downloaded from PDB (7). The amino acid residue in the crystal
structure did not mutate. Crystal structure resolution was
<2.6.
Ligand preparation
Ligand preparation was designed by chemical drug
experience and utilized ChemBioDraw Ultra 11.0 in the ChemOffice
2010 software package (PerkinElmer, Inc., Waltham, MA, USA) to plot
the micromolecule planar construction. The plotted planar
construction was then imported into ChemBio3D Ultra 11.0 to
generate a spatial structure, which was then saved in *.mol2
format. The file in *.mol2 format was imported into Discovery
Studio 3.5 software (BIOVIA, San Diego, CA, USA) for docking and
Erwin Schrodinger 2009 software (Schrodinger, LLC, Portland, OR,
USA) for absorption, distribution, metabolism and excretion (ADME)
analysis, and the LigPrep module was adopted to optimize the
micromolecule, desalt and add an electric charge to produce
ionization consistent with human body pH. The force field of the
optimization molecule was OPLS_2005, which is the same as that of
the optimization receptor, thereby forming a tautomeride of the
molecule. The alloisomerism treatment adopted the input spatial
structure to perform the chiral method of molecular docking.
Molecular docking
Molecular docking was conducted using the ‘docking’
module in the Discovery Studio 3.5 software, with ‘receptor box
producing’ selected and centered on the original ligand
micromolecule to automatically generate the receptor box file.
Flexible docking with standard accuracy was adopted. All
calculations were conducted using the Dell Precision T5500
workstation (Dell Inc., Round Rock, TX, USA) with the Red Hat
Enterprise Linux 6.0 operating system (Red Hat, Raleigh, NC,
USA).
The compound structure was imported into Discovery
Studio 3.5, and the preparation work for the micromolecule prior to
docking was completed using the prepare ligands tool module in the
software package, including restoring valence, generating 3D
conformation and minimizing the energy of the micromolecule.
Molecular docking was performed by the CDOCKER module in Discovery
Studio 3.5, with the original ligand molecule as the center and 9Å
as the radius, to automatically generate docking region. The result
used ‘CDOCKER ENERGY’ as the scoring index. The larger the value
was, the larger the affinity of receptor docking.
ADME analysis
ADME was an important part in the medicine discovery
process to select and optimize the compound. The downloaded AGPS
target crystal structure (2UUV) was imported into Schrodinger 2009
for ADME analysis in order to first correct the chemical bond in
the protein structure, treat the metallic ions, hydrogenate, make
the amino acid residue at end N and end C neutral, reserve the
original ligand and protein, and remove impurity atoms and water
molecules. The OPLS_2005 position was selected to optimize the
protein structure, and sample water orientations served as the
parameters to optimize the hydrogen bond, with the convergence
standard being RMSD0.5Å.
Toxicity analysis
Toxicity prediction was also important in the
selection of a potential drug. Toxicity analysis was conducted
using the ‘TOPKAT’ module in Discovery Studio 3.5 software.
Toxicity parameters included rodent carcinogenicity, mutagenicity,
the ames test, skin irritancy, ocular irritation, aerobic
biodegradability, and whether the molecule was a non-carcinogen,
non-mutagen and non-degradable.
Cell culture
AGPS was demonstrated to be overexpressed in glioma
in a previous study (8), so the
glioma U251 cell line was used to explored the activity of
compounds at first, provided by the Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China). The cells were
cultured in Dulbecco's modified Eagle's medium with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) in a cell incubator with 5% CO2 at 37°C.
MTS assay
A total of 5×103 U251 cells in suspension
with Dulbecco's modified Eagle's medium with 10% fetal bovine serum
were added to 96-well plates at 37°C for 24 h, and N1-N12 (designed
by the School of Pharmacy, Tianjin Medical University, Tianjin,
China; synthesized by Werian Biotech Co., Jinan, China) at
different concentrations (0, 2, 5, 10, 20, 50, 100 and 200 µM) were
added at 37°C for 72 h. Then MTS (Promega Corporation, Madison, WI,
USA) solvent was added and incubated at 37°C for 1 h and the OD
value was measured using a Multiskan FC Microplate Reader (Thermo
Fisher Scientific Inc.). The cell proliferation was then measured
by MTS assay. Inhibition rate was calculated as follows: Inhibition
rate (%)=(1-optical density valuetreatment group/optical
density valuecontrol group) ×100. The half maximal
inhibitory concentration (IC50) was calculated by
Graphpad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA,
USA).
High-content screening (HCS)
assay
The cell cycle and apoptosis were measured by HCS
platform CellInsight CX5 (Thermo Fisher Scientific, Inc.) via
fluorescence intensity. For the cell cycle measurement,
8×103 U251 cells in suspension were added to 96-well
plates with Premo Cdt1-red fluorescent protein and Premo
geminin-green fluorescent protein (both Thermo Fisher Scientific,
Inc.) at 37°C for 24 h. N1-N12 (100 µM) [with 100 µl
phosphate-buffered saline (PBS) as the negative control] were added
for 24 h, and then the cell cycle was measured and analyzed by HCS
platform at 488 nm excitation.
For the cell apoptosis measurements,
8×103 U251 cells in suspension were added to 96-well
plates for 24 h, and 100 µM compounds (with PBS as the negative
control) were added for 24 h. The cells were then incubated with
Annexin V and Hoechst 33258 (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) in the dark at room temperature for 15 min, and the cell
apoptosis was measured and analyzed by HCS platform at 488 nm
excitation.
Statistical analysis
The experimental data were statistically analyzed
with SPSS 11.0 statistical software (SPSS, Inc., Chicago, IL, USA)
and are expressed as the mean ± standard deviation. The statistical
analysis was performed using one-way analysis of variance with the
Tukey-Kramer post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Analysis on affinity
A total of 12 compounds were designed for the
analysis on affinity, which is represented by the docking score (a
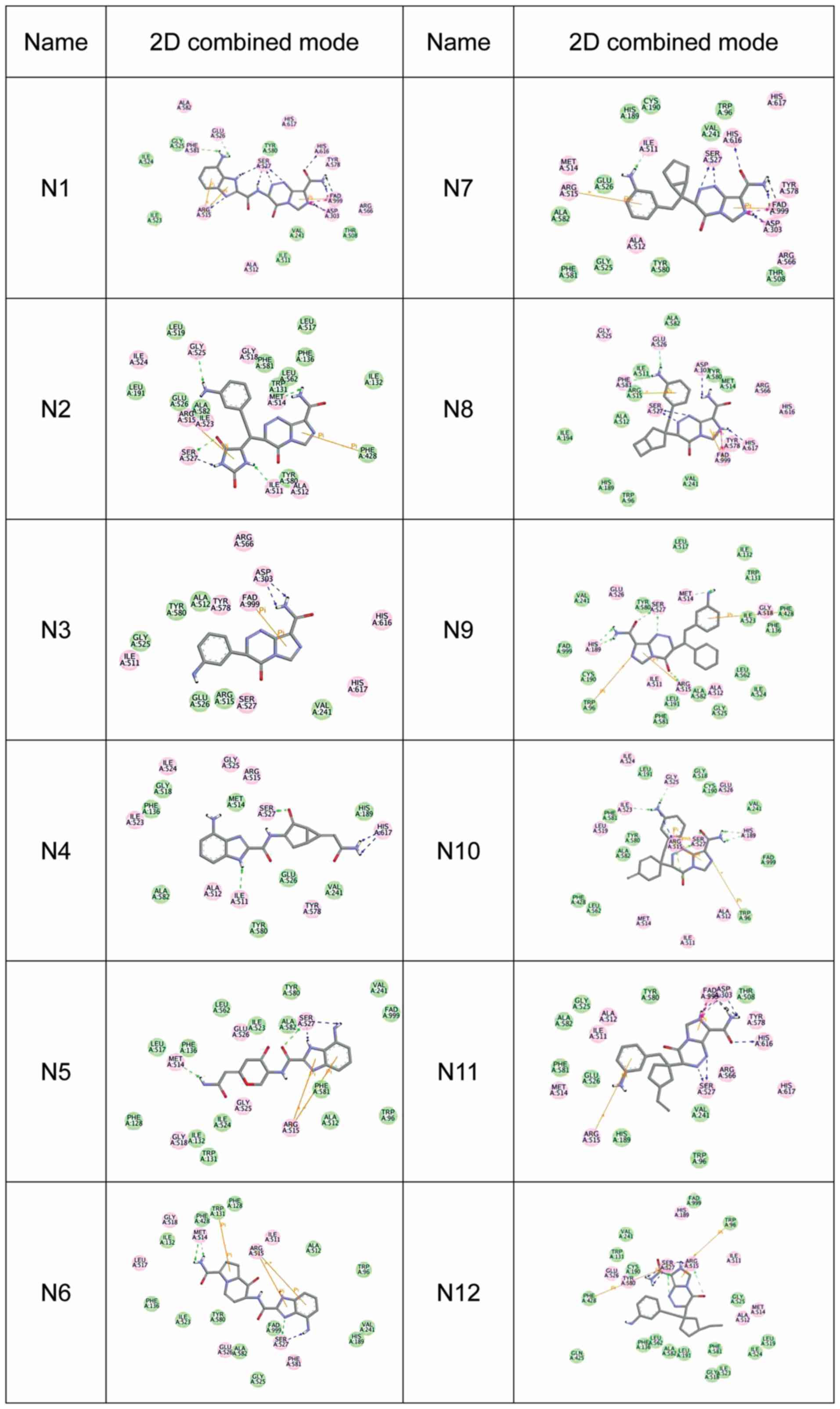
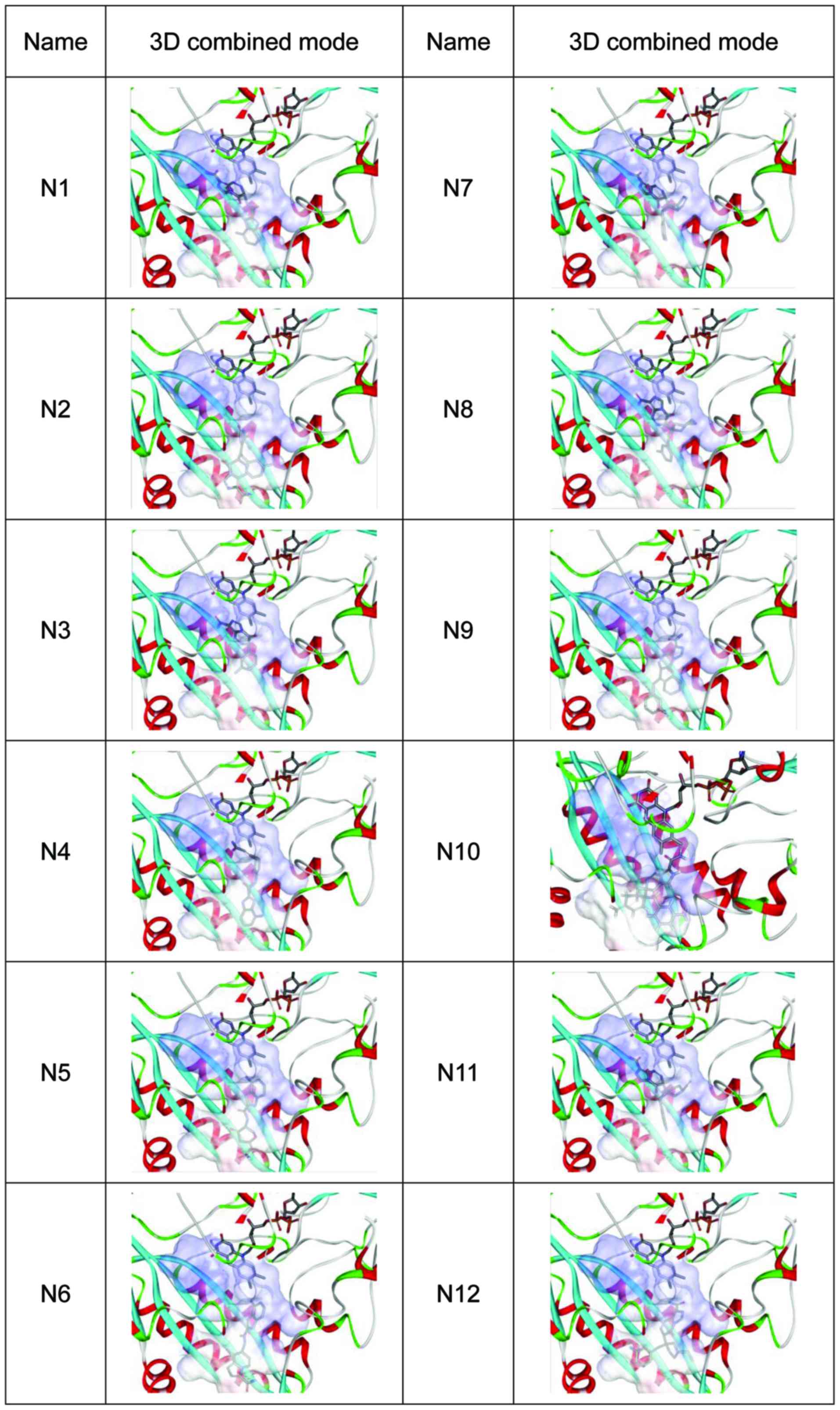
high docking score indicates a high affinity) (Fig. 1). The 2D combined mode with amino acid
residue (Fig. 2) and the 3D combined
mode (Fig. 3) with AGPS show the
hydrophobic bond (green circle and green line), hydrogen (pink
circle and blue line) and pi bond (a covalent bond, orange line)
interactions of the compounds (the dotted lines in Fig. 2).
ADME prediction
ADME properties are an important index to check
whether clinical candidates can reach the required standard
(9). Bad properties imply a high risk
of failure for this candidate, which may become a less than ideal
drug. The ADME of an ideal medicine is as follows: Hydrogen bond
donor, ≤5; hydrogen bond receptor, <10; molecular weight,
<500 Da; lipid water partition coefficient, <5;
water-solubility partition coefficient, −6.5<logS<-0.5; and
polar surface area, 7.0–200.0 (10).
Table I shows the ADME
results of the nitrogenous heterocyclic compounds, with some that
satisfied the aforementioned qualifications. N4, N5, N6, N7, N8 and
N12 were considered to exhibit potential drug properties.
 | Table I.Absorption, distribution, metabolism
and excretion prediction of nitrogenous heterocyclic compounds. |
Table I.
Absorption, distribution, metabolism
and excretion prediction of nitrogenous heterocyclic compounds.
| Compound | MW (Da) | donorHB | accptHB | QPlogPo/w | QPlogS | PSA |
|---|
| N1 | 353.299 | 4.75 | 13.75 | −2.350 | −2.809 | 215.170 |
| N2 | 394.349 | 5.50 | 14.00 | −2.247 | −2.459 | 230.122 |
| N3 | 270.250 | 3.50 | 10.50 | −1.464 | −2.210 | 148.217 |
| N4 | 323.310 | 4.75 | 8.75 | −0.409 | −3.568 | 170.861 |
| N5 | 329.315 | 4.75 | 9.50 | −0.617 | −3.307 | 178.267 |
| N6 | 350.336 | 4.75 | 8.75 | 0.100 | −3.810 | 172.853 |
| N7 | 362.390 | 3.50 | 9.50 | 0.008 | −2.991 | 144.227 |
| N8 | 376.417 | 3.50 | 9.50 | 0.259 | −3.435 | 143.247 |
| N9 | 376.417 | 3.50 | 10.50 | 0.488 | −2.838 | 142.237 |
| N10 | 376.417 | 3.50 | 9.50 | 0.505 | −2.489 | 135.360 |
| N11 | 376.417 | 4.00 | 10.50 | 0.340 | −2.651 | 139.990 |
| N12 | 380.449 | 3.50 | 9.50 | 0.506 | −2.749 | 139.842 |
Toxicity prediction
Table II shows the
toxicity prediction results of the nitrogenous heterocyclic
compounds. All compounds were found to exhibit suitable toxicity
for drug development.
| Table II.Toxicity prediction of nitrogenous
heterocyclic compounds. |
Table II.
Toxicity prediction of nitrogenous
heterocyclic compounds.
| Compound | Mouse female NTP
prediction | Mouse male NTP
prediction | Rat female NTP
prediction | Rat male NTP
prediction | Ames prediction | Skin irritancy | Ocular irritancy | Aerobic
biodegradability prediction |
|---|
| N1 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Mutagen | None | Mild | Non-degradable |
| N2 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N3 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N4 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N5 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N6 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Mutagen | None | Mild | Non-degradable |
| N7 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Mutagen | None | Mild | Non-degradable |
| N8 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Mutagen | None | Mild | Non-degradable |
| N9 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Mutagen | None | Mild | Non-degradable |
| N10 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N11 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
| N12 | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-carcinogen | Non-mutagen | None | Mild | Non-degradable |
Effect of nitrogenous heterocyclic
compounds on the proliferation of U251 cells
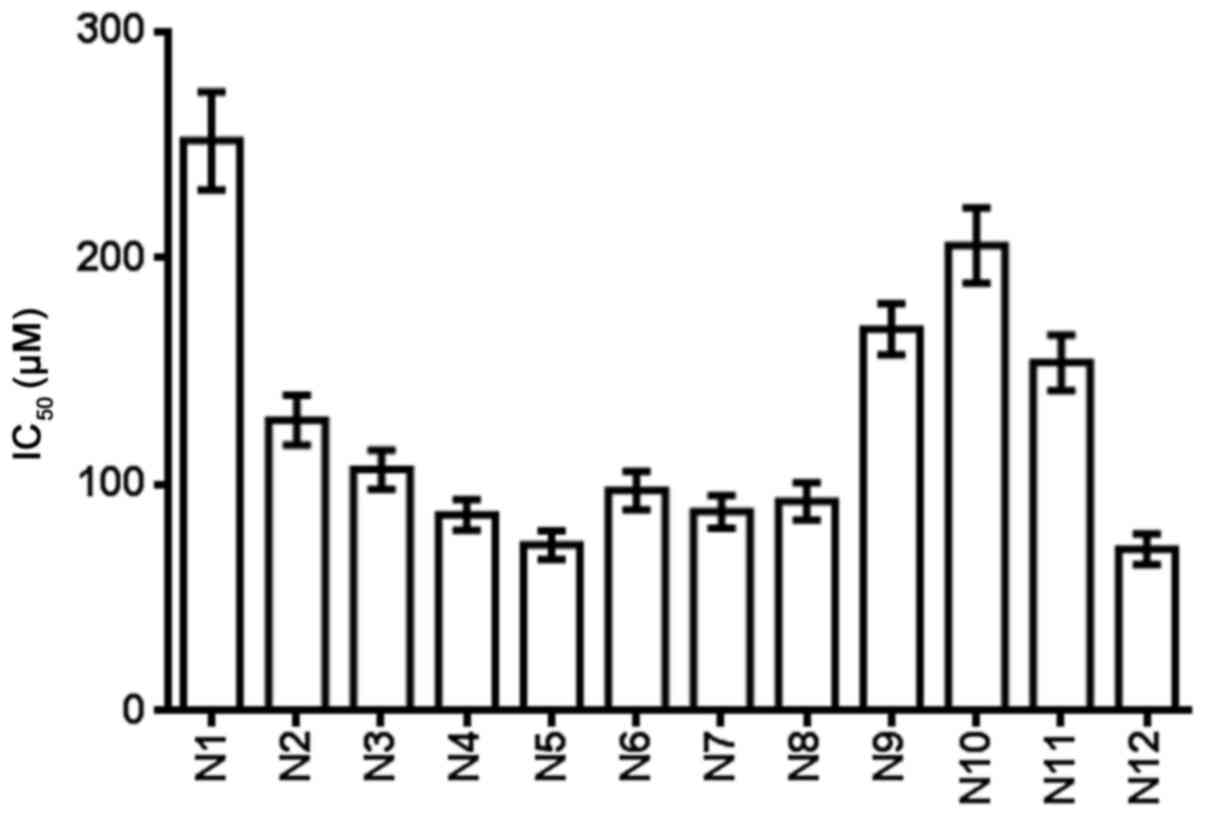
Table III and
Fig. 4 show the IC50 of
the nitrogenous heterocyclic compounds for 72 h. It was found that
compounds N4, N5, N6, N7, N8 and N12, which had IC50
values of <100 µM, were suitable for drug development.
| Table III.IC50 of nitrogenous
heterocyclic compounds in U251 cells. |
Table III.
IC50 of nitrogenous
heterocyclic compounds in U251 cells.
| Compound | IC50,
µM |
|---|
| N1 | 251.7 |
| N2 | 128.5 |
| N3 | 106.8 |
| N4 | 86.7 |
| N5 | 73.5 |
| N6 | 97.5 |
| N7 | 88.2 |
| N8 | 92.8 |
| N9 | 168.7 |
| N10 | 205.6 |
| N11 | 153.8 |
| N12 | 71.7 |
Effect of nitrogenous heterocyclic
compounds on the cell apoptosis of U251 cells
Table IV shows the
effect of the nitrogenous heterocyclic compounds on cell apoptosis
for 24 h. Compounds N4, N5, N6, N7, N8 and N12 induced a >10%
cell apoptosis rate at a 100 µM concentration (Fig. 5).
| Table IV.Apoptosis rate of nitrogenous
heterocyclic compounds in U251 cells. |
Table IV.
Apoptosis rate of nitrogenous
heterocyclic compounds in U251 cells.
| Compound | Apoptosis rate,
% |
|---|
| PBS control | 1.1 |
| N1 | 8.7 |
| N2 | 6.2 |
| N3 | 6.3 |
| N4 | 18.7 |
| N5 | 24.5 |
| N6 | 15.6 |
| N7 | 21.8 |
| N8 | 17.2 |
| N9 | 6.3 |
| N10 | 8.7 |
| N11 | 5.5 |
| N12 | 23.7 |
Effect of nitrogenous heterocyclic
compounds on the cell cycle of U251 cells
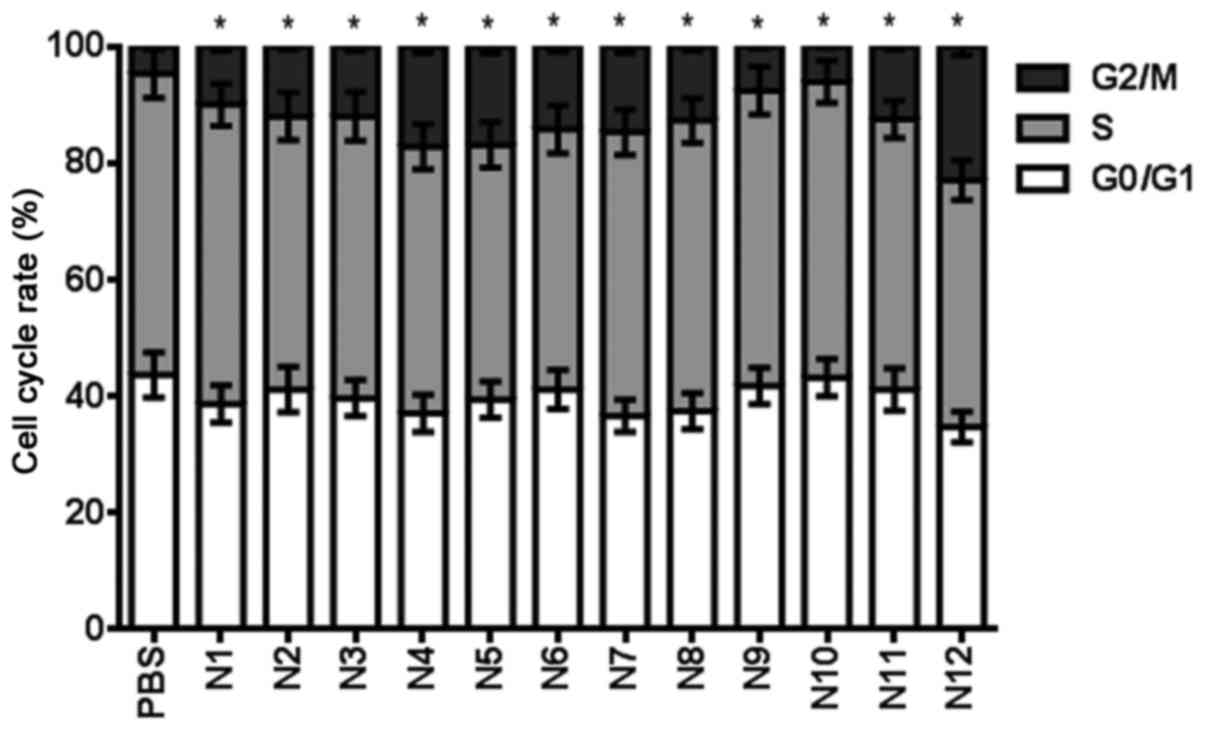
Table V shows the
effect of nitrogenous heterocyclic compounds on the cell cycle for
24 h. It was found that compounds N2, N3, N4, N5, N6, N7, N8, N11
and N12 induced a >10% cell cycle arrest at a 100 µM
concentration (Fig. 6).
| Table V.Effect of nitrogenous heterocyclic
compounds on the cell cycle of U251 cells. |
Table V.
Effect of nitrogenous heterocyclic
compounds on the cell cycle of U251 cells.
| Cell cycle stage
(%) | PBS | N1 | N2 | N3 | N4 | N5 | N6 | N7 | N8 | N9 | N10 | N11 | N12 |
|---|
|
G0/G1 | 43.7 | 38.7 | 41.2 | 39.7 | 37.1 | 39.5 | 41.2 | 36.7 | 37.5 | 41.8 | 43.2 | 41.2 | 34.8 |
| S | 51.6 | 51.3 | 46.8 | 48.3 | 45.7 | 43.6 | 44.6 | 48.6 | 49.8 | 50.6 | 50.7 | 46.3 | 42.3 |
|
G2/M | 4.7 | 10.0 | 12.0 | 12.0 | 17.2 | 16.9 | 14.2 | 14.7 | 12.7 | 7.6 | 6.1 | 12.5 | 22.9 |
Discussion
Malignant glioma is a disease that seriously damages
human health; it accounts for ~70% of primary malignant brain
tumors, with an annual incidence of ~5/100,000 individuals
(11,12). The number of new cases every year
exceeds 14,000. As for glioblastoma (World Health Organization
stage IV) patients, median survival time is only between 14.6 and
17 months, and the annual number of associated mortalities reaches
30,000 (13–15). The chemical features comprise: The
hydrogen bond receptor, hydrogen bond donor, hydrophobic center,
positive charge center, negative charge center and aromatic ring
center (16–18). In the present study, compounds N1-N12
were first docked, resulting in high docking scores with the target
AGPS. This implies their potential ability to inhibit AGPS.
In previous years, the medicine discovery phase
mainly focused on the discovery of active compounds, and other
problems, including pharmacokinetics, toxicity, solubility and
stability, were not considered until the development phase. Thus,
solely analyzing the affinity of the compound and target cannot
ascertain the possibility of these compounds becoming potential
medicines (19). ADME properties are
an important index to check whether clinical candidates can reach
the required standard. Bad properties imply a high risk of failure
for this candidate, which may then become a less than ideal drug.
According to a previous study, ~40% of failures to develop a
medicine in the development phase are due to poor biopharmaceutical
properties (PK and bioavailability) (20). High development expenses make such
failures a primary economic loss in medicine development. Thus,
ADME has become an indispensable part of the medicine discovery
process, and it can be used to supervise the selection and
optimization of precious lead compounds. The QikProp module (in
part of the Erwin Schrodinger 2009 software) can predict the
following properties of compounds: the logarithm value of brain to
plasma concentration ratio, water solubility, liposolubility, MDCK
cell line and Caco-2 cell line for permeability, general central
nervous system activity, K ion channel blocking of human
ether-a-go-go related gene log IC50 and the bonding
activity of serum albumin (log Khsa). The present study experiments
predicted water solubility, polar surface area, MDCK cell
permeability, and oil and water partition coefficient. It was found
that N4, N5, N6, N7, N8 and N12 exhibited potential drug
properties. The toxicity predictions of the compounds were also
investigated with Discovery Studio using the TOPKAT protocol, and
the toxicity of the compounds was found to be suitable for the
development into a medical drug.
HCS was the novel technology used for the drug
activity screening in the present study, and it was able to analyze
the effect on cell proliferation, cell apoptosis and the cell cycle
by high throughput and quick screening using the HCS platform. The
effect on proliferation, apoptosis and the cell cycle by 12
nitrogenous heterocyclic compounds was analyzed, and N4, N5, N6,
N7, N8 and N12 were shown to be most active in vitro
according the ADME prediction, showing the accuracy of the CADD
screening model.
In conclusion, 12 nitrogenous heterocyclic compounds
were designed in the present study, 6 of which presented with
suitable ADME parameters and toxicity predictions; these 6 may be
suitable for development into novel glioma therapeutic drugs. This
study provides a foundation for the study into novel nitrogenous
heterocyclic anti-glioma drugs and future studies will investigate
the toxicity in animal models.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31501159 and
81601047), the Tianjin Public Health Key Research Project (grant
no. 15KG108), the Tianjin Science and Technology Key Project on
Chronic Disease Prevention and Treatment (grant no. 16ZXMJSY00020),
the Special Program of Talent Development for Excellent Youth
Scholars in Tianjin, China (grant no. TJTZJH-QNBJRC-2-9) and the
Tianjin 131 Creative Talents Cultivation Project (1st Class,
2016).
Availability of data and materials
All data and materials relevant to the present study
are described in this published article or available from the
corresponding author on reasonable request.
Authors' contributions
YZ performed the statistical analysis, YZ and YH
designed the study and performed the cell experiments, YM designed
and synthesized the computer-aided drug, and PY performed the
statistical analysis.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ames H, Halushka MK and Rodriguez FJ:
miRNA regulation in gliomas: Usual suspects in glial tumorigenesis
and evolving clinical applications. J Neuropathol Exp Neurol.
76:246–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cives M, Ghayouri M, Morse B, Brelsford M,
Black M, Rizzo A, Meeker A and Strosberg J: Analysis of potential
response predictors to capecitabine/temozolomide in metastatic
pancreatic neuroendocrine tumors. Endocr Relat Cancer. 23:759–767.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dilillo M, Ait-Belkacem R, Esteve C,
Pellegrini D, Nicolardi S, Costa M, Vannini E, Graaf EL, Caleo M
and McDonnell LA: Ultra-high mass resolution MALDI imaging mass
spectrometry of proteins and metabolites in a mouse model of
glioblastoma. Sci Rep. 7:6032017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Piano V, Benjamin DI, Valente S, Nenci S,
Marrocco B, Mai A, Aliverti A, Nomura DK and Mattevi A: Discovery
of inhibitors for the ether lipid-generating enzyme AGPS as
anti-cancer agents. ACS Chem Biol. 10:2589–2597. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu Y, Liu XJ, Yang P, Zhao M, Lv LX,
Zhang GD, Wang Q and Zhang L: Alkylglyceronephosphate synthase
(AGPS) alters lipid signaling pathways and supports chemotherapy
resistance of glioma and hepatic carcinoma cell lines. Asian Pac J
Cancer Prev. 15:3219–3226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu Y, Liu A, Zhang X, Qi L, Zhang L, Xue
J, Liu Y and Yang P: The effect of benzyl isothiocyanate and its
computer-aided design derivants targeting alkylglycerone phosphate
synthase on the inhibition of human glioma U87MG cell line. Tumour
Biol. 36:3499–3509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Razeto A, Mattiroli F, Carpanelli E,
Aliverti A, Pandini V, Coda A and Mattevi A: The crucial step in
ether phospholipid biosynthesis: Structural basis of a noncanonical
reaction associated with a peroxisomal disorder. Structure.
15:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu Y, Zhu L, Lu L, Zhang L, Zhang G, Wang
Q and Yang P: Role and mechanism of the alkylglycerone phosphate
synthase in suppressing the invasion potential of human glioma and
hepatic carcinoma cells in vitro. Oncol Rep. 32:431–436. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Battu MB, Chandra AM, Sriram D and
Yogeeswari P: Pharmacophore-based 3DQSAR and molecular docking
studies to identify new non-peptidic inhibitors of cathepsin S.
Curr Med Chem. 21:1910–1921. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Verma SK and Thareja S: Structure based
comprehensive modelling, spatial fingerprints mapping and ADME
screening of curcumin analogues as novel ALR2 inhibitors. PLoS One.
12:e01753182017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Silginer M, Weller M, Stupp R and Roth P:
Biological activity of tumor-treating fields in preclinical glioma
models. Cell Death Dis. 8:e27532017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin CY and Huang HM: Unilateral malignant
optic glioma following glioblastoma multiforme in the young: A case
report and literature review. BMC Ophthalmol. 17:212017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Louis DN, Perry A, Reifenberger G, et al:
The 2016 World Health Organization classification of tumors of the
central nervous system: A summary. Acta Neuropathol. 131:803–820.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang N, Zhang Q, Ning B, Luo L and Fang Y:
β-Asarone promotes Temozolomide's entry into glioma cells and
decreases the expression of P-glycoprotein and MDR1. Biomed
Pharmacother. 90:368–374. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Holdhoff M, Ye X, Supko JG, Nabors LB,
Desai AS, Walbert T, Lesser GJ, Read WL, Lieberman FS, Lodge MA, et
al: Timed sequential therapy of the selective T-type calcium
channel blocker mibefradil and temozolomide in patients with
recurrent high-grade gliomas. Neuro Oncol. 19:845–852. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai Z, Zhang G, Zhang X, Liu Y and Fu X:
Current insights into computer-aided immunotherapeutic design
strategies. Int J Immunopathol Pharmacol. 28:278–285. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Medeiros Turra K, Pineda Rivelli D,
Berlanga de Moraes Barros S and Mesquita Pasqualoto KF:
Constructing and validating 3D-pharmacophore models to a set of
MMP-9 inhibitors for designing novel anti-melanoma agents. Mol
Inform. 35:238–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valasani KR, Chaney MO, Day VW and Shidu
Yan S: Acetylcholinesterase inhibitors: Structure based design,
synthesis, pharmacophore modeling, and virtual screening. J Chem
Inf Model. 53:2033–2046. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao D, Wang J, Zhou R, Li Y, Yu H and Hou
T: ADMET evaluation in drug discovery. 11. PharmacoKinetics
Knowledge Base (PKKB): A comprehensive database of pharmacokinetic
and toxic properties for drugs. J Chem Inf Model. 52:1132–1137.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gomeni R, Bani M, D'Angeli C, Corsi M and
Bye A: Computer-assisted drug development (CADD): An emerging
technology for designing first-time-in-man and proof-of-concept
studies from preclinical experiments. Eur J Pharm Sci. 13:261–270.
2001. View Article : Google Scholar : PubMed/NCBI
|