Introduction
Pancreatic cancer (PC) is the fourth leading cause
of cancer mortality globally with an estimated 5-year survival rate
of only ~7% in 2010 (1,2), and its clinical course is aggressive.
When diagnosed, 80–95% of patients present locally advanced or
metastatic disease (3) as symptoms
are unobservable at the early stages of PC (4). Thus, few patients undergo surgical
resection at an early stage, whilst the majority of patients are
treated with chemotherapy, which often combines gemcitabine (GEM)
with other chemotherapeutics, at advanced stages (5,6).
GEM, as an antitumor drug, is used for the treatment
of advanced PC (7). Chemotherapy
always results in toxicity, yet counteracting this by using low
concentrations of GEM limits its efficacy, resulting in reduced
overall survival rates of patients with PC (8,9).
Therefore, methods for enhancing GEM efficacy must be explored. It
has been demonstrated that heating a tumor up to a temperature of
~42°C may synergistically enhance the antitumor effect of GEM
(10,11); however the underlying mechanism
remains unclear.
In the present study, the PC cell line SW1990 was
used to investigate the effect of hyperthermia on GEM antitumor
activity. When treated with hyperthermia at a temperature of 42°C,
SW1990 cells exposed to GEM demonstrated decreased viability and
increased apoptosis with the upregulation of apoptosis-activating
genes. Additionally, a higher number of cells treated using
hyperthermia combined with GEM were arrested in the S-phase stage
of the cell cycle in comparison with the control group treated with
GEM alone, a process in which survivin may be involved (12). Furthermore, the underlying mechanism
of apoptosis caused by hyperthermia combined with GEM in PC cells
was examined, and it was revealed that reactive oxygen species
(ROS)/c-Jun N-terminal kinase (JNK) signaling was involved.
Materials and methods
Cell culture
The human PC SW1990 cell line was obtained from the
Cell Resource Centre at Peking Union Medical College (Beijing,
China). Cells were cultured in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (Lonza Group, Ltd., Basel, Switzerland), 5 mM
L-glutamine, 5 mM non-essential amino acids, 100 U/ml
penicillin-streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.), in a humidified 5% CO2 incubator at 37°C.
Viability assay
Cell viability was evaluated using an MTT assay
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). A total of
1×104 SW1990 cells were seeded into each well of a
96-well plate in 100 µl RPMI-1640 medium and incubated with 0, 1,
5, 10 and 20 µM GEM (Selleck Chemicals, Houston, TX, USA) for 12,
24, 48 and 72 h at 37°C in a 5% CO2 incubator. Then,
cells were incubated with 20 µl of 5 mg/ml MTT (Sigma-Aldrich, St.
Louis, MO, USA) at 37°C for 4 h, and then cells were lysed at room
temperature for 10 min following the addition of 200 µl dimethyl
sulfoxide (DMSO; OriGene Technologies, Inc., Rockville, MD, USA),
which was used to dissolve the formazan crystals. Absorbance was
measured at 490 nm using a Rainbow microplate reader (Tecan Group,
Ltd., Männedorf, Switzerland). Cell viability was expressed as a
percentage of the untreated control.
Apoptosis assay
A total of 1×106 SW1990 cells were seeded
on 60 mm dishes and grown to 80% confluence, and then treated with
or without hyperthermia at 42°C for 90 min in the presence or
absence of 10 µM JNK inhibitor Sp600125 (Selleck Chemicals) or 5 mM
ROS inhibitor N-acetyl-L-cysteine (NAC; Selleck Chemicals),
followed by 5 µM GEM treatment at 37°C for 24 h. Cells were
collected, washed three times with PBS and resuspended in PBS.
Apoptosis was analyzed using an Annexin V-fluorescein
isothiocyanate (FITC)/propidium iodide (PI) detection kit (cat. no.
556547; BD Biosciences, Franklin Lakes, NJ, USA), according to the
manufacturer's protocol. Briefly, the cells were washed three times
with PBS and then incubated for 15 min at room temperature in the
dark in 100 µl of 1X Annexin binding buffer containing 5 µl Annexin
V-FITC and 2 µl PI. The quantity of phosphatidylserine on the outer
surface of the plasma membrane (a biochemical alteration unique to
the membranes of apoptotic cells) and the quantity of PI, a dye
that easily enters dead cells or cells in the late stages of
apoptosis and binds DNA, but does not bind to the plasma membrane
of viable cells, was detected. Fluorescence was detected using a
FACSCalibur flow cytometer by fluorescence-activated cell sorting
(FACS; Accuri C6 Flow Cytometer; BD Biosciences) analysis, and data
were analyzed using CellQuestPro version 5.2 software (BD
Biosciences). Cells with phosphatidylserine on their surface were
considered to be apoptotic (early apoptotic cells in addition to
late apoptotic cells). Cell cycle distribution was analyzed using
FACS and CFlow Plus software version 1.0 (BD Biosciences) (13). Briefly, 1×105 cells were
seeded into 12-well plates to reach 80% confluence by the next day.
Cells were treated with or without hyperthermia at 42°C for 90 min,
followed by 5 µM GEM treatment at 37°C for 24 h. Cells were
collected, washed three times with PBS and incubated in staining
buffer (0.1% sodium citrate, 0.1% Triton X-100 and 50 µg/ml
propidium iodide) at 4°C for 1 h. Cell cycle phases were determined
according to the content of DNA in cells, as follows: Phase
G1, DNA replication has not started and the content of
DNA is the lowest; phase G0 cells contain the least
amount of DNA; phase S, cells replicate and progress from 1×DNA to
2×DNA; phase G2, DNA doubles to 2×DNA; phase M, cell
division and 2×DNA in cells. DNA was presented using PI staining
and thus cell cycle phases were determined. Cell cycle distribution
was measured using CFlow Plus software version 1.0.
Morphological analysis of cells
A total of 1×106 SW1990 cells were seeded
into four culture flasks at a density of 1×106/ml and
allowed to reach 75–80% confluence. PC SW1990 cells were treated
with or without hyperthermia at 42°C for 90 min, followed by 5 µM
GEM treatment at 37°C for 24 h. The control and treated cells were
dislodged using EDTA/bovine serum albumin solution, made up into a
suspension and washed twice with PBS. The cell fixation was
performed as previously described by Hobot et al (14). Briefly, each cell suspension was mixed
rapidly with an equal volume of 2% glutaraldehyde solution in 0.1 M
cacodylate buffer and fixed at 4°C for 2 h. The pellet of cells,
following centrifugation at 1,000 × g at 4°C for 5 min, was
resuspended twice in an excess of 0.1 M cacodylate buffer for a
15-min interval. Cells were resuspended in 1% osmium tetroxide in
0.1 M cacodylate buffer and fixed at 4°C for 30 min, and then
centrifuged at 1,000 × g at 4°C for 5 min. Then, 0.1 M cacodylate
buffer was added and fixed at 4°C for 30 min, and then centrifuged
at 1,000 × g at 4°C for 5 min. To the pellet, an equal volume of 2%
agar in 0.1 M sodium cacodylate buffer (heated to 40°C) was added,
and then the agar and cells were mixed rapidly. Following cooling,
the pellet was cut with a razor blade into 2-mm cubic pieces and
stored in 2% glutaraldehyde solution at 4°C until further
processing and viewing using a transmission electron microscope
(OPTON EM 900; Zeiss AG, Oberkochen, Germany; magnification,
×6,000).
Western blot analysis
A total of 1×106 SW1990 cells were seeded
on 60 mm dishes and grown to 80% confluence. Cells were collected
and washed twice with PBS, and then lysed 4°C for 15 min using
lysis buffer [50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1% NP40, 150 mM
NaCl, 10 mM NaF, 1 mM Na3VO4)] containing a
protease inhibitor cocktail (Roche Molecular Diagnostics,
Branchburg, NJ, USA). Following centrifugation at 12,000 × g for 10
min at 4°C, the supernatant was collected and quantified using a
bicinchoninic acid quantification kit (Beyotime Institute of
Biotechnology, Haimen, China). A total of 50 µg protein per lane
samples were separated via 10% SDS-PAGE (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) and transferred to polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). The membranes were
blocked using 5% non-fat dried milk in Tris-buffered saline with
0.05% Tween-20 for 1 h at room temperature, and incubated with the
following specific primary antibodies overnight at 4°C: Mouse
monoclonal immunoglobulin G (IgG) for B-cell lymphoma 2 (Bcl-2;
1:500; cat no. sc7382), rabbit polyclonal IgG for Bcl-2-associated
X protein (Bax; 1:500; cat no. sc493), mouse monoclonal IgG for
β-actin (1:1,000; cat no. sc47778), rabbit polyclonal IgG for
Survivin (1:500; cat no. sc-10811; all Santa Cruz Biotechnology,
Inc., Dallas, CA, USA), rabbit polyclonal IgG for cleaved-caspase-3
(1:1,000; cat no. 9661S) and for cleaved-caspase-9 (1:1,000; cat
no. 9505; both Cell Signaling Technology, Inc., Danvers, MA, USA).
Rabbit polyclonal antibodies specific for phosphorylated (p-)JNK
(1:1,000; cat no. 9251) and JNK (1:1,000; cat no. 9252; both Cell
Signaling Technology, Inc.) were used for detection, followed by
the horseradish peroxidase-conjugated goat anti-mouse secondary
antibody (1:2,000; cat no. sc-2005) and anti-rabbit IgG antibody
(1:2,000; cat no. sc-2004) (both from Santa Cruz Biotechnology,
Inc.) for 2 h at room temperature. Enhanced
chemiluminescence-detecting reagent (GE Healthcare, Chicago, IL,
USA) was used for development. The gray value of the targeted bands
was analyzed using QuantityOne software version 4.6.2 (Bio-Rad
Laboratories, Inc.) subsequent to incubation; β-actin was detected
as the internal reference.
Detection of ROS production
A total of 5×105 SW1990 cells were seeded
in 6-well plates overnight and then treated with or without
hyperthermia at 42°C for 90 min, followed by 5 µM GEM treatment at
37°C for 24 h. Following treatment, the cells were collected and
stained with 10 µM 2′,7′-dichlorofluorescin diacetate at 37°C for
30 min in the dark. Fluorescence was detected using a FACSCalibur
flow cytometer (BD Biosciences) using a FITC channel, and data were
analyzed using CellQuestPro version 5.2 software.
Statistical analysis
Data were obtained from at least three experiments.
Statistical analysis was preformed using SPSS 13.0 for Windows
(SPSS, Inc., Chicago, IL, USA). Values are expressed as the mean ±
standard error of the mean. One-way analysis of variance was used
to assess differences between groups. The Duncan method was
employed for pairwise comparison and was followed by Bonferroni's
correction. P<0.05 was considered to indicate a statistically
significant difference.
Results
Hyperthermia increases the inhibition
of GEM on the viability of PC SW1990 cells
To investigate the effect of GEM on PC cell
viability, the human SW1990 PC cell line with high metastatic
ability was used in the present study. SW1990 cells were exposed to
various concentrations of GEM for different durations (Fig. 1A). An MTT assay was performed in order
to evaluate cell viability. The results revealed that GEM treatment
inhibited cellular viability in a dose-dependent manner at 12, 24,
48 and 72 h treatment durations and that the viability decrease
trends were similar amongst the 24, 48 and 72 h groups, which
demonstrated a sharper decrease compared with the 12 h group
(Fig. 1A). Of the treatment
concentrations, 5 µM GEM exerted a slight inhibitory effect on the
viability of SW1990 cells and was selected to be used in subsequent
experiments due to a lower cytotoxicity compared with relatively
high concentrations of GEM. SW1990 cells were treated with
different temperatures (40°C, 42°C or 44°C) for 90 min with or
without GEM. The results revealed that 42°C treatment alone did not
significantly result in heat-induced cytotoxicity compared with the
44°C alone treatment, whereas the 42°C hyperthermia treatment may
mostly enhance the role of GEM on inhibiting cell viability
relative to 40°C treatment (data not shown). Hence, the temperature
of the hyperthermia treatment was selected to be 42°C. Following
this, SW1990 cells were treated with mild hyperthermia at 42°C for
30, 60 or 90 min with and without GEM, followed by GEM treatment
for 24 h at 37°C, and then cell viability was assessed. The results
indicated that 42°C hyperthermia for 30, 60 and 90 min
significantly enhanced the inhibitory effect of GEM on cell
viability compared with GEM alone (P<0.05, P<0.01 and
P<0.01, respectively), whilst hyperthermia treatment alone
without GEM resulted in low heat-induced toxicity and did not
significantly affect cell viability compared with GEM alone
(Fig. 1B). These results suggested
that hyperthermia served a promoting function on the inhibitory
effect of GEM on PC cell viability.
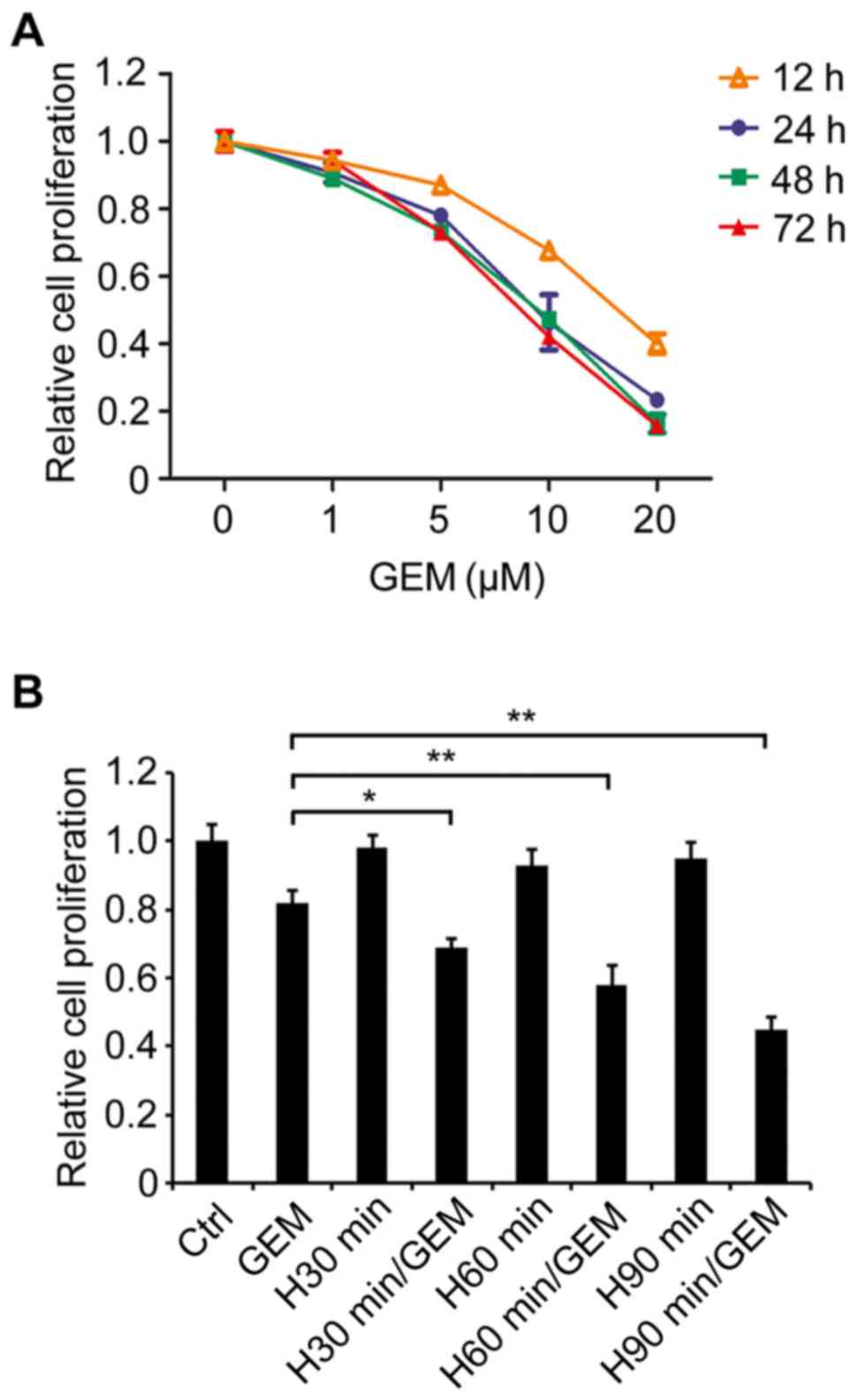 | Figure 1.Hyperthermia enhances the inhibitory
effect of GEM on viability in SW1990 cells. (A) SW1990 cells were
treated with 0, 1, 5, 10 and 20 µM GEM for 12, 24, 48 and 72 h.
Cell viability was assessed using an MTT assay. (B) Cells were
treated with hyperthermia at 42°C for different durations (30, 60
or 90 min), and then were treated using 5 µM GEM for 24 h. Cell
viability was assessed using an MTT assay. *P<0.05 and
**P<0.01 with comparisons shown by lines. Ctrl, untreated group;
GEM, treated with gemcitabine alone; H30 min, treated with
hyperthermia alone for 30 min; H30 min/GEM, treated with
hyperthermia for 30 min in addition to gemcitabine; H60 min,
treated with hyperthermia alone for 60 min; H60 min/GEM, treated
with hyperthermia for 60 min in addition to gemcitabine; H90 min,
treated with hyperthermia alone for 90 min; H90 min/GEM, treated
with hyperthermia for 90 min in addition to gemcitabine. |
Hyperthermia enhances GEM-induced
apoptosis in SW1990 cells
To confirm the effect of hyperthermia on GEM-induced
apoptosis in PC SW1990 cells, an Annexin V-FITC/PI assay was
performed. Cells with phosphatidylserine on their surface
(indicating that they were either early apoptotic cells or late
apoptotic cells) were considered to be apoptotic. The result
revealed that hyperthermia treatment alone did not significantly
increase the cell apoptosis compared with the untreated control
cells, whilst GEM-treated cells revealed significantly upregulated
apoptosis compared with the control group (P<0.01). However,
hyperthermia significantly increased GEM-induced cell apoptosis
compared with the cells treated with GEM alone (P<0.01; Fig. 2A). Additionally, apoptotic bodies
could be clearly observed in PC cells treated with hyperthermia
combined with GEM (Fig. 2B).
Meanwhile, SW1990 cells at the G0/G1 stage, S
stage and G2/M stage were analyzed using flow cytometry
(Fig. 2C). The results indicated that
the percentage of PC cells at the S-phase stage was significantly
increased following treatment with 42°C hyperthermia combined with
GEM compared with cells treated with GEM alone (P<0.05), whilst
the percentage of PC cells at the G2/M stage was
significantly decreased following treatment with 42°C hyperthermia
combined with GEM compared with cells treated with GEM alone
(P<0.05), implying that hyperthermia combined with GEM
potentially blocked cells at the S-phase stage of cell cycle and
thus accelerated apoptosis (Fig. 2C).
To further determine the effect of hyperthermia on GEM-induced cell
apoptosis, western blot analysis was performed to detect the levels
of apoptosis-associated proteins. The results revealed that the
expression of the anti-apoptotic protein Bcl-2 in GEM-treated cells
was downregulated when combined with hyperthermia treatment. By
contrast, the pro-apoptosis proteins Bax, cleaved caspase-3 and
cleaved caspase-9 demonstrated a notable increase in protein
expression following treatment with hyperthermia combined with GEM
compared with GEM treatment alone (Fig.
2D). The data indicated that hyperthermia increased the
inhibitory effect of GEM on the viability of SW1990 cells,
potentially by stimulating apoptosis, which may be mediated by the
modulation of Bax, Bcl-2, cleaved caspase-3 and cleaved caspase-9
apoptotic factors.
 | Figure 2.Hyperthermia increases GEM-induced
cell apoptosis. (A) SW1990 cells were treated with hyperthermia at
42°C for 90 min, and then were treated with 5 µM GEM for 24 h. Cell
apoptosis was assessed using an Annexin V-fluorescein
isothiocyanate/propidium iodide assay. (B) Morphological changes of
SW1990 cells were observed under a transmission electron microscope
(magnification, ×6,000), including the following groups: (a) Normal
cells, (b) cells treated with hyperthermia alone, (c) cells treated
with GEM alone and (d) cells treated with hyperthermia combined
with GEM. (C) Cell cycle distributions of PC cells were detected
using flow cytometry. (D) Protein expression of the
apoptosis-associated proteins, B-cell lymphoma 2, Bcl-2-associated
X protein, cleaved caspase-3 and cleaved caspase-9 were evaluated
using western blot analysis using respective antibodies. β-actin
was detected as internal reference. *P<0.05 and **P<0.01 with
comparisons shown by lines. NS, no significance; Ctrl, untreated
group; H, treated with hyperthermia alone; GEM, treated with
gemcitabine alone; H+GEM, treated with hyperthermia combined with
gemcitabine. |
Hyperthermia enhances GEM-induced
apoptosis of SW1990 cells through ROS/JNK signaling
To further investigate the mechanism of hyperthermia
enhancing GEM-induced apoptosis in PC cells, western blot analysis
was performed to detect the protein expression of survivin, a
protein specifically expressed at the G2/M stage
(15), JNK and p-JNK, and a flow
cytometry assay was used to detect the generation of ROS. It was
revealed that the expression of survivin was downregulated in cells
treated with 42°C hyperthermia combined with GEM treated cells
compared with that in cells treated with GEM alone (Fig. 3A), suggesting that hyperthermia
combined with GEM may affect the cell cycle by downregulating
survivin. ROS/JNK signaling has been reported to be activated in a
hyperthermic perfused amphibian heart and thus mediate cell
apoptosis (16). In the present
study, p-JNK was demonstrated to be notably upregulated following
treatment with hyperthermia combined with GEM, relative to GEM
treatment alone (Fig. 3A).
Furthermore, it was revealed that the release of ROS during
apoptosis in cells treated with hyperthermia in combination with
GEM was significantly elevated compared with that in cells treated
with GEM alone (P<0.01; Fig. 3B).
These data suggested that ROS/JNK signaling may be involved in the
effects of hyperthermia on GEM-induced apoptosis in SW1990
cells.
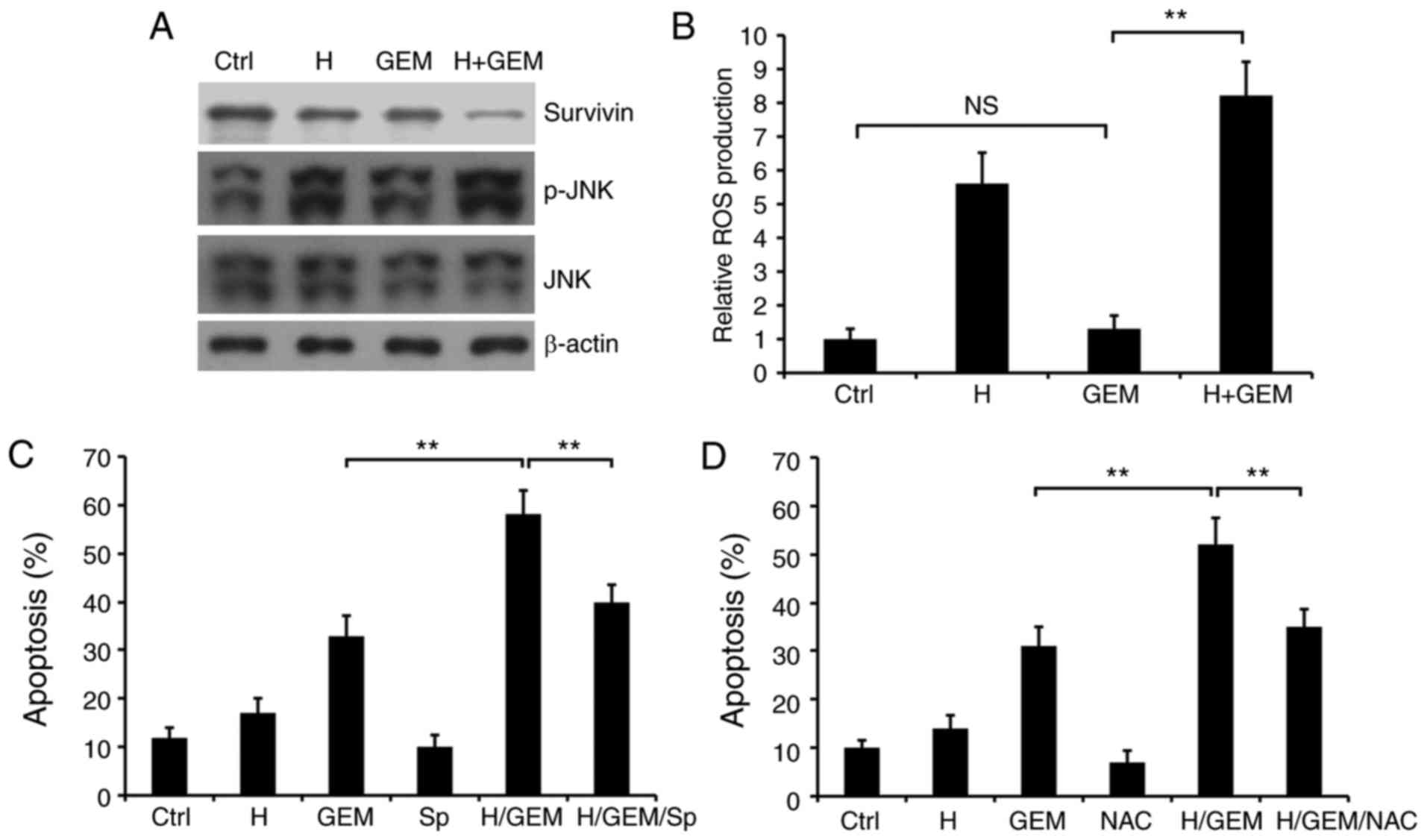 | Figure 3.Involvement of survivin and ROS/JNK
signaling in hyperthermia enhancing GEM-induced cell apoptosis. (A)
Western blot analysis of survivin, JNK and p-JNK. β-actin was
detected as internal reference. (B) Cells were treated with or
without 42°C hyperthermia for 90 min, followed by treatment with 5
µM GEM for 24 h, and then ROS production was analyzed using
2′,7′-dichlorodihydrofluorescein diacetate staining. (C) SW1990
cells were treated with or without hyperthermia for 90 min at 42°C
in the presence or absence of the JNK inhibitor Sp600125 (10 µM),
and then were treated with 5 µM GEM for 24 h. Apoptosis was
measured using an Annexin V-FITC/PI assay. (D) SW1990 cells were
treated with or without hyperthermia for 90 min at 42°C in the
presence or absence of the ROS inhibitor NAC (5 mM), and then were
treated with 5 µM GEM for 24 h. Apoptosis was measured using an
Annexin V-FITC/PI assay. **P<0.01 with comparisons shown by
lines. Ctrl, untreated normal cells; H, treated with hyperthermia
alone; GEM, treated with gemcitabine alone; NAC, treated with
N-acetyl-L-cysteine alone; H/GEM, treated with hyperthermia in
addition to gemcitabine; H/GEM/NAC, treated with hyperthermia in
the presence of N-acetyl-L-cysteine in addition to gemcitabine; Sp,
treated with Sp600125 alone H/GEM/Sp, treated with hyperthermia in
the presence of Sp600125 plus GEM; NS, no significance; ROS,
reactive oxygen species; JNK, c-Jun-N-terminal kinase; p-,
phosphorylated; FITC/PI, fluorescein isothiocyanatae/propidium
iodide. |
To verify the function of ROS/JNK signaling in
hyperthermia increasing GEM-induced apoptosis of PC SW1990 cells, a
JNK inhibitor, Sp600125, was introduced in order to suppress JNK
activation. It was revealed that Sp600125 significantly decreased
the apoptotic effect of hyperthermia combined with GEM on SW1990
cells (P<0.01; Fig. 3C),
indicating a positive effect of JNK on PC cell apoptosis.
Furthermore, the ROS inhibitor NAC was applied to SW1990 cells in
addition to hyperthermia treatment combined with GEM, and revealed
that NAC was also able to significantly decrease the apoptotic
effect of hyperthermia combined with GEM on SW1990 cells
(P<0.01; Fig. 3C), which implies a
function of ROS in PC cell apoptosis mediated by hyperthermia
treatment combined with GEM. In summary, it was demonstrated that
hyperthermia enhanced GEM-induced cell apoptosis, potentially
through ROS/JNK signaling.
Discussion
PC is an aggressive type of human cancer with a low
5-year survival rate (1,17). One of the drugs used for the treatment
of advanced PC is GEM (7). Although
GEM is used as a first-line chemotherapeutic treatment for advanced
PC, its efficacy still requires improvement (7,8,18).
Hyperthermia treatment applied in a clinical setting
includes whole body hyperthermia, local hyperthermia and
intracellular hyperthermia (19,20).
Infrared radiation and extracorporeal circulation heating are
commonly used for whole body hyperthermia; microwave thermotherapy,
super-sound focusing thermotherapy, radiofrequency thermotherapy
and endogenic field thermotherapy are commonly used for local
hyperthermia (19,20). In the present study, cells were
treated with mild hyperthermia at 42°C in a CO2
incubator and it was revealed that mild hyperthermia at 42°C
treatment may effectively enhance the sensitivity of PC SW1990
cells to GEM by inhibiting cell viability.
Apoptosis serves a critical function in
chemotherapies in various cancer types (21). Whether hyperthermia increases the
inhibitory effect of GEM on SW1990 cells viability by inducing
apoptosis still requires further study. It has been demonstrated
that the overexpression of heat shock protein 27 (HSP27) increases
GEM sensitivity in PC cells through S-phase arrest and apoptosis
(22) and HSP27 expression may be
upregulated by hyperthermia in osteosarcoma cells and PC cells
(22,23). Therefore, it may be that HSP27 is
involved in the effect of hyperthermia on the sensitivity of PC
SW1990 cells to GEM, which requires further investigation.
Additionally, in the present study, it was revealed that
hyperthermia increased GEM-induced cell apoptosis. To explore the
molecular mechanism underlying the effect of hyperthermia combined
with GEM on the inhibition of viability of and the induction of
apoptosis in SW1990 cells, the present study detected the
expression levels of apoptosis-associated proteins (including
Bcl-2, Bax, cleaved caspase-3 and cleaved caspase-9).
Anti-apoptosis protein Bcl-2 and pro-apoptosis protein Bax, which
are Bcl-2 family proteins, regulate mitochondrial permeability to
affect apoptosis through an intrinsic pathway (24). Cleaved caspase-3 and caspase-9, which
are known as mature or activated caspase-3 and caspase-9, are
critical mediators of cell apoptosis (25). All caspases require cleavage adjacent
to aspartates to liberate one large and one small subunit, which
associate with an a2b2 tetramer to form the active enzyme (26). Caspase enzymes are able to cleave the
death substrate poly ADP-ribose polymerase to a specific 85 kDa
form observed during apoptosis (25).
The results of the expression of these apoptosis-associated
proteins in the present study were consistent with the promotion of
apoptosis and indicated that hyperthermia combined with GEM induced
apoptosis in SW1990 cells by downregulating the expression of Bcl-2
and upregulating the expression of Bax, caspase 3 and caspase
9.
Survivin is a type of anti-apoptosis protein, which
is specifically expressed in cells at the G2/M stage
(12). It has been reported that
survivin knockdown PC stem cells demonstrated a greater sensitivity
to GEM compared with normal PC stem cells, and GEM-induced cell
death was significantly promoted (27), which suggested an association between
survivin and the effect of hyperthermia on GEM-induced apoptosis.
In the present study, it was revealed that the majority of PC cells
treated with hyperthermia combined with GEM were arrested at the
S-phase stage instead of entering the G2/M stage, and
possessed a larger percentage of apoptotic cells compared with the
control group. Additionally, the downregulated expression of
survivin may indicate that hyperthermia treatment combined with GEM
affects the cycle of PC cells by downregulating survivin, thereby
accelerating the apoptosis of PC cells.
ROS are active forms of oxygen, produced as
by-products of cellular metabolism (28). Previous studies have revealed that the
slight upregulation of ROS may enhance cell viability, whereas
excessive amounts of ROS may result in cell apoptosis (29). A majority of cancer therapeutic
methods damage cells due to the generation of a substantial
quantity of ROS resulting in cell death (30,31). ROS
function as intracellular messengers and affect protein structure
and function by oxidizing crucial amino acid residues (32). It was hypothesized as to whether
hyperthermia combined with GEM resulted in redundant ROS thereby
triggering PC cell apoptosis. It was revealed that ROS production
was significantly increased by hyperthermia treatment combined with
GEM compared with GEM treatment alone, suggesting that ROS may be
associated with cell apoptosis induced by hyperthermia combined
with GEM treatment. It is widely known that ROS may affect numerous
signaling pathways (33).
Mitogen-activated protein kinase (MAPK) signaling transduction is
one of the signaling pathways modulated by ROS (34,35).
Extracellular signal-regulated kinases (ERKs), p38 MAPKs and JNKs
are three critical protein kinases involved in MAPK signaling
pathways (36). ERK cascades are
commonly associated with cell survival and may be activated by
growth or survival factors. Whereas, p38 MAPK and JNK pathways are
often associated with pro-apoptotic effects (37,38). ROS
has been demonstrated to be able to activate apoptosis
signal-regulated kinase 1 and induce the activation of the JNK
cascade and apoptosis (39,40). JNK signaling initiates apoptosis
potentially through two different methods. One method is to
directly regulate apoptosis-associated proteins including Bcl-2
proteins (41), whereas an indirect
method is to translocate to the nucleus where it regulates the
activity of transcription factors, including c-Jun, and then
regulates the expression of apoptotic genes (42,43). As a
member of MAPK family, JNK is involved in cell apoptosis (44,45). In
the present study, the expression of JNK and p-JNK were detected,
and demonstrated upregulation following treatment with hyperthermia
combined with GEM compared with GEM treatment alone. Additionally,
the upregulation of p-JNK is involved in the activation of JNK
signaling (46). Furthermore, the JNK
inhibitor, Sp600125, was used, and significantly decreased the
apoptosis of PC SW1990 cells compared with cells treated with
hyperthermia and GEM. Therefore, it was considered that
hyperthermia combined with GEM enhanced PC cell apoptosis through
JNK signaling. Finally, a ROS inhibitor, NAC, was introduced in
order to verify the association between JNK and PC cell apoptosis.
It was concluded that hyperthermia increased the GEM sensitivity of
PC SW1990 cells by inhibiting cell growth and inducing apoptosis
via ROS/JNK signaling, which may be significant for improving
therapy for patients with PC. However, the exact association
between hyperthermia and GEM mediated ROS production, apoptosis and
activation of JNK signal transduction requires further
investigation.
Acknowledgements
Not applicable.
Funding
This project was supported by Hangzhou Science and
Technology Development Plan project (grant no. 20162013A01).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
HJ and SM made major contributions in the conception
and design of the research and revision of the manuscript for
important intellectual content and the funding was obtained by SM.
Acquisition of data was performed by YZ and JY. YZ and XZ were the
major contributors in the analysis and interpretation of data and
statistical analysis. Drafting the manuscript and revising it
critically for important intellectual content were performed by
HJ.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Worni M, Guller U, White RR, Castleberry
AW, Pietrobon R, Cerny T, Gloor B and Koeberle D: Modest
improvement in overall survival for patients with metastatic
pancreatic cancer: A trend analysis using the surveillance,
epidemiology, and end results registry from 1988 to 2008. Pancreas.
42:1157–1163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ottaiano A, Capozzi M, De Divitiis C, De
Stefano A, Botti G, Avallone A and Tafuto S: Gemcitabine
mono-therapy versus gemcitabine plus targeted therapy in advanced
pancreatic cancer: A meta-analysis of randomized phase III trials.
Acta Oncol. 56:377–383. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bednar F and Simeone DM: Recent advances
in pancreatic surgery. Curr Opin Gastroenterol. 30:518–523. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ghaneh P, Smith R, Tudor-Smith C, Raraty M
and Neoptolemos JP: Neoadjuvant and adjuvant strategies for
pancreatic cancer. Eur J Surg Oncol. 34:297–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Viaud J, Brac C, Artru P, Le Pabic E,
Leconte B, Bodère A, Pracht M, Le Sourd S, Edeline J and Lièvre A:
Gemcitabine as second-line chemotherapy after Folfirinox failure in
advanced pancreatic adenocarcinoma: A retrospective study. Dig
Liver Dis. 49:692–696. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Massari F, Santoni M, Ciccarese C,
Brunelli M, Conti A, Santini D, Montironi R, Cascinu S and Tortora
G: Emerging concepts on drug resistance in bladder cancer:
Implications for future strategies. Crit Rev Oncol Hematol.
96:81–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Immordino ML, Brusa P, Rocco F, Arpicco S,
Ceruti M and Cattel L: Preparation, characterization, cytotoxicity
and pharmacokinetics of liposomes containing lipophilic gemcitabine
prodrugs. J Control Release. 100:331–346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim CE, Lim SK and Kim JS: In vivo
antitumor effect of cromolyn in PEGylated liposomes for pancreatic
cancer. J Control Release. 157:190–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Maeda H, Wu J, Sawa T, Matsumura Y and
Hori K: Tumor vascular permeability and the EPR effect in
macromolecular therapeutics: A review. J Control Release.
65:271–284. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kirui DK, Celia C, Molinaro R, Bansal SS,
Cosco D, Fresta M, Shen H and Ferrari M: Mild hyperthermia enhances
transport of liposomal gemcitabine and improves in vivo therapeutic
response. Adv Healthc Mater. 4:1092–1103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shamir ER and Ewald AJ: Adhesion in
mammary development: Novel roles for E-cadherin in individual and
collective cell migration. Curr Top Dev Biol. 112:353–382. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nicoletti I, Migliorati G, Pagliacci MC,
Grignani F and Riccardi C: A rapid and simple method for measuring
thymocyte apoptosis by propidium iodide staining and flow
cytometry. J Immunol Methods. 139:271–279. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hobot J, Carlemalm E, Villiger W and
Kellenberger E: Periplasmic Gel: New concept resulting from the
reinvestigation of bacterial cell envelope ultrastructure by new
methods. J Bacteriol. 160:143–152. 1984.PubMed/NCBI
|
|
15
|
Li E, Ambrosini G, Chu EY, Plescia J,
Tognin S, Marchisio PC and Altieri DC: Control of apoptosis and
mitotic spindle checkpoint by survivin. Nature. 396:580–584. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gaitanaki C, Mastri M, Aggeli IK and Beis
I: Differential roles of p38-MAPK and JNKs in mediating early
protection or apoptosis in the hyperthermic perfused amphibian
heart. J Exp Biol. 211:2524–2532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Sousa Cavalcante L and Monteiro G:
Gemcitabine: Metabolism and molecular mechanisms of action,
sensitivity and chemoresistance in pancreatic cancer. Eur J
Pharmacol. 741:8–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
van der Zee J: Heating the patient: A
promising approach? Ann Oncol. 13:1173–1184. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Falk MH and Issels RD: Hyperthermia in
oncology. Int J Hyperthermia. 17:1–18. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
San-Miguel ICB, Vallejo D, Álvarez M,
Prieto J, González-Gallego J and Tuñón MJ: Melatonin modulates the
autophagic response in acute liver failure induced by the rabbit
hemorrhagic disease virus. J Pineal Res. 56:313–321. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo Y, Ziesch A, Hocke S, Kampmann E, Ochs
S, De Toni EN, Göke B and Gallmeier E: Overexpression of heat shock
protein 27 (HSP27) increases gemcitabine sensitivity inpancreatic
cancer cells through S-phase arrest and apoptosis. J Cell Mol Med.
19:340–350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakajima K, Yanagawa T, Watanabe H and
Takagishi K: Hyperthermia reduces migration of osteosarcoma by
suppression of autocrine motilityfactor. Oncol Rep. 28:1953–1958.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin S, Shen JN, Wang J, Huang G and Zhou
JG: Oridonin induced apoptosis through Akt and MAPKs signaling
pathways in human osteosarcoma cells. Cancer Biol Ther. 6:261–268.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera
S and Ikejima T: Oridonin induces G2/M arrest and apoptosis via
activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK
survival pathway in murine fibrosarcoma L929 cells. Arch Biochem
Biophys. 490:70–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi Y: Caspase activation: Revisiting the
induced proximity model. Cell. 117:855–858. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Takeda H, Okada M, Suzuki S, Kuramoto K,
Sakaki H, Watarai H, Sanomachi T, Seino S, Yoshioka T and Kitanaka
C: Rho-associated protein kinase (ROCK) inhibitors inhibit survivin
expression and sensitize pancreatic cancer stem cells to
gemcitabine. Anticancer Res. 36:6311–6318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Renschler MF: The emerging role of
reactive oxygen species in cancer therapy. Eur J Cancer.
40:1934–1940. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Toler SM, Noe D and Sharma A: Selective
enhancement of cellular oxidative stress by chloroquine:
Implications for the treatment of glioblastoma multiforme.
Neurosurg Focus. 21:E102006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scandalios JG: Oxidative stress: Molecular
perception and transduction of signals triggering antioxidant gene
defenses. Braz J Med Biol Res. 38:995–1014. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct 2011. 7926392011.
|
|
33
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2017. View Article : Google Scholar
|
|
34
|
Liu QR, Liu JM, Chen Y, Xie XQ, Xiong XX,
Qiu XY, Pan F, Liu D, Yu SB and Chen XQ: Piperlongumine inhibits
migration of glioblastoma cells via activation of ROS-dependent p38
and JNK signaling pathways. Oxid Med Cell Longev. 2014:6537322014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Goldsmith EJ, Min X, He H and Zhou T:
Structural studies of MAP kinase cascade components. Methods Mol
Biol. 661:223–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lencioni R, Chen XP, Dagher L and Venook
AP: Treatment of intermediate/advanced hepatocellular carcinoma in
the clinic: How can outcomes be improved? Oncologist. 15 Suppl
4:S42–S52. 2010. View Article : Google Scholar
|
|
38
|
Katz M, Amit I and Yarden Y: Regulation of
MAPKs by growth factors and receptor tyrosine kinases. Biochim
Biophys Acta. 1773:1161–1176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tobiume K, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Takeda K, Noguchi T, Naguro I and Ichijo
H: Apoptosis signal-regulating kinase 1 in stress and immune
response. Annu Rev Pharmacol Toxicol. 48:199–225. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu CC and Bratton SB: Regulation of the
intrinsic apoptosis pathway by reactive oxygen species. Antioxid
Redox Signal. 19:546–558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Suzuki S, Okada M, Shibuya K, Seino M,
Sato A, Takeda H, Seino S, Yoshioka T and Kitanaka C: JNK
suppression of chemotherapeutic agents-induced ROS confers
chemoresistance on pancreatic cancer stem cells. Oncotarget.
6:458–470. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sun ZL, Dong JL and Wu J: Juglanin induces
apoptosis and autophagy in human breast cancer progression via
ROS/JNK promotion. Biomed Pharmacother. 85:303–312. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Genet Dev. 12:14–21. 2002.
View Article : Google Scholar : PubMed/NCBI
|














