Introduction
Prostate cancer (PCa) is one of the most prevalent
cancer types in men; in 2015, there were 60,300 newly diagnosed
cases of PCa in China, resulting in 26,000 mortalities (1). Disease recurrence has been reported in a
large proportion of patients following radical prostatectomy
(2), and castration-resistant disease
typically develops as a result (3,4). Although
prognostic and clinical indicators were implemented, the prognostic
effect was not fully understood (5).
Thus, clinical biomarkers for PCa biochemical recurrence are
required. Huang et al (6) used
long non-coding RNAs to develop a prediction model for biochemical
recurrence; however, the analysis lacked validation datasets.
Over the previous decade, single biomarkers have
been identified for the prognosis of PCa (7–9); however,
the utilization of these biomarkers requires further investigation
owing to the heterogeneity of PCa (10). Multiple gene-based studies of
prognostic biomarkers are currently prevalent owing to their
robustness in multiple different cancer types (11–17).
By associating gene expression and survival
information from The Cancer Genome Atlas (TCGA),
survival-associated genes were identified. Using a random
forest-based variable hunting approach, eight genes were selected
and a risk score staging system was developed. Patients with
high-risk scores had significantly poorer survival rates compared
with patients with low-risk scores. This result was further
validated using an independent dataset from the National Center for
Biotechnology Information (NCBI), GSE70769 (18). Analysis of clinicopathological factors
revealed that the risk score was independent of age but was
significantly associated with Tumor Node Metastasis (TNM) stage
(19), lymph node invasion and
Gleason score. Cox multivariate regression and the 5-year
biochemical recurrence area under the receiver operating curve
(ROC) reveal that the risk score was an important indicator for
prediction of biochemical recurrence.
Materials and methods
Data pre-processing
Raw microarray data of the NCBI dataset GSE70769 was
downloaded from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) (20). Subsequent to background correction and
normalization using the Robust Multi-Array Averaging (RMA) approach
(21), the data was used for further
analysis. The probe names were annotated according to the
manufacturer's annotation file. For genes matching multiple probes,
the average values were calculated and used as the expression
values for the corresponding genes. TCGA gene expression
(https://cancergenome.nih.gov/) data was
downloaded from University of California Santa Cruz Xena and
converted to fragments per kilobase of transcript sequence per
million base pairs sequenced (FPKM) values. The log 2-transformed
RNA-Sequence by expectation-maximization values were retained for
model development.
Prediction gene selection, Cox
multivariate regression model and validation
Cox univariate regression was performed on TCGA
dataset. Genes with relative expression levels associated with
biochemical recurrence-free survival (BFS) were retained for a
further forest-based variable hunting approach, performed as
previously described (22,23). Following 100 repeats and 100
iterations, genes from the top of the list were selected for
further analysis. Finally, eight genes were identified as the most
frequently present in the repeats and iterations, thus these eight
genes were selected for model development. Next, multivariate Cox
regression was performed using the aforementioned genes to
construct a linear risk-score model. In the validation datasets,
coefficients were locked and the risk score for each sample was
calculated. The risk score was calculated using the following
formula; where βi indicates the coefficients evaluated
with gene expression and xi refers to the relative gene
expression level.
Risk score=∑inβi*xi
For the training dataset, the samples were divided
into low- and high-risk groups according to the median risk score
using R software (v3.0.1; http://cran.r-project.org/doc/FAQ/R-FAQ.html) and
packages (24,25).
Statistical analysis
Background correction and RMA normalization of raw
Affymetrix CEL data were performed using the ‘RMA’ function in the
‘affy’ package (v1.56.0) (26). The
survival difference between the high-risk and low-risk groups,
univariate regression in the training dataset, multivariate Cox
proportional hazard model development and multivariate regression
with risk score and other clinical indicators were performed using
the ‘survival’ function in the R package (v1.4–8). The ROCs were
drawn and the area under curve (AUC) calculation was performed
using the R package, ‘pROC’ (v1.11.0) (27). All statistical analysis was performed
using R software and packages. P<0.05 was considered to indicate
a statistically significant difference.
Results
Identification of survival-associated
genes
Univariate Cox regression was performed on TCGA
dataset, following filtering of the non-primary PCa tissues, by
associating BFS and gene expression. Detailed information of the
samples enrolled in TCGA dataset are presented in Table I. Genes significantly associated with
BFS (P<0.01) were retained for further analysis. As the
identified gene panel was relatively large, a random forest-based
variable hunting approach was implemented to retrieve the best
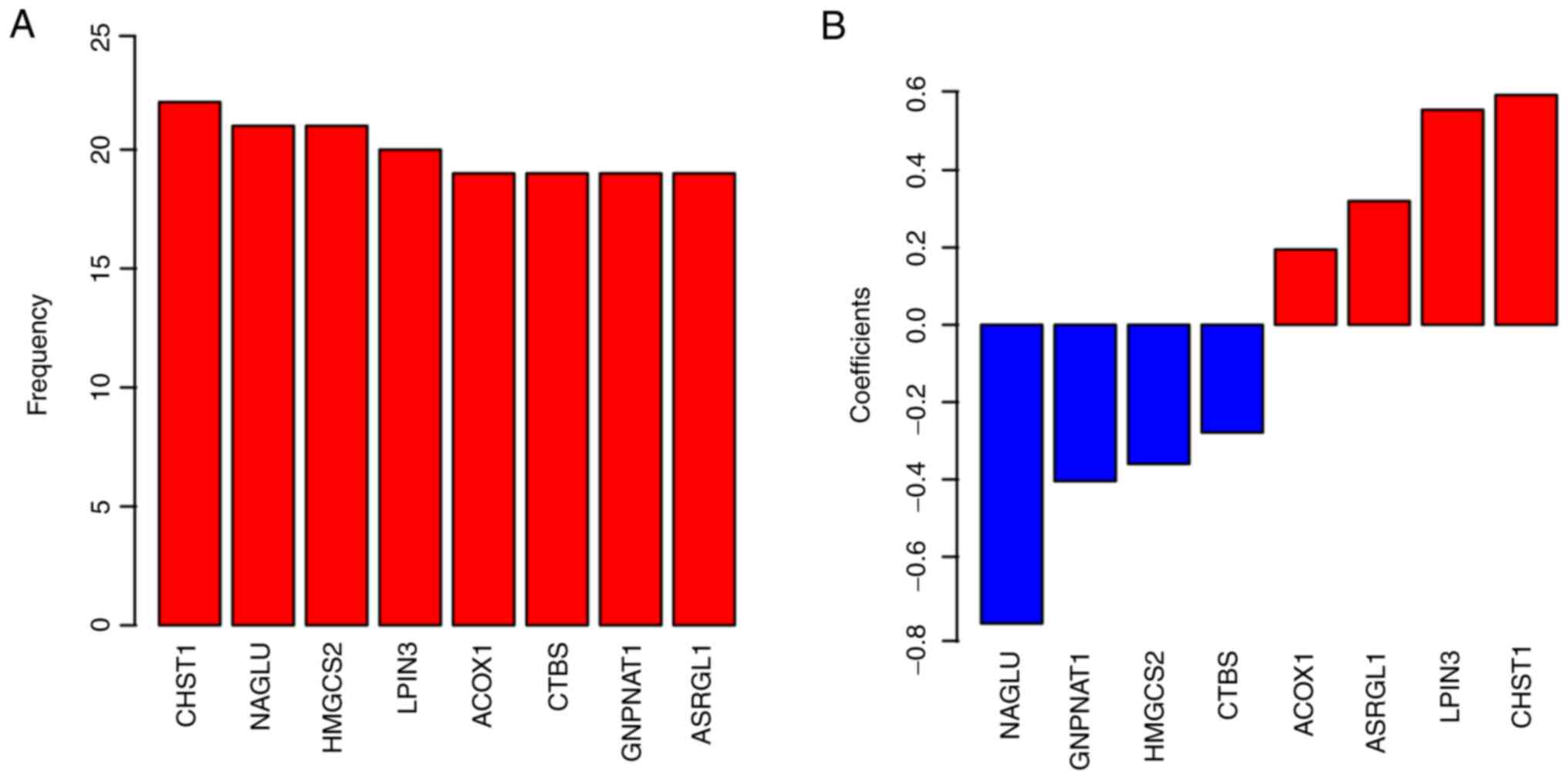
combination of biomarkers. Eight genes were selected for further
model development (Fig. 1A; Table II). Finally, the coefficients are
presented in Fig. 1B. The positive
coefficients suggest that the genes are oncogenes, while the
negative coefficients indicate tumor suppressor genes.
 | Table I.The Cancer Genome Atlas sample
information. |
Table I.
The Cancer Genome Atlas sample
information.
| Variables | Samples, n |
|---|
| Age, years |
|
|
<60 | 138 |
|
>60 | 170 |
| Tumor stage |
|
| T2 | 131 |
|
T3-T4 | 177 |
| Gleason score |
|
| 1 | 21 |
| 2 | 115 |
| 3 | 72 |
| 4 | 38 |
| 5 | 62 |
| Table II.Univariate and multivariate Cox
regression analysis of candidate genes. |
Table II.
Univariate and multivariate Cox
regression analysis of candidate genes.
|
| Cox univariate
regression | Cox multivariate
regression |
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value | HR | 95% CI | P-value |
|---|
| CHST1 | 1.800 | 1.300–2.600 | 0.001 | 1.380 | 0.940–2.020 | 0.100 |
| ACOX1 | 0.300 | 0.130–0.680 | 0.004 | 1.740 | 0.630–4.850 | 0.286 |
| CTBS | 0.400 | 0.250–0.640 | <0.001 | 0.700 | 0.370–1.320 | 0.270 |
| GNPNAT1 | 0.390 | 0.220–0.710 | 0.002 | 0.460 | 0.220–0.990 | 0.047 |
| NAGLU | 0.550 | 0.360–0.840 | 0.006 | 0.670 | 0.440–1.010 | 0.058 |
| LPIN3 | 2.000 | 1.300–3.000 | 0.001 | 1.220 | 0.760–1.950 | 0.419 |
| ASRGL1 | 1.600 | 1.200–2.200 | 0.002 | 1.810 | 1.300–2.520 | <0.001 |
| HMGCS2 | 0.740 | 0.640–0.860 | <0.001 | 0.760 | 0.630–0.900 | 0.002 |
Performance of risk score in the
training dataset
To assess the prognostic value of the risk score
model, the survival difference between high- and low-risk scores
(using the median value as the cut-off) was compared to evaluate
the performance of the risk score. According to the results, the
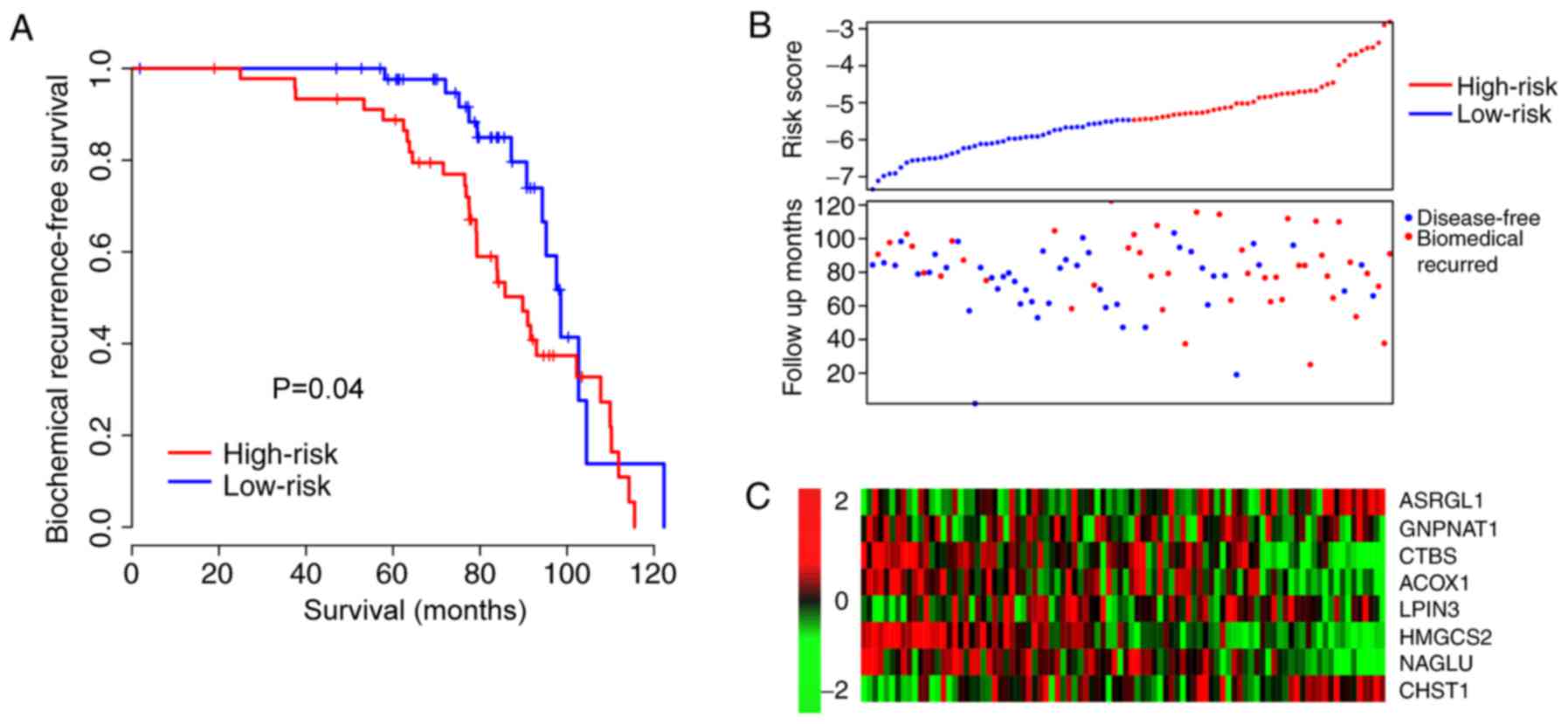
BFS in the high-risk-score group was significantly shorter compared
with the low-risk score group (P=5×10−7; Fig. 2A). As presented in Fig. 2A, samples with early biomedical
recurrence were characterized with a high expression of
asparaginase like 1 (ASRGL1), lipin 3 and carbohydrate
sulfotransferase 1. However, patients without biochemical
recurrence presented with a high expression of
glucosamine-phosphate N-acetyltransferase 1 (GNPNAT1), chitobiase,
acyl-CoA oxidase 1 (ACOX1), 3-hydroxy-3-methylglutaryl-CoA synthase
2 (HMGCS2) and N-acetyl-α-glucosaminidase (NAGLU), which was
consistent with the coefficients (Figs.
1B and 2B). Disease-free survival
time was additionally compared between the high- and low-risk
groups and the result was similar to the BFS pattern as the
survival of the high-risk group was notably lower compared with
that of the low-risk group (Fig. 2C).
The 5-year BFS ROC was identified to be an effective method to
compare the prognostic value of the risk score and other
clinicopathological observations (Fig.
2D). The AUCs of age, Gleason index, primary tumor stage, lymph
node invasion and risk score were 0.597, 0.647, 0.628, 0.578 and
0.819, respectively. Specifically, it is indicated that the
mortality risk of patients with the highest risk scores was very
high. These results indicate that the risk score is better at
predicting BFS than the other clinical observations.
 | Figure 2.Risk score for prognosis in the
training dataset. (A) Biochemical recurrence-free survival rate of
high- and low-risk groups. (B) Heat maps of gene expression for
each dataset. Blue/red dots in the first panel refer to the low and
high-risk groups, respectively. (C) Disease-free survival rates of
high- and low-risk groups. (D) The 5-year survival receiver
operating curve of risk score and other clinical observations and
their AUC. *P<0.001, risk score AUV vs. other clinical
observations. AUC, area under the curve; T stage, tumor stage;
CHST1, carbohydrate sulfotransferase 1; ACOX1, acyl-CoA oxidase 1;
CTBS, chitobiase; GNPNAT1, glucosamine-phosphate
N-acetyltransferase 1; NAGLU, N-acetyl-α-glucosaminidase; LPIN3,
lipin 3; ASRGL1, asparaginase like 1; HMGCS2,
3-hydroxy-3-methylglutaryl-CoA synthase 2. |
Validation of risk score
performance
The high performance of the risk score may have
resulted from the over-fit dataset. To test if over-fitness
existed, the coefficients were locked in order to evaluate the
robustness of this model, and the risk scores were calculated for
an independent NCBI dataset (GSE70769). The samples from the
independent dataset were additionally divvied into high- and
low-risk groups, as with the training dataset. The results were
similar to the BFS profile of the training dataset. The BFS of
patients in the high-risk-score group were significantly shorter
than the low-risk-score group (P=0.04; Fig. 3A) and associated with early biomedical
recurrence (Fig. 3B). The expression
profile was additionally similar to that of the training dataset
(Fig. 3C). These results indicate
that the risk score is a robust indicator for PCa prognosis.
Association between risk score and
other clinical information
Analyses of risk score and clinical information were
performed. The results indicated that the risk score was
significantly associated with primary tumor stage (P<0.05),
Gleason score (P<0.01) and lymph invasion (P<0.01), but not
with age (Fig. 4A). Cox multivariate
regression was performed using the risk score and the
aforementioned clinical observations. The risk score was the only
prognostic indicator identified to be significantly associated with
biochemical recurrence (P=3×10−5; Fig. 4B). In summary, these results indicate
that risk score is an important clinical indicator of PCa
prognosis.
Discussion
Despite the low rate of progression, biomedical
recurrence and metastasis continue to be observed in a large
proportion of patients with PCa (28). Thus, prognostic biomarkers are
urgently required. Over the previous decade, single biomarkers have
been reported to predict the survival of patients with PCa
(3,9,29).
However, the single-biomarker approach to cancer prognosis
assessment is less robust compared with the more widely reported
multiple-biomarker-based models (30–32). Using
machine learning and gene expression, the present study developed a
Cox multivariate regression-based risk score model. The model was
then further evaluated for performance and robustness. The risk
score staging system performed well in predicting survival in two
datasets from different microarray platforms.
Among the candidate genes selected, serum NAGLU has
been reported to be associated with the clinical indicators and
survival of gastrointestinal adenocarcinoma (33); and the expression of another gene,
GNPNAT1, had been demonstrated to be associated with the
progression of castration-resistant PCa (34) via the
phosphatidylinositol3-kinase/protein kinase B signaling pathway.
Proteomics analysis revealed that HMGCS2 expression is altered in
PCa, and that the expression of this gene is associated with the
survival of squamous cell carcinoma following surgery (35,36). It
has additionally been revealed to affect the extracellular
signal-regulated kinase/c-Jun signaling pathway in hepatocellular
carcinoma (37). In addition, ACOX1
has been reported to be associated with migration and metastasis in
the xenografts of colorectal carcinoma (38), and associated with the
mitogen-activated protein kinase signaling pathway in
hepatocellular carcinoma (39). A
similar function was detected for ASRGL1 in endometrial carcinoma
(39), although the underlying
mechanism remains unclear. Collectively, these results indicate
that the candidate genes used in the model are reliable, thus
reinforcing the robustness of the model.
In a previous study, Huang et al (6) used gene expression to predict
biochemical recurrence using TCGA expression data, the study lacked
a validation dataset. The present study was novel as it developed a
robust prediction model for PCa that was validated using another
platform. Indeed, the RNA-sequencing data was presented with
log2-transformed FPKM values, whereas microarray data
was presented as log2-transformed intensity values. The
formula was calculated using the relative gene expression level,
regardless of its unit. This may explain why this model is
functional across different platforms.
However, limitations of the present study exist.
Firstly, the present study is a retrospective study. The clinical
information and long-term follow-up are unavailable, and detailed
clinical information are unavailable. Thus, bias may have resulted.
Secondly, although the robustness of the risk score was validated
using another dataset, the clinical utilization of the risk score
requires further studies in order to fully confirm its efficiency.
The present findings may provide novel insights for predicting the
biochemical recurrence of patients with PCa.
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in china,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brockman JA, Alanee S, Vickers AJ,
Scardino PT, Wood DP, Kibel AS, Lin DW, Bianco FJ Jr, Rabah DM,
Klein EA, et al: Nomogram predicting prostate cancer-specific
mortality for men with biochemical recurrence after radical
prostatectomy. Eur Urol. 67:1160–1167. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Torrecilla Lopez J, Hervas A, Zapatero A,
Gómez Caamaño A, Macías V, Herruzo I, Maldonado X, Iturriaga Gómez
A, Casas F and Segundo González San C: Uroncor consensus statement:
Management of biochemical recurrence after radical radiotherapy for
prostate cancer: From biochemical failure to castration resistance.
Rep Pract Oncol Radiother. 20:259–272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Algarra R, Hevia M, Tienza A, Merino I,
Velis JM, Zudaire J, Robles JE and Pascual I: Survival analysis of
patients with biochemical relapse after radical prostatectomy
treated with androgen deprivation: Castration-resistance
influential factors. Can Urol Assoc J. 8:E333–E341. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Epstein JI, Zelefsky MJ, Sjoberg DD,
Nelson JB, Egevad L, Magi-Galluzzi C, Vickers AJ, Parwani AV,
Reuter VE, Fine SW, et al: A contemporary prostate cancer grading
system: A validated alternative to the gleason score. Eur Urol.
69:428–435. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang TB, Dong CP, Zhou GC, Lu SM, Luan Y,
Gu X, Liu L and Ding XF: A potential panel of four-long noncoding
RNA signature in prostate cancer predicts biochemical
recurrence-free survival and disease-free survival. Int Urol
Nephrol. 49:825–835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang H, Cheng S, Wang A, Ma H, Yao B, Qi
C, Liu R, Qi S and Xu Y: Expression of RABEX-5 and its clinical
significance in prostate cancer. J Exp Clin Cancer Res. 33:312014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chiang YT, Wang K, Fazli L, Qi RZ, Gleave
ME, Collins CC, Gout PW and Wang Y: GATA2 as a potential
metastasis-driving gene in prostate cancer. Oncotarget. 5:451–461.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Guo F, Gao X and Wu Y: Golgi
phosphoprotein 3 expression predicts poor prognosis in patients
with prostate cancer undergoing radical prostatectomy. Mol Med Rep.
12:1298–1304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Amaro A, Esposito AI, Gallina A, Nees M,
Angelini G, Albini A and Pfeffer U: Validation of proposed prostate
cancer biomarkers with gene expression data: A long road to travel.
Cancer Metastasis Rev. 33:657–671. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Samra Bou E, Klein B, Commes T and Moreaux
J: Development of gene expression-based risk score in
cytogenetically normal acute myeloid leukemia patients. Oncotarget.
3:824–832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ito H, Mo Q, Qin LX, Viale A, Maithel SK,
Maker AV, Shia J, Kingham P, Allen P, DeMatteo RP, et al: Gene
expression profiles accurately predict outcome following liver
resection in patients with metastatic colorectal cancer. PloS One.
8:e816802013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang W, Gao X, Han Y, Du Y, Liu Q, Wang
L, Tan X, Zhang Q, Liu Y, Zhu Y, et al: Gene expression
profiling-derived immunohistochemistry signature with high
prognostic value in colorectal carcinoma. Gut. 63:1457–1467. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salazar R, Roepman P, Capella G, Moreno V,
Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga
J, et al: Gene expression signature to improve prognosis prediction
of stage II and III colorectal cancer. J Clin Oncol. 29:17–24.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Samra Bou E, Klein B, Commes T and Moreaux
J: Identification of a 20-gene expression-based risk score as a
predictor of clinical outcome in chronic lymphocytic leukemia
patients. Biomed Res Int. 2014:4231742014.PubMed/NCBI
|
|
16
|
Kim SK, Kim SY, Kim JH, Roh SA, Cho DH,
Kim YS and Kim JC: A nineteen gene-based risk score classifier
predicts prognosis of colorectal cancer patients. Mol Oncol.
8:1653–1666. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang ZL, Zhao LJ, Chai L, Zhou SH, Wang
F, Wei Y, Xu YP and Zhao P: Seven LncRNA-mRNA based risk score
predicts the survival of head and neck squamous cell carcinoma. Sci
Rep. 7:3092017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ross-Adams H, Lamb AD, Dunning MJ, Halim
S, Lindberg J, Massie CM, Egevad LA, Russell R, Ramos-Montoya A,
Vowler SL, et al: Integration of copy number and transcriptomics
provides risk stratification in prostate cancer: A discovery and
validation cohort study. EBioMedicine. 2:1133–1144. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim KH, Yang SS, Yoon YS, Lim SB, Yu CS
and Kim JC: Validation of the seventh edition of the american joint
committee on cancer tumor-node-metastasis (AJCC TNM) staging in
patients with stage II and stage III colorectal carcinoma: Analysis
of 2511 cases from a medical centre in korea. Colorectal Dis.
13:e220–e226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Edgar R, Domrachev M and Lash AE: Gene
expression omnibus: NCBI gene expression and hybridization array
data repository. Nucleic Acids Res. 30:207–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Katz S, Irizarry RA, Lin X, Tripputi M and
Porter MW: A summarization approach for affymetrix genechip data
using a reference training set from a large, biologically diverse
database. BMC Bioinformatics. 7:4642006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ishwaran H and Kogalur UB: Consistency of
random survival forests. Stat Probab Lett. 80:1056–1064. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishwaran H, Gerds TA, Kogalur UB, Moore
RD, Gange SJ and Lau BM: Random survival forests for competing
risks. Biostatistics. 15:757–773. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of affymetrix genechip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Z and Pounds S: An R package that
automatically collects and archives details for reproducible
computing. BMC Bioinformatics. 15:1382014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Charlop-Powers Z and Brady SF: Phylogeo:
An R package for geographic analysis and visualization of
microbiome data. Bioinformatics. 31:2909–2911. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Robin X, Turck N, Hainard A, Tiberti N,
Lisacek F, Sanchez JC and Müller M: pROC: An open-source package
for R and S+ to analyze and compare ROC curves. BMC Bioinformatics.
12:772011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim R: Anesthetic technique and cancer
recurrence in oncologic surgery: Unraveling the puzzle. Cancer
Metastasis Rev. 36:159–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Miao Z, Xue B, Shan Y, Weng G and
Shen B: Long non-coding RNAs in urologic malignancies: Functional
roles and clinical translation. J Cancer. 7:1842–1855. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nimse SB, Sonawane MD, Son KS and Kim T:
Biomarker detection technologies and future directions. Analyst.
141:740–755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian X, Zhu X, Yan T, Yu C, Shen C, Hong
J, Chen H and Fang JY: Differentially expressed lncrnas in gastric
cancer patients: A potential biomarker for gastric cancer
prognosis. J Cancer. 8:2575–2586. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bensalah K, Lotan Y, Karam JA and Shariat
SF: New circulating biomarkers for prostate cancer. Prostate Cancer
Prostatic Dis. 11:112–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gupta D and Lis CG: Pretreatment serum
albumin as a predictor of cancer survival: A systematic review of
the epidemiological literature. Nutr J. 9:692010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaushik AK, Shojaie A, Panzitt K, Sonavane
R, Venghatakrishnan H, Manikkam M, Zaslavsky A, Putluri V, Vasu VT,
Zhang Y, et al: Inhibition of the hexosamine biosynthetic pathway
promotes castration-resistant prostate cancer. Net Commun.
7:116122016. View Article : Google Scholar
|
|
35
|
Tang H, Wu Y, Qin Y, Wang H, Jia Y, Yang
S, Luo S and Wang Q: Predictive significance of HMGCS2 for
prognosis in resected Chinese esophageal squamous cell carcinoma
patients. OncoTargets Ther. 10:2553–2560. 2017. View Article : Google Scholar
|
|
36
|
Saraon P, Cretu D, Musrap N, Karagiannis
GS, Batruch I, Drabovich AP, van der Kwast T, Mizokami A, Morrissey
C, Jarvi K and Diamandis EP: Quantitative proteomics reveals that
enzymes of the ketogenic pathway are associated with prostate
cancer progression. Mol Cell Proteomics. 12:1589–1601. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su SG, Yang M, Zhang MF, Peng QZ, Li MY,
Liu LP and Bao SY: miR-107-mediated decrease of HMGCS2 indicates
poor outcomes and promotes cell migration in hepatocellular
carcinoma. Int J Biochem Cell Biol. 91:53–59. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun LN, Zhi Z, Chen LY, Zhou Q, Li XM, Gan
WJ, Chen S, Yang M, Liu Y, Shen T, et al: SIRT1 suppresses
colorectal cancer metastasis by transcriptional repression of
miR-15b-5p. Cancer Lett. 409:104–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Edqvist PH, Huvila J, Forsstrom B, Talve
L, Carpén O, Salvesen HB, Krakstad C, Grénman S, Johannesson H,
Ljungqvist O, et al: Loss of ASRGL1 expression is an independent
biomarker for disease-specific survival in endometrioid endometrial
carcinoma. Gynecol Oncol. 137:529–537. 2015. View Article : Google Scholar : PubMed/NCBI
|